
Sperm Transcriptome Analysis Accurately Reveals Male Fertility Potential in Livestock

 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
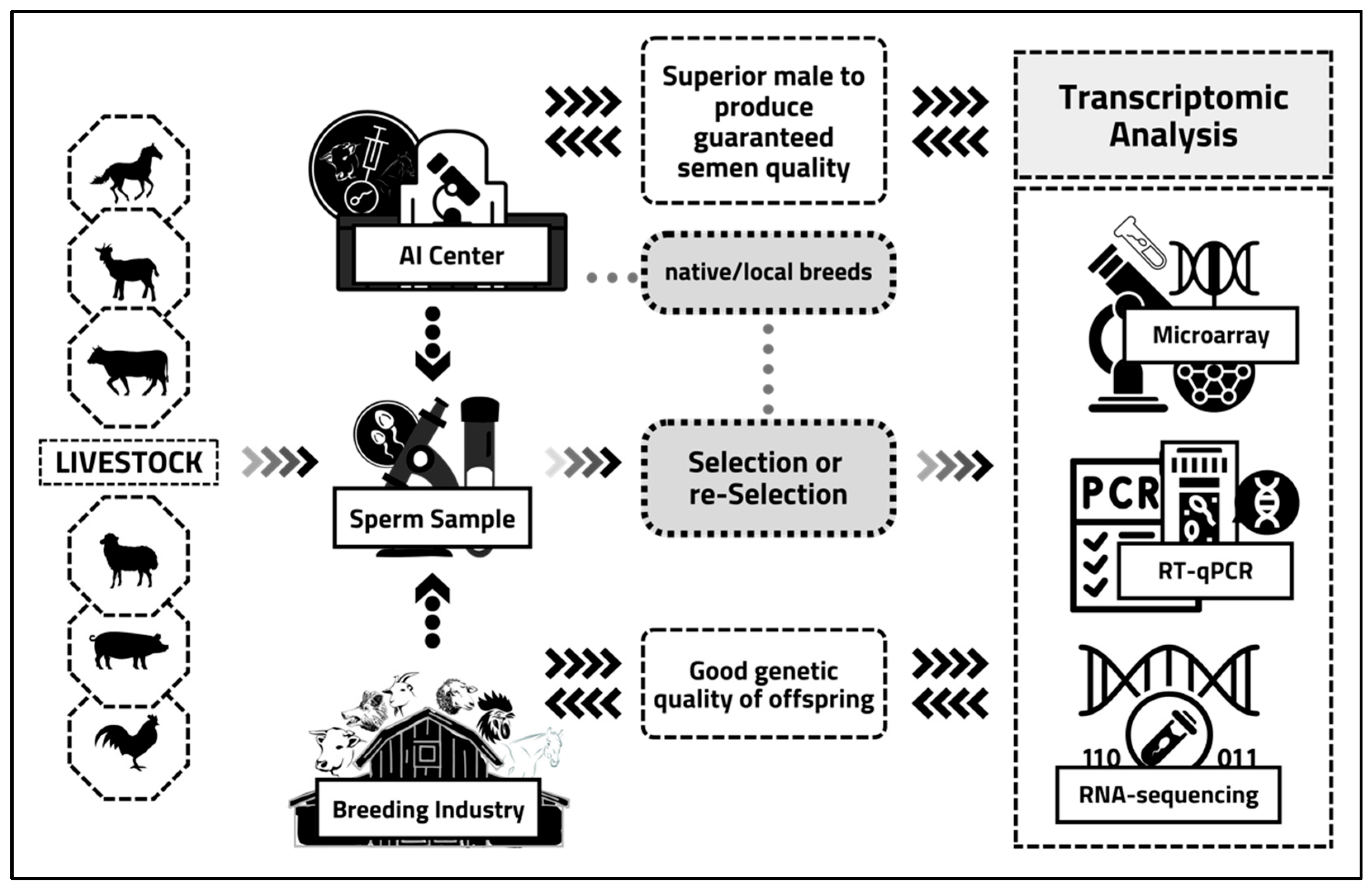
2. Sperm Transcriptome Analysis: Principles, Regulation, Applications, and Experimental Tools
3. Sperm Transcriptome Analysis Approaches in the Reproduction Process
4. Sperm Transcriptome Analysis of Reproductive Functions in Livestock
5. Future Challenges and Opportunities in the Livestock-Breeding Industry
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Turri, F.; Capra, E.; Lazzari, B.; Cremonesi, P.; Stella, A.; Pizzi, F. A combined flow cytometric semen analysis and miRNA profiling as a tool to discriminate between high- and low-fertility bulls. Front. Vet. Sci. 2021, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kirkman-Brown, J.; Björndahl, L. Evaluation of a disposable plastic Neubauer counting chamber for semen analysis. Fertil. Steril. 2009, 91, 627–631. [Google Scholar] [CrossRef] [PubMed]
- van der Horst, G.; Maree, L.; du Plessis, S.S. Current perspectives of CASA applications in diverse mammalian spermatozoa. Reprod. Fertil. Dev. 2018, 30, 875–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samplaski, M.K.; Dimitromanolakis, A.; Lo, K.C.; Grober, E.D.; Mullen, B.; Garbens, A.; Jarvi, K.A. The relationship between sperm viability and DNA fragmentation rates. Reprod. Biol. Endocrinol. 2015, 13, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrell, J.M.; Nongbua, T.; Valeanu, S.; Lima Verde, I.; Lundstedt-Enkel, K.; Edman, A.; Johannisson, A. Sperm quality variables as indicators of bull fertility may be breed dependent. Anim. Reprod. Sci. 2017, 185, 42–52. [Google Scholar] [CrossRef]
- Indriastuti, R.; Ulum, M.F.; Arifiantini, R.I.; Purwantara, B. Individual variation in fresh and frozen semen of Bali bulls (Bos sondaicus). Vet. World 2020, 13, 840–846. [Google Scholar] [CrossRef]
- Rickard, J.P.; Schmidt, R.E.; Maddison, J.W.; Bathgate, R.; Lynch, G.W.; Druart, X.; de Graaf, S.P. Variation in seminal plasma alters the ability of ram spermatozoa to survive cryopreservation. Reprod. Fertil. Dev. 2016, 28, 516–523. [Google Scholar] [CrossRef] [Green Version]
- Enciso, M.; Cisale, H.; Johnston, S.D.; Sarasa, J.; Fernández, J.L.; Gosálvez, J. Major morphological sperm abnormalities in the bull are related to sperm DNA damage. Theriogenology 2011, 76, 23–32. [Google Scholar] [CrossRef]
- Fortes, M.R.; Satake, N.; Corbet, D.H.; Corbet, N.J.; Burns, B.M.; Moore, S.S.; Boe-Hansen, G.B. Sperm protamine deficiency correlates with sperm DNA damage in Bos indicus bulls. Andrology 2014, 2, 370–378. [Google Scholar] [CrossRef]
- Khalil, W.A.; El-Harairy, M.A.; Zeidan, A.E.B.; Hassan, M.A.E.; Mohey-Elsaeed, O. Evaluation of bull spermatozoa during and after cryopreservation: Structural and ultrastructural insights. Int. J. Vet. Sci. Med. 2017, 6, S49–S56. [Google Scholar] [CrossRef]
- Evenson, D.P. The Sperm Chromatin Structure Assay (SCSA(®)) and other sperm DNA fragmentation tests for evaluation of sperm nuclear DNA integrity as related to fertility. Anim. Reprod. Sci. 2016, 169, 56–75. [Google Scholar] [CrossRef] [Green Version]
- Ugur, M.R.; Kutchy, N.A.; de Menezes, E.B.; Ul-Husna, A.; Haynes, B.P.; Uzun, A.; Kaya, A.; Topper, E.; Moura, A.; Memili, E. Retained acetylated histone four in bull sperm associated with fertility. Front. Vet. Sci. 2019, 6, 223. [Google Scholar] [CrossRef] [Green Version]
- Imrat, P.; Hernandez, M.; Rittem, S.; Thongtip, N.; Mahasawangkul, S.; Gosálvez, J.; Holt, W.V. The dynamics of sperm DNA stability in Asian elephant (Elephas maximus) spermatozoa before and after cryopreservation. Theriogenology 2012, 77, 998–1007. [Google Scholar] [CrossRef]
- Fraser, L. Markers for Sperm Freezability and Relevance of Transcriptome Studies in Semen Cryopreservation: A Review. In Theriogenology; Carreira, R.P., Ed.; IntechOpen: London, UK, 2017. [Google Scholar]
- García-Macías, V.; de Paz, P.; Martinez-Pastor, F.; Alvarez, M.; Gomes-Alves, S.; Bernardo, J.; Anel, E.; Anel, L. DNA fragmentation assessment by flow cytometry and Sperm-Bos-Halomax (bright-field microscopy and fluorescence microscopy) in bull sperm. Int. J. Androl. 2007, 30, 88–98. [Google Scholar] [CrossRef]
