
Melatonin Targets Metabolism in Head and Neck Cancer Cells by Regulating Mitochondrial Structure and Function

 , , , , , ,
, , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatment
2.2. Metabolomic Analysis
2.3. Pyruvate Measurement
2.4. Mitochondrial Respiration
2.5. Measurement of Redox Status
2.6. Glycolysis Capacity
2.7. Electron Microscopy Analysis
2.8. Quantification of Mitochondrial Mass
2.9. Mitochondrial DNA Copy Number
2.10. Cell Proliferation Assay
2.11. Cell Cycle Analysis
2.12. Western Blot Analysis
2.13. Statistical Analysis
3. Results
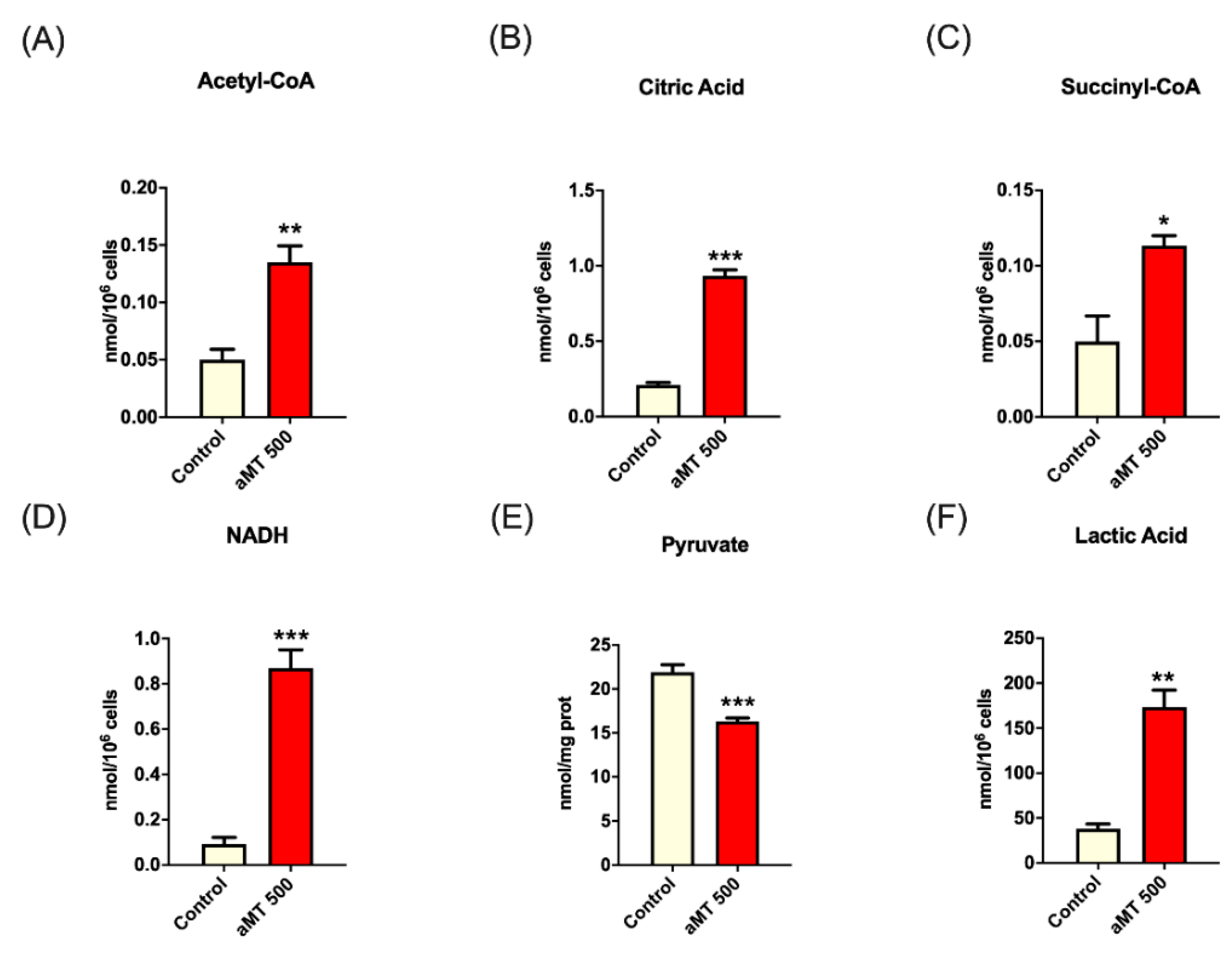
3.1. Melatonin Up-Regulates Key TCA Cycle Metabolites in HNSCC Cells