- Pardede, B.P.; Maulana, T.; Kaiin, E.M.; Agil, M.; Karja, N.W.K.; Sumantri, C.; Supriatna, I. The potential of sperm bovine protamine as a protein marker of semen production and quality at the National Artificial Insemination Center of Indonesia. Vet. World 2021, 14, 2473–2481. [Google Scholar] [CrossRef]
- Franken, D.R.; Oehninger, S. Semen analysis and sperm function testing. Asian J. Androl. 2012, 14, 6–13. [Google Scholar] [CrossRef] [Green Version]
- Simon, L.; Liu, L.; Murphy, K.; Ge, S.; Hotaling, J.; Aston, K.I.; Emery, B.; Carrell, D.T. Comparative analysis of three sperm DNA damage assays and sperm nuclear protein content in couples undergoing assisted reproduction treatment. Hum. Reprod. 2014, 29, 904–917. [Google Scholar] [CrossRef]
- Sá, R.; Cunha, M.; Rocha, E.; Barros, A.; Sousa, M. Sperm DNA fragmentation is related to sperm morphological staining patterns. Reprod. Biomed. Online 2015, 31, 506–515. [Google Scholar] [CrossRef] [Green Version]
- McSwiggin, H.M.; O’Doherty, A.M. Epigenetic reprogramming during spermatogenesis and male factor infertility. Reproduction 2018, 156, R9–R21. [Google Scholar] [CrossRef]
- Pardede, B.P.; Agil, M.; Yudi, Y.; Supriatna, I. Relationship of frozen-thawed semen quality with the fertility rate after being distributed in the Brahman Cross Breeding Program. Vet. World 2020, 13, 2649–2657. [Google Scholar] [CrossRef]
- Pardede, B.P.; Supriatna, I.; Yudi, Y.; Agil, M. Decreased bull fertility: Age-related changes in sperm motility and DNA fragmentation. E3S Web Conf. 2020, 151, 01010. [Google Scholar] [CrossRef]
- Özbek, M.; Hitit, M.; Kaya, A.; Jousan, F.D.; Memili, E. Sperm Functional Genome Associated with Bull Fertility. Front. Vet. Sci. 2021, 8, 610888. [Google Scholar] [CrossRef] [PubMed]
- Lowe, R.; Shirley, N.; Bleakley, M.; Dolan, S.; Shafee, T. Transcriptomics technologies. PLoS Comput. Biol. 2017, 13, e1005457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suliman, Y.; Becker, F.; Wimmers, K. Implication of transcriptome profiling of spermatozoa for stallion fertility. Reprod. Fertil. Dev. 2018, 30, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- Selvaraju, S.; Parthipan, S.; Somashekar, L.; Binsila, B.K.; Kolte, A.P.; Arangasamy, A.; Ravindra, J.P.; Krawetz, S.A. Current status of sperm functional genomics and its diagnostic potential of fertility in bovine (Bos taurus). Syst. Biol. Reprod. Med. 2018, 64, 484–501. [Google Scholar] [CrossRef]
- Pang, W.K.; Kang, S.; Ryu, D.Y.; Rahman, M.S.; Park, Y.J.; Pang, M.G. Optimization of sperm RNA processing for developmental research. Sci. Rep. 2020, 10, 11606. [Google Scholar] [CrossRef]
- Das, P.J.; McCarthy, F.; Vishnoi, M.; Paria, N.; Gresham, C.; Li, G.; Kachroo, P.; Sudderth, A.K.; Teague, S.; Love, C.C.; et al. Stallion sperm transcriptome comprises functionally coherent coding and regulatory RNAs as revealed by microarray analysis and RNA-seq. PLoS ONE 2013, 8, e56535. [Google Scholar] [CrossRef] [Green Version]
- Sendler, E.; Johnson, G.D.; Mao, S.; Goodrich, R.J.; Diamond, M.P.; Hauser, R.; Krawetz, S.A. Stability, delivery and functions of human sperm RNAs at fertilization. Nucleic Acids Res. 2013, 41, 4104–4117. [Google Scholar] [CrossRef]
- Rando, O.J. Intergenerational Transfer of Epigenetic Information in Sperm. Cold Spring Harb. Perspect. Med. 2016, 6, a022988. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.P.; Shafeeque, C.M.; Sharma, S.K.; Singh, R.; Mohan, J.; Sastry, K.V.; Saxena, V.K.; Azeez, P.A. Chicken sperm transcriptome profiling by microarray analysis. Genome 2016, 59, 185–196. [Google Scholar] [CrossRef]
- El Fekih, S.; Nguyen, M.H.; Perrin, A.; Beauvillard, D.; Morel, F.; Saad, A.; Ben Ali, H.; De Braekeleer, M. Sperm RNA preparation for transcriptomic analysis: Review of the techniques and personal experience. Andrologia 2017, 49, 1–8. [Google Scholar] [CrossRef]
- Jodar, M.; Selvaraju, S.; Sendler, E.; Diamond, M.P.; Krawetz, S.A.; Reproductive Medicine Network. The presence, role and clinical use of spermatozoal RNAs. Hum. Reprod. Update 2013, 19, 604–624. [Google Scholar] [CrossRef]
- Sahoo, B.; Choudhary, R.K.; Sharma, P.; Choudhary, S.; Gupta, M.K. Significance and Relevance of Spermatozoal RNAs to Male Fertility in Livestock. Front. Genet. 2021, 12, 768196. [Google Scholar] [CrossRef]
- Inoue, M.; Horimoto, K. Relationship between regulatory pattern of gene expression level and gene function. PLoS ONE 2017, 12, e0177430. [Google Scholar] [CrossRef] [Green Version]
- Mitsis, T.; Efthimiadou, A.; Bacopoulou, F.; Vlachakis, D.; Chrousos, G.P.; Eliopoulos, E. Transcription factors and evolution: An integral part of gene expression (Review). World Acad. Sci. J. 2020, 2, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Gibcus, J.H.; Dekker, J. The context of gene expression regulation. F1000 Biol. Rep. 2012, 4, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Payne, J.L.; Khalid, F.; Wagner, A. RNA-mediated gene regulation is less evolvable than transcriptional regulation. Proc. Natl. Acad. Sci. USA 2018, 115, E3481–E3490. [Google Scholar] [CrossRef] [Green Version]
- Stahl, F.; Hitzmann, B.; Mutz, K.; Landgrebe, D.; Lübbecke, M.; Kasper, C.; Walter, J.; Scheper, T. Transcriptome analysis. Adv. Biochem. Eng. Biotechnol. 2012, 127, 1–25. [Google Scholar]
- Kuang, J.; Yan, X.; Genders, A.J.; Granata, C.; Bishop, D.J. An overview of technical considerations when using quantitative real-time PCR analysis of gene expression in human exercise research. PLoS ONE 2018, 13, e0196438. [Google Scholar]
- Roszkowski, M.; Mansuy, I.M. High Efficiency RNA Extraction From Sperm Cells Using Guanidinium Thiocyanate Supplemented With Tris (2-Carboxyethyl) Phosphine. Front. Cell Dev. Biol. 2021, 9, 648274. [Google Scholar] [CrossRef]
- Pedersen, K.B.; Williams, A.; Watt, J.; Ronis, M.J. Improved method for isolating high-quality RNA from mouse bone with RNAlater at room temperature. Bone Rep. 2019, 11, 100211. [Google Scholar] [CrossRef] [PubMed]
- Georgiadis, A.P.; Kishore, A.; Zorrilla, M.; Jaffe, T.M.; Sanfilippo, J.S.; Volk, E.; Rajkovic, A.; Yatsenko, A.N. High quality RNA in semen and sperm: Isolation, analysis and potential application in clinical testing. J. Urol. 2015, 193, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Parthipan, S.; Selvaraju, S.; Somashekar, L.; Kolte, A.P.; Arangasamy, A.; Ravindra, J.P. Spermatozoa input concentrations and RNA isolation methods on RNA yield and quality in bull (Bos taurus). Anal. Biochem. 2015, 482, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Chiang, P.W.; Song, W.J.; Wu, K.Y.; Korenberg, J.R.; Fogel, E.J.; Van, K.M.L.; LAshkari, D.; Kurnit, D.M. Use of a fluorescent-PCR reaction to detect genomic sequence copy number and transcriptional abundance. Genome Res. 1996, 6, 1013–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saiki, R.K.; Scharf, S.; Faloona, F.; Mullis, K.B.; Horn, G.T.; Erlich, H.A.; Arnheim, N. Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science 1985, 230, 1350–1354. [Google Scholar] [CrossRef] [PubMed]
- Van Guilder, H.D.; Vrana, K.E.; Freeman, W.M. Twenty-five years of quantitative PCR for gene expression analysis. Biotechniques 2008, 44, 619–626. [Google Scholar] [CrossRef] [Green Version]
- Fassbinder-Orth, C.A. Methods for quantifying gene expression in ecoimmunology: From qPCR to RNA-Seq. Integr. Comp. Biol. 2014, 54, 396–406. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Riesgo, A.; Andrade, S.C.; Sharma, P.P.; Novo, M.; Pe’rez-Porro, A.R.; Vahtera, V.; Gonza’lez, V.L.; Kawauchi, G.Y.; Giribet, G. Comparative description of ten transcriptomes of newly sequenced invertebrates and efficiency estimation of genomic sampling in non-model taxa. Front. Zool. 2012, 9, 33. [Google Scholar] [CrossRef] [Green Version]
- Tulin, S.; Aguiar, D.; Istrail, S.; Smith, J. A quantitative reference transcriptome for Nematostella vectensis early embryonic development: A pipeline for de novo assembly in emerging model systems. EvoDevo 2013, 4, 16. [Google Scholar] [CrossRef] [Green Version]
- Okoniewski, M.F.; Miller, C.J. Hybridization interactions between probe sets in short oligo microarrays lead to spurious correlations. BMC Inform. 2006, 7, 276. [Google Scholar]
- Roy, N.C.; Altermann, E.; Park, Z.A.; McNabb, W.C. A comparison of analog and next-generation transcriptomic tools for mammalian studies. Briefings Funct. Genom. 2011, 10, 135–150. [Google Scholar] [CrossRef] [Green Version]
- Lalancette, C.; Thibault, C.; Bachand, I.; Caron, N.; Bissonnette, N. Transriptome analysis of bull semen with extreme nonreturn rate: Use of suppression-subtrative hybridization to identify functional markers for fertility. Boil Reprod. 2008, 78, 618–635. [Google Scholar] [CrossRef]