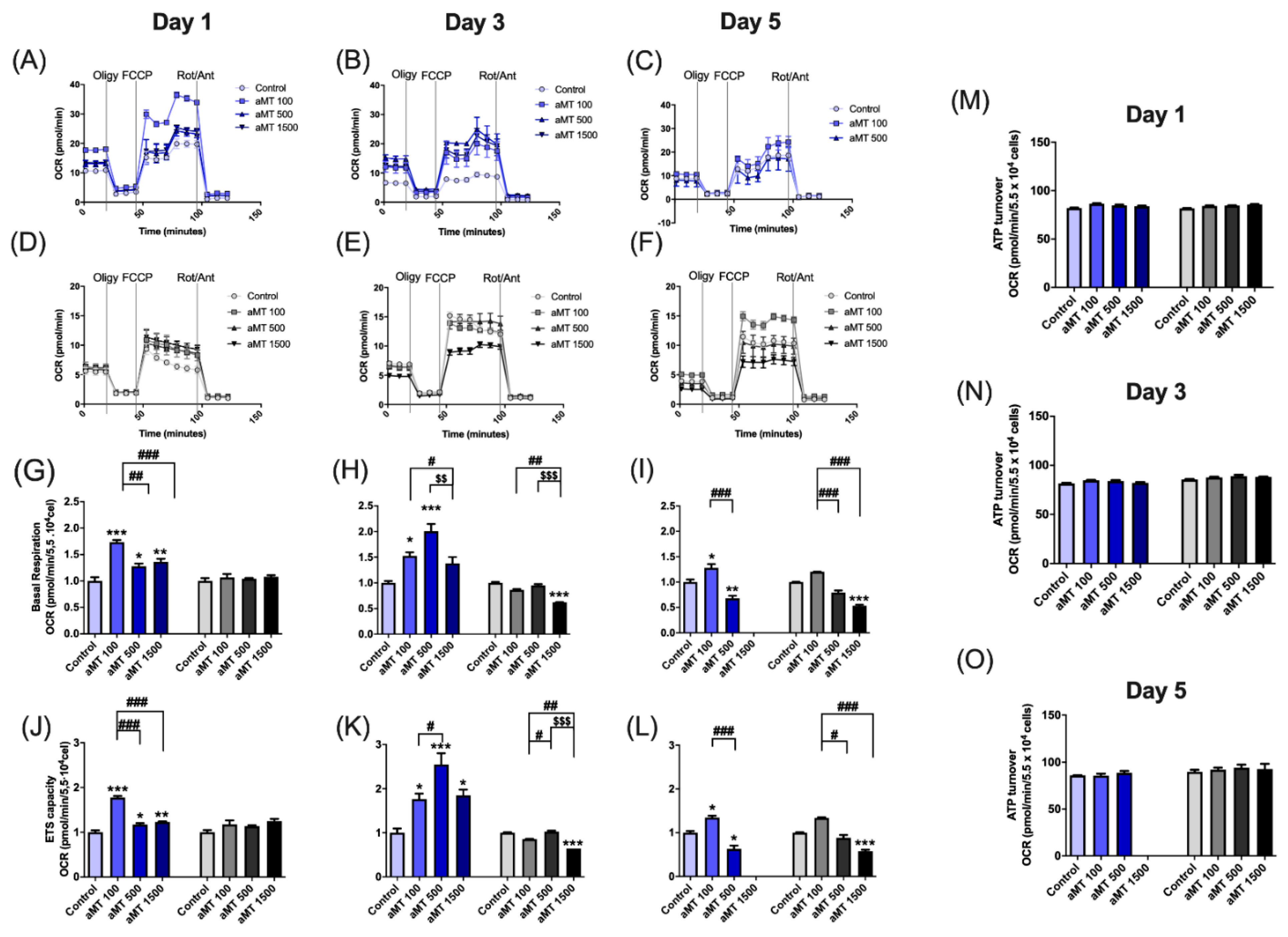
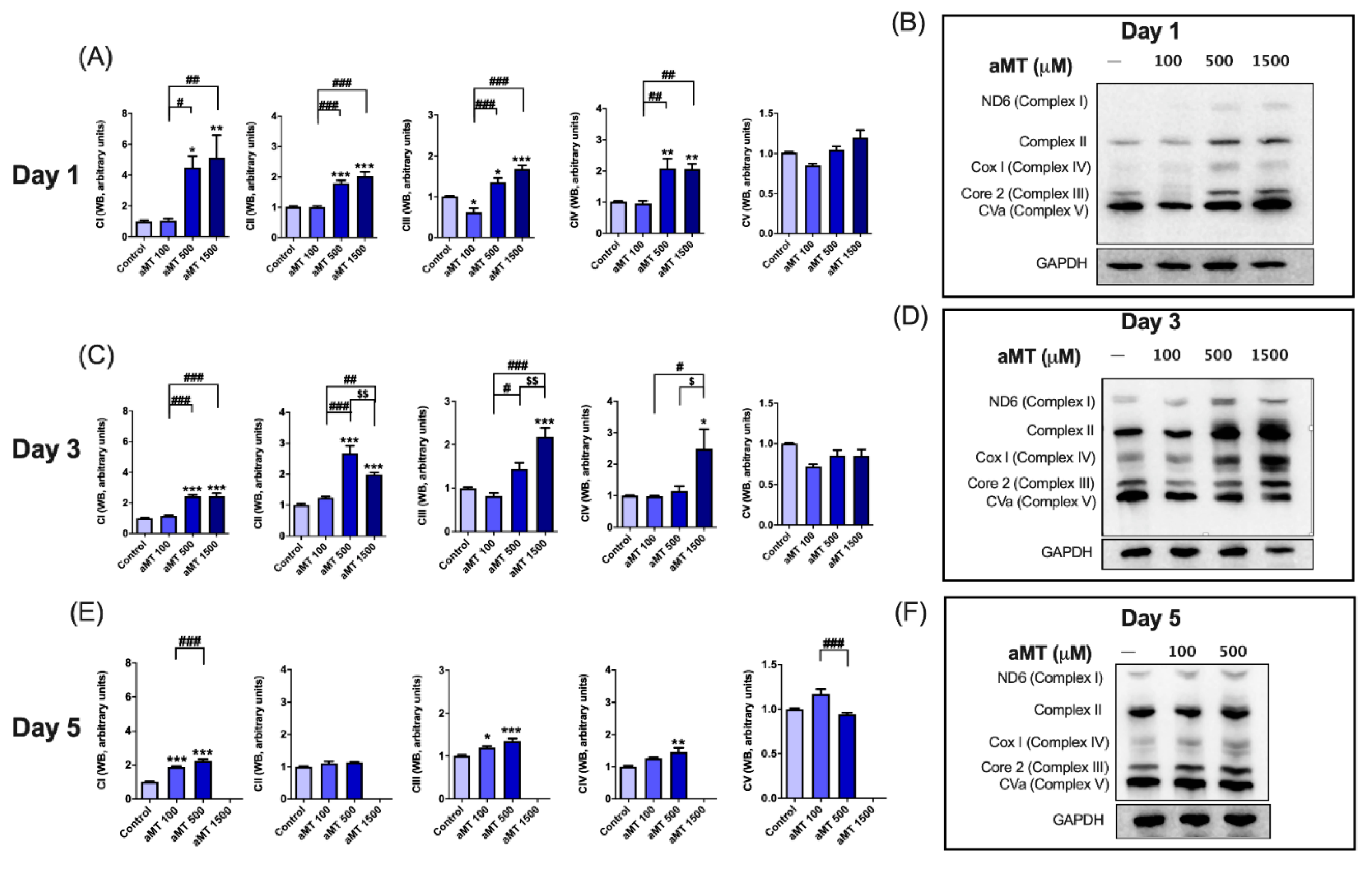
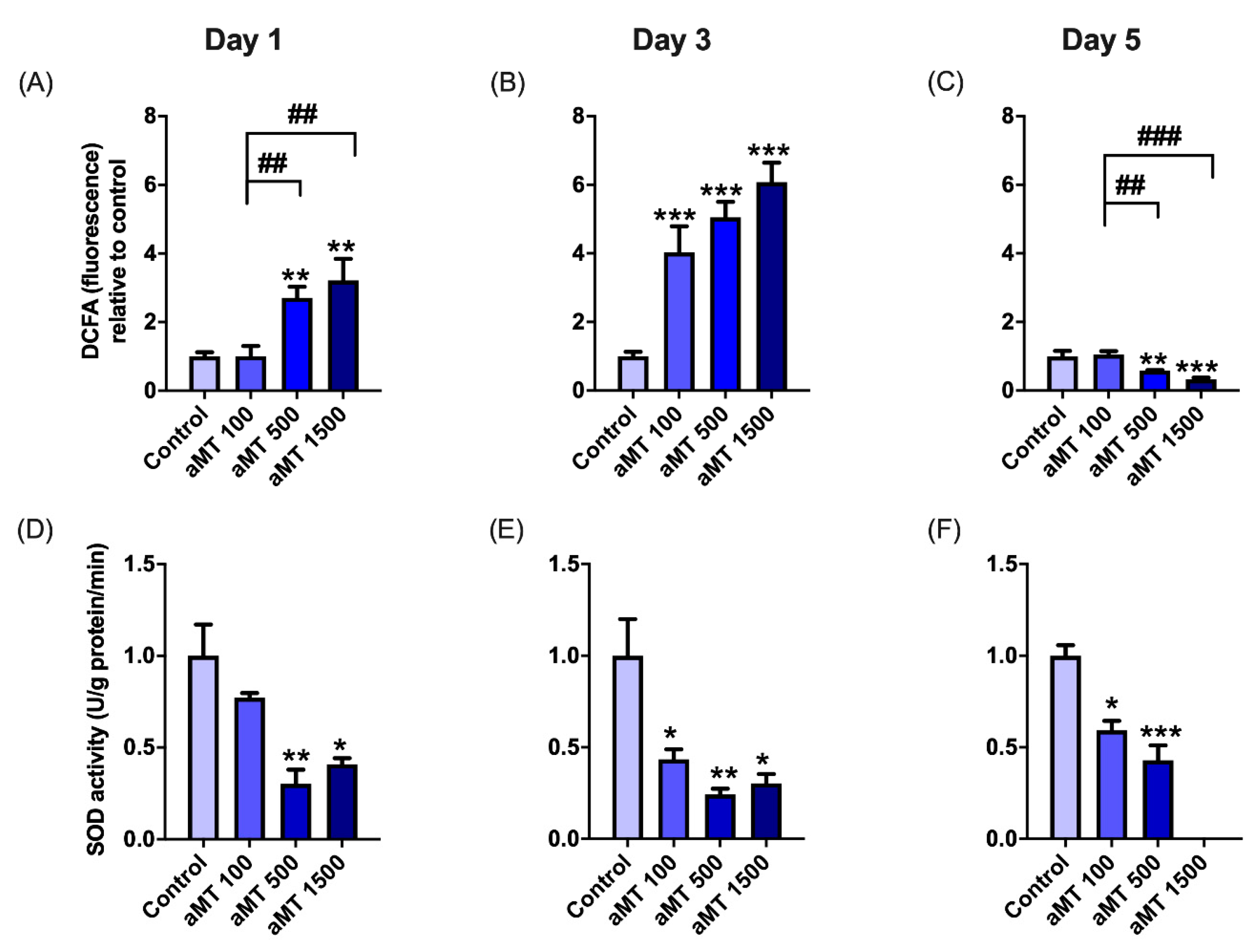
3.2. Melatonin Treatment Induces Uncoupling between Respiration and Phosphorylation in Mitochondria, Correlating with Increased ROS Production in HNSCC Cells
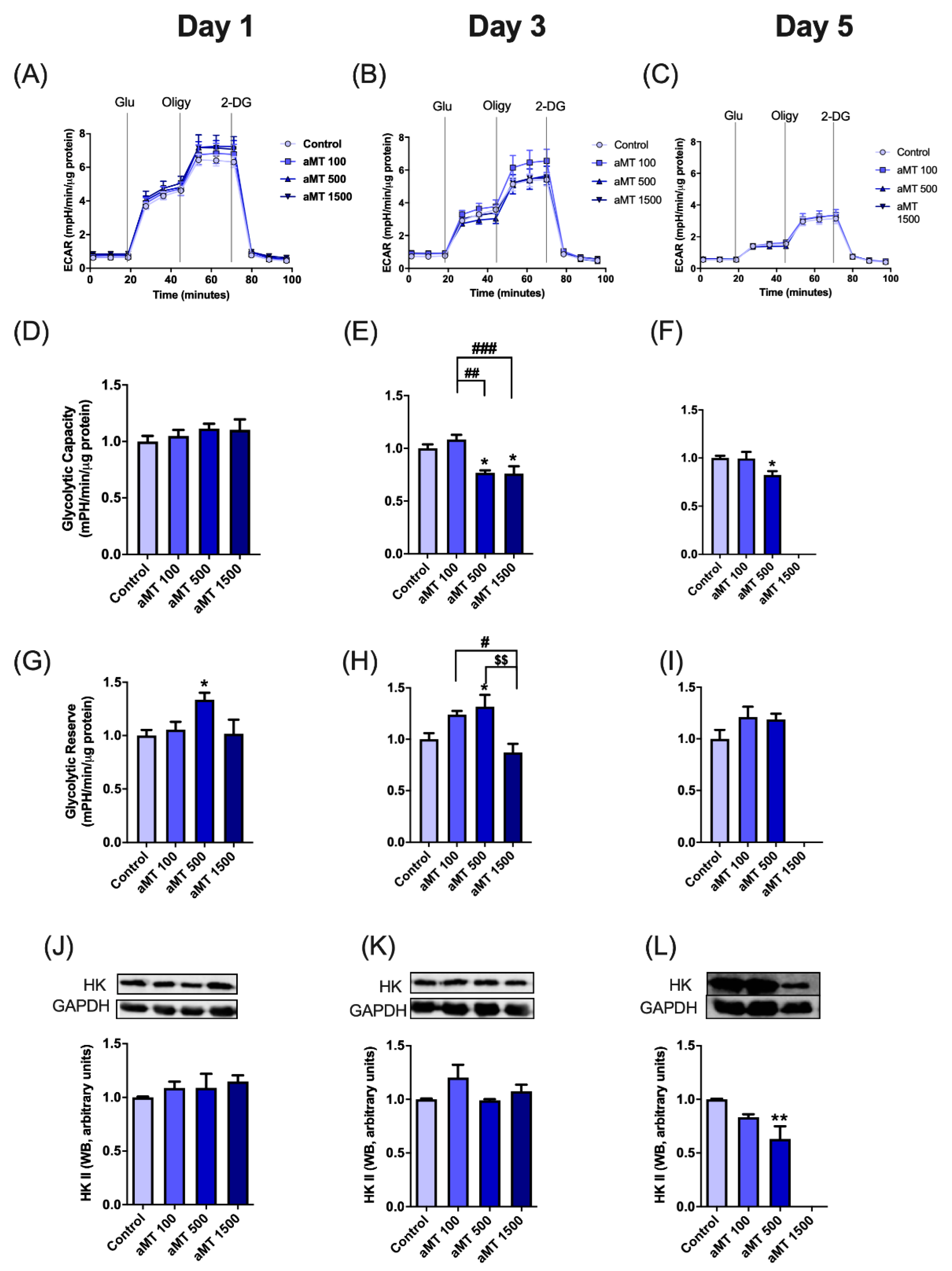
3.3. Effects of Melatonin on Glycolytic Activity in HNSCC Cells
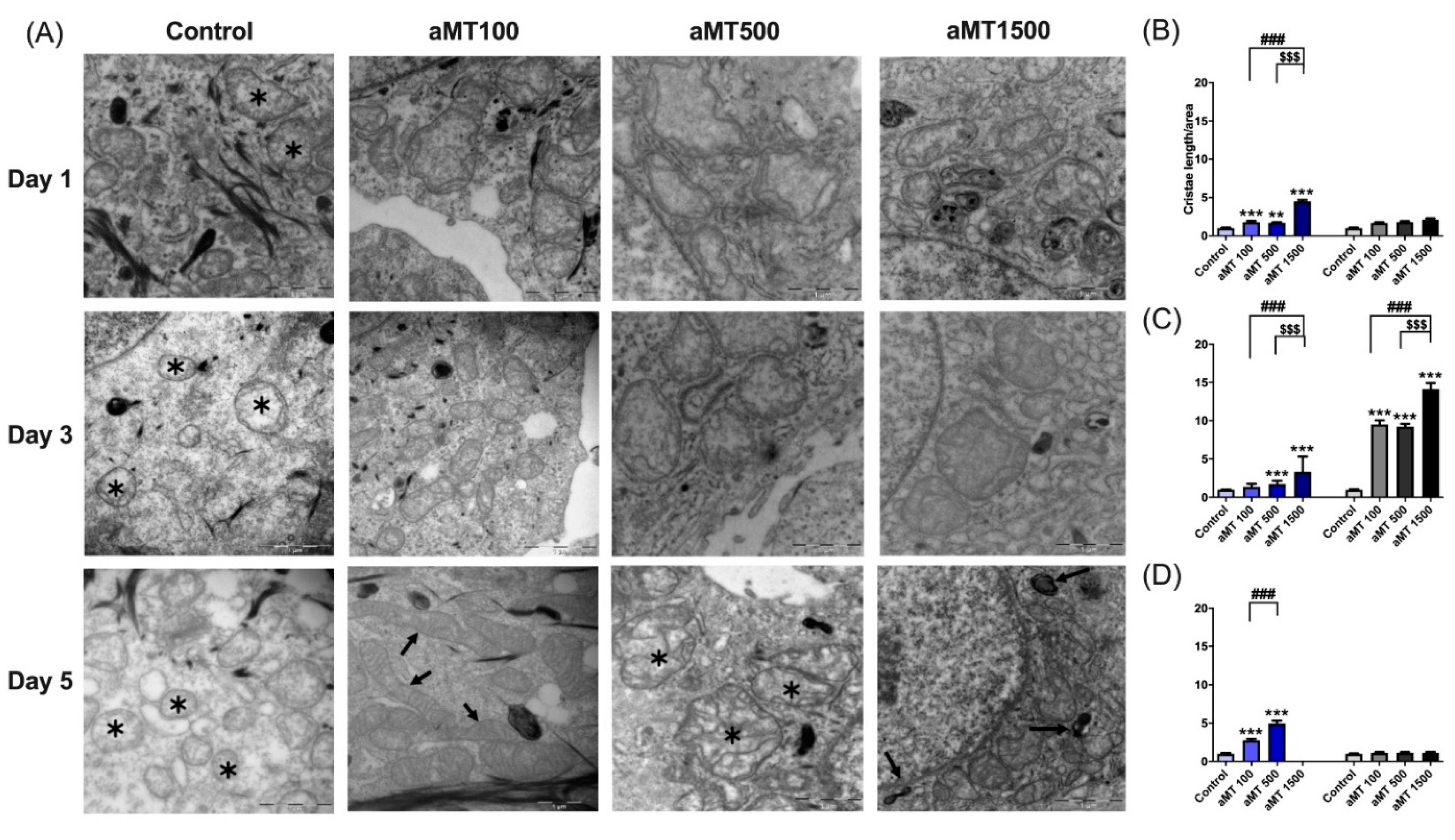
3.4. Melatonin Treatment Modifies the Mitochondrial Morphology of HNSCC Cells
3.5. Melatonin Alters Mitochondrial Fission and Fusion in HNSCC Cells
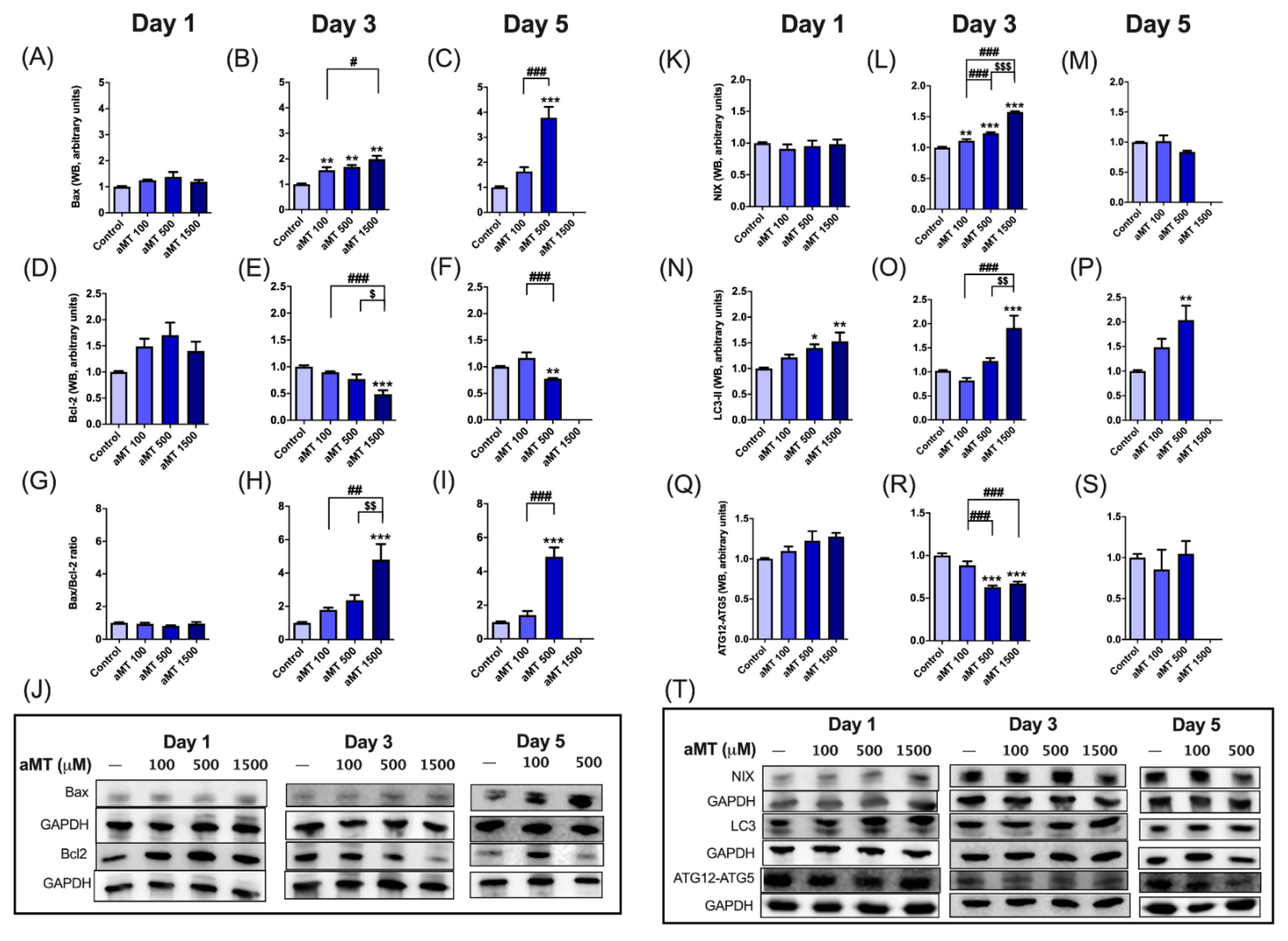
3.6. Melatonin Increases Apoptosis and Autophagy in HNSCC Cells
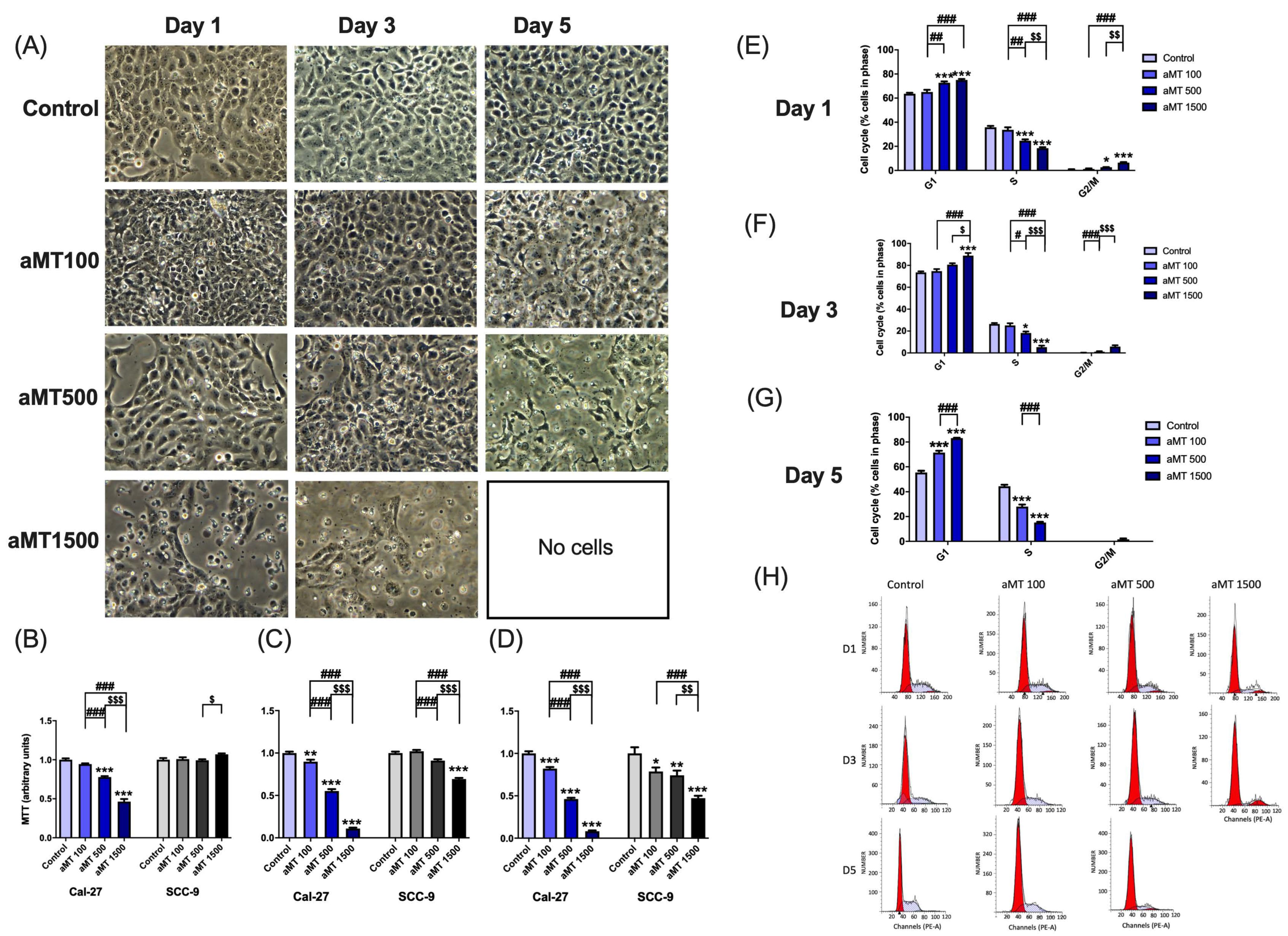
3.7. Melatonin Decreases HNSCC Cell Proliferation and Induces Cell Cycle Arrest in the G1 and G2/M Phases
3.8. High Doses of Melatonin Change Melatonin Receptor Protein Levels
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Johnson, N.W.; Jayasekara, P.; Amarasinghe, A.A.; Hemantha, K. Squamous cell carcinoma and precursor lesions of the oral cavity: Epidemiology and aetiology. Periodontology 2000 2011, 57, 19–37. [Google Scholar] [CrossRef] [Green Version]
- Kumar, D. Regulation of glycolysis in head and neck squamous cell carcinoma. Postdoc J. 2017, 5, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Guo, Y.; Seo, W.; Zhang, R.; Lu, C.; Wang, Y.; Luo, L.; Paul, B.; Yan, W.; Saxena, D.; et al. Targeting cellular metabolism to reduce head and neck cancer growth. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, Y.T.; Chen, Y.F.; Lin, S.C.; Chang, K.W.; Li, W.C. Targeting cellular metabolism modulates head and neck oncogenesis. Int. J. Mol. Sci. 2019, 20, 3960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, T.; Wang, C.; Tian, Y.; Dai, C.; Zhu, Y.; Xu, F. Mitochondrial metabolic reprogramming: An important player in liver cancer progression. Cancer Lett. 2020, 470, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Audet-Walsh, É.; Papadopoli, D.J.; Gravel, S.P.; Yee, T.; Bridon, G.; Caron, M.; Bourque, G.; Giguère, V.; St-Pierre, J. The