- Qi, X.L.; Xing, K.; Huang, Z.; Chen, Y.; Wang, L.; Zhang, L.C.; Sheng, X.H.; Wang, X.G.; Ni, H.M.; Guo, Y. Comparative transcriptome analysis digs out genes related to antifreeze between fresh and frozen-thawed rooster sperm. Poult. Sci. 2020, 99, 2841–2851. [Google Scholar] [CrossRef]
- Ablondi, M.; Gòdia, M.; Rodriguez-Gil, J.E.; Sánchez, A.; Clop, A. Characterisation of sperm piRNAs and their correlation with semen quality traits in swine. Anim. Genet. 2021, 52, 114–120. [Google Scholar] [CrossRef]
- Eddy, E.M. Regulation of gene expression during spermatogenesis. Cell Dev. Biol. 1998, 9, 451–457. [Google Scholar] [CrossRef] [Green Version]
- Somashekar, L.; Selvaraju, S.; Parthipan, S.; Patil, S.K.; Binsila, B.K.; Venkataswamy, M.M.; Karthik Bhat, S.; Ravindra, J.P. Comparative sperm protein profiling in bulls differing in fertility and identification of phosphatidylethanolamine-binding protein 4, a potential fertility marker. Andrology 2017, 5, 1032–1051. [Google Scholar] [CrossRef] [Green Version]
- Hosken, D.J.; Hodgson, D.J. Why do sperm carry RNA? Relatedness, conflict, and control. Trends Ecol. Evol. 2014, 29, 451–455. [Google Scholar] [CrossRef] [Green Version]
- Card, C.J.; Krieger, K.E.; Kaproth, M.; Sartini, B.L. Oligo-dT selected spermatozoal transcript profiles differ among higher and lower fertility dairy sires. Anim. Reprod. Sci. 2017, 177, 105–123. [Google Scholar] [CrossRef] [Green Version]
- Gòdia, M.; Swanson, G.; Krawetz, S.A. A history of why fathers’ RNA matters. Biol. Reprod. 2018, 99, 147–159. [Google Scholar] [CrossRef] [Green Version]
- Arangasamy, A.; Kasimanickam, V.R.; DeJarnette, J.M.; Kasimanickam, R.K. Association of CRISP2, CCT8, PEBP1 mRNA abundance in sperm and sire conception rate in Holstein bulls. Theriogenology 2011, 76, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Prakash, M.A.; Kumaresan, A.; Ebenezer Samuel King, J.P.; Nag, P.; Sharma, A.; Sinha, M.K.; Kamaraj, E.; Datta, T.K. Comparative Transcriptomic Analysis of Spermatozoa From High- and Low-Fertile Crossbred Bulls: Implications for Fertility Prediction. Front. Cell Dev. Biol. 2021, 9, 647717. [Google Scholar] [CrossRef] [PubMed]
- Selvaraju, S.; Parthipan, S.; Somashekar, L.; Kolte, A.P.; Krishnan Binsila, B.; Arangasamy, A.; Ravindra, J.P. Occurrence and functional significance of the transcriptome in bovine (Bos taurus) spermatozoa. Sci. Rep. 2017, 7, 42392. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, C.; Guo, F.; Zhang, Y.; Ju, Z.; Jiang, Q.; Zhao, X.; Liu, Y.; Zhao, H.; Wang, J.; et al. Integrated analysis of mRNAs and long non-coding RNAs in the semen from Holstein bulls with high and low sperm motility. Sci. Rep. 2019, 9, 2092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, D.H.; Qazi, I.H.; Ran, M.X.; Liang, K.; Zhang, Y.; Zhang, M.; Zhou, G.B.; Angel, C.; Zeng, C.J. Exploration of miRNA and mRNA Profiles in Fresh and Frozen-Thawed Boar Sperm by Transcriptome and Small RNA Sequencing. Int. J. Mol. Sci. 2019, 20, 802. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Hu, Z.; Qin, Y.; Dong, J.; Dai, J.; Lu, C.; Zhang, W.; Shen, H.; Xia, Y.; Wang, X. Seminal plasma microRNAs: Potential biomarkers for spermatogenesis status. Mol. Hum. Reprod. 2012, 18, 489–497. [Google Scholar] [CrossRef]
- Keles, E.; Malama, E.; Bozukova, S.; Siuda, M.; Wyck, S.; Witschi, U.; Bauersachs, S.; Bollwein, H. The micro-RNA content of unsorted cryopreserved bovine sperm and its relation to the fertility of sperm after sex-sorting. BMC Genom. 2021, 22, 30. [Google Scholar] [CrossRef]
- Selvaraju, S.; Swathi, D.; Ramya, L.; Lavanya, M.; Archana, S.S.; Sivaram, M. Orchestrating the expression levels of sperm mRNAs reveals CCDC174 as an important determinant of semen quality and bull fertility. Syst. Biol. Reprod. Med. 2021, 67, 89–101. [Google Scholar] [CrossRef]
- Pardede, B.P.; Agil, M.; Karja, N.W.K.; Sumantri, C.; Supriatna, I.; Purwantara, B. PRM1 Gene Expression and Its Protein Abundance in Frozen-Thawed Spermatozoa as Potential Fertility Markers in Breeding Bulls. Vet. Sci. 2022, 9, 111. [Google Scholar] [CrossRef]
- Fagerlind, M.; Stålhammar, H.; Olsson, B.; Klinga-Levan, K. Expression of miRNAs in Bull Spermatozoa Correlates with Fertility Rates. Reprod. Domest. Anim. 2015, 50, 587–594. [Google Scholar] [CrossRef] [Green Version]
- Capra, E.; Turri, F.; Lazzari, B.; Cremonesi, P.; Gliozzi, T.M.; Fojadelli, I.; Stella, A.; Pizzi, F. Small RNA sequencing of cryopreserved semen from single bull revealed altered miRNAs and piRNAs expression between High- and Low-motile sperm populations. BMC Genom. 2017, 18, 14. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Ju, Z.; Wang, L.; Zhang, Y.; Huang, J.; Li, Q.; Li, J.; Zhong, J.; An, L.; Wang, C. Six novel single-nucleotide polymorphisms in SPAG11 gene and their association with sperm quality traits in Chinese Holstein bulls. Anim. Reprod. Sci. 2011, 129, 14–21. [Google Scholar] [CrossRef]
- Gao, Q.; Ju, Z.; Zhang, Y.; Huang, J.; Zhang, X.; Qi, C.; Li, J.; Zhong, J.; Li, G.; Wang, C. Association of TNP2 gene polymorphisms of the bta-miR-154 target site with the semen quality traits of Chinese Holstein bulls. PLoS ONE 2014, 9, e84355. [Google Scholar] [CrossRef]
- Elango, K.; Kumaresan, A.; Sharma, A.; Nag, P.; Prakash, M.A.; Sinha, M.K.; Manimaran, A.; Peter, E.S.K.J.; Jeyakumar, S.; Selvaraju, S.; et al. Sub-fertility in crossbred bulls: Deciphering testicular level transcriptomic alterations between zebu (Bos indicus) and crossbred (Bos taurus × Bos indicus) bulls. BMC Genom. 2020, 21, 502. [Google Scholar] [CrossRef]
- Li, X.; Duan, C.; Li, R.; Wang, D. Insights into the Mechanism of Bovine Spermiogenesis Based on Comparative Transcriptomic Studies. Animals 2021, 11, 80. [Google Scholar] [CrossRef]
- Kadivar, A.; Esfandabadi, N.S.; Nazhvani, E.D.; Shirazi, A.; Ahmadi, E. Effects of cryopreservation on stallion sperm protamine messenger RNAs. Reprod. Domest. Anim. 2020, 55, 274–282. [Google Scholar] [CrossRef]
- Paul, N.; Kumaresan, A.; Das Gupta, M.; Nag, P.; Guvvala, P.R.; Kuntareddi, C.; Sharma, A.; Selvaraju, S.; Datta, T.K. Transcriptomic Profiling of Buffalo Spermatozoa Reveals Dysregulation of Functionally Relevant mRNAs in Low-Fertile Bulls. Front. Vet. Sci. 2021, 7, 609518. [Google Scholar] [CrossRef]
- Verma, A.; Rajput, S.; Kumar, S.; De, S.; Chakravarty, A.K.; Kumar, R.; Datta, T.K. Differential histone modification status of spermatozoa in relation to fertility of buffalo bulls. J. Cell. Biochem. 2015, 116, 743–753. [Google Scholar] [CrossRef]
- Xu, Z.; Xie, Y.; Zhou, C.; Hu, Q.; Gu, T.; Yang, J.; Zheng, E.; Huang, S.; Xu, Z.; Cai, G.; et al. Expression Pattern of Seminal Plasma Extracellular Vesicle Small RNAs in Boar Semen. Front. Vet. Sci. 2020, 7, 585276. [Google Scholar] [CrossRef]
- Gòdia, M.; Estill, M.; Castelló, A.; Balasch, S.; Rodríguez-Gil, J.E.; Krawetz, S.A.; Sánchez, A.; Clop, A. A RNA-Seq Analysis to Describe the Boar Sperm Transcriptome and Its Seasonal Changes. Front Genet. 2019, 10, 299. [Google Scholar] [CrossRef] [Green Version]
- Mańkowska, A.; Brym, P.; Paukszto, Ł.; Jastrzębski, J.P.; Fraser, L. Gene Polymorphisms in Boar Spermatozoa and Their Associations with Post-Thaw Semen Quality. Int. J. Mol. Sci. 2020, 21, 1902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunawan, A.; Kaewmala, K.; Uddin, M.J.; Cinar, M.U.; Tesfaye, D.; Phatsara, C.; Tholen, E.; Looft, C.; Schellander, K. Association study and expression analysis of porcine ESR1 as a candidate gene for boar fertility and sperm quality. Anim. Reprod. Sci. 2011, 128, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Gòdia, M.; Castelló, A.; Rocco, M.; Cabrera, B.; Rodríguez-Gil, J.E.; Balasch, S.; Lewis, C.; Sánchez, A.; Clop, A. Identification of circular RNAs in porcine sperm and evaluation of their relation to sperm motility. Sci. Rep. 2020, 10, 7985. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Rodriguez, M.; Martinez, C.; Wright, D.; Barranco, I.; Roca, J.; Rodriguez-Martinez, H. The Transcriptome of Pig Spermatozoa, and Its Role in Fertility. Int. J. Mol. Sci. 2020, 21, 1572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lian, Y.; Gòdia, M.; Castello, A.; Rodriguez-Gil, J.E.; Balasch, S.; Sanchez, A.; Clop, A. Characterization of the Impact of Density Gradient Centrifugation on the Profile of the Pig Sperm Transcriptome by RNA-Seq. Front. Vet. Sci. 2021, 8, 668158. [Google Scholar] [CrossRef]
- Hwang, J.Y.; Mulligan, B.P.; Kim, H.M.; Yang, B.C.; Lee, C.K. Quantitative analysis of sperm mRNA in the pig: Relationship with early embryo development and capacitation. Reprod. Fertil. Dev. 2013, 25, 807–817. [Google Scholar] [CrossRef]
- Nikbin, S.; Panandam, J.M.; Yaakub, H.; Murugaiyah, M.; Sazili, A.Q. Novel SNPs in heat shock protein 70 gene and their association with sperm quality traits of Boer goats and Boer crosses. Anim. Reprod. Sci. 2014, 146, 176–181. [Google Scholar] [CrossRef] [Green Version]