PGC-1α/ERRα axis represses one-carbon metabolism and promotes sensitivity to anti-folate therapy in breast cancer. Cell Rep. 2016, 14, 920–931. [Google Scholar] [CrossRef] [Green Version]
- Tan, A.S.; Baty, J.W.; Berridge, M.V. The role of mitochondrial electron transport in tumorigenesis and metastasis. Biochim. Biophys. Acta (BBA). 2014, 1840, 1454–1463. [Google Scholar] [CrossRef]
- Yoshida, G.J. Metabolic reprogramming: The emerging concept and associated therapeutic strategies. J. Exp. Clin. Cancer Res. 2015, 34, 111. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Karakhanova, S.; Hartwig, W.; D’Haese, J.G.; Philippov, P.P.; Werner, J.; Bazhin, A.V. Mitochondria and mitochondrial ROS in cancer: Novel targets for anticancer therapy. J. Cell. Physiol. 2016, 231, 2570–2581. [Google Scholar] [CrossRef]
- Shen, Y.Q.; Guerra-Librero, A.; Fernandez-Gil, B.I.; Florido, J.; García-López, S.; Martinez-Ruiz, L.; Mendivil-Perez, M.; Soto-Mercado, V.; Acuña-Castroviejo, D.; Ortega-Arellano, H.; et al. Combination of melatonin and rapamycin for head and neck cancer therapy: Suppression of AKT/mTOR pathway activation, and activation of mitophagy and apoptosis via mitochondrial function regulation. J. Pineal. Res. 2018, 64, 1–18. [Google Scholar] [CrossRef]