- Sahoo, B.; Guttula, P.K.; Gupta, M.K. Comparison of spermatozoal RNA extraction methods in goats. Anal. Biochem. 2021, 614, 114059. [Google Scholar] [CrossRef]
- Ureña, I.; González, C.; Ramón, M.; Gòdia, M.; Clop, A.; Calvo, J.H.; Carabaño, M.J.; Serrano, M. Exploring the ovine sperm transcriptome by RNAseq techniques. I Effect of seasonal conditions on transcripts abundance. PLoS ONE 2022, 17, e0264978. [Google Scholar] [CrossRef]
- Hodge, M.J.; de Las Heras-Saldana, S.; Rindfleish, S.J.; Stephen, C.P.; Pant, S.D. Characterization of Breed Specific Differences in Spermatozoal Transcriptomes of Sheep in Australia. Genes 2021, 12, 203. [Google Scholar] [CrossRef]
- Yang, H.; Ma, J.; Wan, Z.; Wang, Q.; Wang, Z.; Zhao, J.; Wang, F.; Zhang, Y. Characterization of sheep spermatogenesis through single-cell RNA sequencing. FASEB J. 2021, 35, e21187. [Google Scholar] [CrossRef]
- Serrano, M.; Ramón, M.; Calvo, J.H.; Jiménez, M.Á.; Freire, F.; Vázquez, J.M.; Arranz, J.J. Genome-wide association studies for sperm traits in Assaf sheep breed. Animal 2021, 15, 100065. [Google Scholar] [CrossRef]
- Yang, L.; Zheng, X.; Mo, C.; Li, S.; Liu, Z.; Yang, G.; Zhao, Q.; Li, S.; Mou, C. Transcriptome analysis and identification of genes associated with chicken sperm storage duration. Poult. Sci. 2020, 99, 1199–1208. [Google Scholar] [CrossRef]
- Brady, K.; Krasnec, K.; Long, J.A. Transcriptome analysis of inseminated sperm storage tubules throughout the duration of fertility in the domestic turkey, Meleagris gallopavo. Poult. Sci. 2022, 101, 101704. [Google Scholar] [CrossRef]
- Shafeeque, C.M.; Singh, R.P.; Sharma, S.K.; Mohan, J.; Sastry, K.V.; Kolluri, G.; Saxena, V.K.; Tyagi, J.S.; Kataria, J.M.; Azeez, P.A. Development of a new method for sperm RNA purification in the chicken. Anim. Reprod. Sci. 2014, 149, 259–265. [Google Scholar] [CrossRef]
- Vijayalakshmy, K.; Kumar, D.; Virmani, M.; Jacob, N.; Kumar, P. Sperm Transcriptomics: An Emerging Technique to Assess Male Fertility. Int. J. Curr. Microbiol. App. Sci. 2018, 7, 1188–1200. [Google Scholar] [CrossRef]
- Zhang, Y.; Dai, D.; Chang, Y.; Li, Y.; Zhang, M.; Zhou, G.; Peng, Z.; Zeng, C. Cryopreservation of boar sperm induces differential microRNAs expression. Cryobiology 2017, 76, 24–33. [Google Scholar] [CrossRef]
- Thippeswamy, V.B.; Layek, S.S.; Kumaresan, A.; Mohanty, T.K.; Gupta, A.K.; Chakravarty, A.K.; Manimaran, A.; Prasad, S. Effects of pedigree and exotic genetic inheritance on semen production traits of dairy bulls. Asian Pac. J. Reprod. 2014, 3, 13–17. [Google Scholar] [CrossRef]
- Gopinathan, A.; Sivaselvam, S.N.; Karthickeyan, S.M.K.; Kulasekar, K. Studies on Fresh Semen Discard Percentage in Crossbred Bulls of Tamil Nadu. Shanlax Int. J. Vet. Sci. 2018, 4, 2321–6387. [Google Scholar]
- Park, Y.J.; Pang, W.K.; Ryu, D.Y.; Song, W.H.; Rahman, M.S.; Pang, M.G. Optimized combination of multiple biomarkers to improve diagnostic accuracy in male fertility. Theriogenology 2019, 139, 106–112. [Google Scholar] [CrossRef]
- Jodar, M. Sperm and seminal plasma RNAs: What roles do they play beyond fertilization? Reproduction 2019, 158, R113–R123. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Haugen, H.S.; Clegg, C.H.; Braun, R.E. Premature translation of protamine 1 mRNA causes precocious nuclear condensation and arrest spermatid differentiation in mice. Proc. Natl. Acad. Sci. USA 1995, 92, 12451–12455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helmi, T.Z.; Hambal, M.; Sugito, S.; Zamzami, R.S.; Rusli, R.; Akmal, M. Identification and Characterization of Protamine1 Gene in Aceh Cattle. E3S Web Conf. 2020, 151, 01039. [Google Scholar] [CrossRef]
- El Debaky, H.A.; Kutchy, N.A.; Ul-Husna, A.; Indriastuti, R.; Akhter, S.; Purwantara, B.; Memili, E. Review: Potential of water buffalo in world agriculture: Challenges and opportunities. Appl. Anim. Sci. 2019, 35, 255–268. [Google Scholar] [CrossRef]
- Laske, C.; Teixeira, B.B.; Dionello, N.; Cardoso, F.F. Breeding objectives and economic values for traits of low input family-based beef cattle production system in the State of Rio Grande do Sul. Rev. Bras. Zootec. 2012, 41, 298–305. [Google Scholar] [CrossRef]


{kind=link}
| Transcriptomes | Transcript Names | Reproductive Functions | References |
|---|---|---|---|
| messenger RNA (mRNA) | PPP1R36 | Drives autophagy during spermatogenesis | [60] |
| CRISP2, PEBP1, BSP3, PRKACA, CAPZA3, HSP70 | Sperm capacitation | [62,63] | |
| MAP7, PTK2, PLK1S1, MYH9, PRKCZ | Spermatogenesis and sperm function | [64] | |
| DNAJC2, TNP1 | Sperm nuclear formation | [64] | |
| CATSPER3, SPAG, COX5, COX1 | Sperm motility seven mitochondrial function | [64] | |
| PLCB1 | Acrosomal reaction | [64] | |
| PRSS37 | Sperm-egg recognition | [64] | |
| HSPA1L, ADAM1B, ADAM2, ADAM32 | Sperm-zona pellucid binding | [64] | |
| PLCZ1 | Egg activation | [64] | |
| BCL2L11, BRCA1 | Embryogenesis | [64] | |
| BSP5, SPADH2 | Fertilization | [64] | |
| TNP1, RAD21, UBE2B, RAN | Chromosomal organization | [64] | |
| PAG5, PAG7, PAG10 BCL2L11, MYH10, RBBP6, UBE2B, YBX1 | Early embryonic development | [63,64] | |
| YBX1 | Embryo survival | [58] | |
| TPT1 | Apoptosis, sperm functions | [63] | |
| PFN1 | Oocyte maturation, fertilization, embryo development, spermatogenesis | [63] | |
| ZNF706 | Sperm function | [63] | |
| TSSK6 | Protein phosphorylation, sperm chromatin condensation, sperm motility, gamete function | [63] | |
| TMSB10 | Sperm capacitation, fertilization | [63] | |
| FABP3 | Spermatogenesis | [63] | |
| IQCF1 | Sperm motility, capacitation, and acrosome reaction | [63] | |
| ODF1, ODF2, SPEM1, MEA1, BCL2L11, PRM2, TNP2 | Spermatogenesis | [63] | |
| RBMX | Spermatogenesis | [65] | |
| SHD | Sperm motility | [65] | |
| EFNA1 | Sperm morphology and membrane functionally | [65] | |
| CDKN1B | DNA damage, sperm apoptosis | [66] | |
| ACADS, SCD | Metabolic activity during cryopreservation | [66] | |
| PKD2 | Sperm movement | [66] | |
| microRNAs (miRNAs) | miR-19b, miR-let7a | Spermatogenic status | [67] |
| miR-34b/c | Sperm chromatin condensation | [68] | |
| miR-34c | Growth, differentiation, and apoptosis of male germ cell Sperm motility | [68] | |
| miR-342 | Sperm capacitation | [68] | |
| miR-19-92, miR-106b-25 | Spermatogenesis, sperm quality | [68] | |
| Let-7a, -7d, -7e, miR-22, let-7d, let-7e | Sperm morphology and motility | [66] | |
| miR-29a, miR-376, miR-125b, miR-490, miR-92b-3p, miR-31a-5p, and miR-100-5p | Sperm freezing ability | [66] | |
| miR-3155, miR-8197, miR-6727, miR-11796, miR-14189, miR-6125, and miR-13659 | Sperm fertilization | [66] | |
| miR-26a, miR-455-5p, miR-1 | Sperm motility, apoptosis | [66] | |
| miR-17-92, miR-106b-25 | Sperm motility, morphology | [68] | |
| Long non-coding RNA (lncRNAs) | 52lncRNAs | Sperm motility | [65] |
| piwi interacting RNA (piRNAs) | piR-ssc-1201188 | Negative correlation with tail and neck abnormalities, proximal droplet | [56] |
| piR-ssc-113649 | Sperm viability, normal acrosome, and tail. | [56] | |
| piR-bta-16647898, piR-ssc-109407 | Some parameters of sperm kinematics (sperm motility) in CASA, Sperm viability, normal acrosome, and tail. | [56] |
| Species | Samples | Phenotypes | Transcriptomic Technologies | Finding | References |
|---|---|---|---|---|---|
| Cattle | Sperm | High vs. Low Fertility | miRNA profiling miRNA sequencing | An amount of 15 miRNA was differentially expressed (9 miRNA are known, and the remaining are new) | [1] |
| Sperm | High vs. Sub-fertility | Gene expression studies by Real-time PCR | CCDC174 was confirmed as a marker for predicting bull fertility | [69] | |
| Sex-sorted sperm vs. unsorted sperm | High vs. Low fertility | miRNA profiling by miRNA sequencing | An amount of 85 miRNA was expressed. | [68] | |
| Sperm | High vs. low sperm motility | Coding and non-coding RNA sequencing | 20875 protein-encoding genes were detected, and 19 were differentially expressed; meanwhile, 11,561 lncRNA were identified, and 2517 were differentially expressed | [65] | |
| Sperm | High vs. low fertility crossbred bulls | Global differential expression using RNA sequencing | 13563 genes were found (2093 were specific in high fertile, and 5454 were specific in the low-fertility group | [63] | |
| Sperm | Higher vs. lower fertility | Sperm transcript profiles using RNA-sequencing | 3227 transcripts (805 are unique) and 5366 transcripts (2944 are unique) were found in higher and lower fertility groups | [60] | |
| Buffalo | Sperm | High vs. low fertility | Transcriptomic analysis using microarray technology | 51282 transcripts were found, while 4050 were DE and 709 were significantly dysregulated | [78] |
| Sheep | Sperm | Sperm sample of Merino vs. Dohne vs. Poll Dorset | Characterize the sperm transcriptome using RNA sequencing | Dohne vs. Merino: 72 DEGs (25 upregulated and 47 downregulated)Dohne vs. Poll Dorset L 73 DEGs (41 upregulated and 31 downregulated) Merino vs. Poll Dorset: 570 DEGs (234 upregulated and 336 downregulated) These genes play an essential role in physiological functions (spermatogenesis, fertilization, conception, embryonic development, and performance of offspring production) | [91] |
| Testis | Germ cells vs. somatic cells | Single-cell transcriptomic analysis of sheep spermatogenesis using single-cell sequencing (scRNA-seq) | Germ cells: several pathways of the cell cycle, gamete generation, protein processing, and mRNA surveillance were enriched Somatic cells: ribosome pathways were enriched Marker genes of germ cells: EZH2, SOX18, SCP2, PCNA, and PRKD | [92] | |
| Sperm | Sperm traits | Exploration of genome mutations associated with sperm traits variability using Genome-Wide Association Study (GWAS) | 76 SNPs in 4 chromosomes for ejaculate concentration, 20 SNPs in 3 for ejaculate volume, 32 SNPs in 1 chromosome for ejaculate number of spermatozoa, and 23 SNPs in 17 chromosomes for spermatozoa mass motility in were found. Candidate genes for sperm traits:
| [93] | |
| Horse | Sperm and testis | Sperm vs. testis | Sperm transcriptome analysis using microarray analysis and RNA-sequencing | Microarray: 6761 gen/EST transcripts were found in sperm and 11,112 in testis, and 6596 of them were shared. RNA-seq: 19,257 sequence tags were mapped. 12,013 transcripts were expressed in testis | [28] |
| Sperm | Fertile vs. sub-fertile | Transcriptome profiling by microarray analysis | 437 DE genes were found | [25] | |
| Pig | Sperm | Fresh vs. frozen sperm | miRNA and mRNA profiling by transcriptome and small RNA sequencing | Amounts of 567 mRNAs and 135 miRNAs were expressed differentially | [66] |
| Sperm | Summer vs. winter boar ejaculates | Sperm transcriptome profiling by RNA sequencing | 14 miRNAs up- and 20 miRNAs are downregulated in summer. Five miRNAs were down, and two miRNAs were upregulated in the winter | [81] | |
| Sperm | Embryo cleavage rate and sperm capacitation | The abundance of mRNA coding analysis using quantitative real-time PCR | MYC, CYP19, ADAM2, PRM1, and PRM2 genes were more significant in the high cleavage group, while MYV mRNA was downregulated in capacitated sperm | [87] | |
| Sperm | Fresh vs. cryopreserved sperm (with vs. without glycerol) | miRNA expression using RT-qPCR analysis | Forty-six miRNAs were expressed. Twenty-three miRNAs were differentially expressed. Seven miRNAs were downregulated, and two were upregulated in sperm cryopreserved without glycerol. Fourteen miRNAs were upregulated, and two were downregulated in sperm cryopreserved with glycerol. Seventeen miRNAs were upregulated, but one miRNA was downregulated in cryopreserved sperm with or without glycerol. | [98] | |
| Chicken | Sperm | Fresh vs. frozen sperm | mRNA expression using microarray analysis | 2115 genes were differentially expressed, while 2086 were downregulated and 29 were upregulated in frozen sperm | [55] |
| Sperm and testis | Sperm vs. testis | RT-PCR verified sperm and testis RNA analysis using microarray | Microarray: 17,524 transcripts were identified in sperm (83.7% were shared with the testis, and 12.7% were detected only in sperm). Upregulated transcripts were responsible for signal transduction (63.20%), embryo development (56.67%), and cell structure (56.25%). | [31] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Indriastuti, R.; Pardede, B.P.; Gunawan, A.; Ulum, M.F.; Arifiantini, R.I.; Purwantara, B. Sperm Transcriptome Analysis Accurately Reveals Male Fertility Potential in Livestock. Animals 2022, 12, 2955. https://doi.org/10.3390/ani12212955
Indriastuti R, Pardede BP, Gunawan A, Ulum MF, Arifiantini RI, Purwantara B. Sperm Transcriptome Analysis Accurately Reveals Male Fertility Potential in Livestock. Animals. 2022; 12(21):2955. https://doi.org/10.3390/ani12212955
Chicago/Turabian StyleIndriastuti, Rhesti, Berlin Pandapotan Pardede, Asep Gunawan, Mokhamad Fakhrul Ulum, Raden Iis Arifiantini, and Bambang Purwantara. 2022. "Sperm Transcriptome Analysis Accurately Reveals Male Fertility Potential in Livestock" Animals 12, no. 21: 2955. https://doi.org/10.3390/ani12212955