- Fernandez-Gil, B.I.; Guerra-Librero, A.; Shen, Y.-Q.; Florido, J.; Martínez-Ruiz, L.; García-López, S.; Adan, C.; Rodríguez-Santana, C.; Acuña-Castroviejo, D.; Quiñones-Hinojosa, A.; et al. Melatonin enhances cisplatin and radiation cytotoxicity in head and neck squamous cell carcinoma by stimulating mitochondrial ROS generation, apoptosis, and autophagy. Oxid. Med. Cell. Longev. 2019, 2019, 7187128. [Google Scholar] [CrossRef] [Green Version]
- Ortiz, F.; Acuña-Castroviejo, D.; Doerrier, C.; Dayoub, J.C.; López, L.C.; Venegas, C.; García, J.A.; López, A.; Volt, H.; Luna-Sánchez, M.; et al. Melatonin blunts the mitochondrial/NLRP3 connection and protects against radiation-induced oral mucositis. J. Pineal. Res. 2015, 58, 34–49. [Google Scholar] [CrossRef]
- Fernández-Gil, B.; Moneim, A.E.A.; Ortiz, F.; Shen, Y.; Soto-Mercado, V.; Mendivil-Perez, M.; Guerra-Librero, A.; Acuña-Castroviejo, D.; Molina-Navarro, M.; García-Verdugo, J.M.; et al. Melatonin protects rats from radiotherapy—Induced small intestine toxicity. PLoS ONE 2017, 12, e0174474. [Google Scholar] [CrossRef]
- Moneim, A.E.A.; Guerra-Librero, A.; Florido, J.; Shen, Y.; Fernández, B.; Acuña-Castroviejo, D.; Escames, G. Oral mucositis: Melatonin gel an effective new treatment. Int. J. Mol. Sci. 2017, 18, 1003. [Google Scholar] [CrossRef]
- Othman, M.S.; Fareid, M.A.; Abdel Hameed, R.S.; Abdel Moneim, A.E. The Protective Effects of Melatonin on Aluminum-Induced Hepatotoxicity and Nephrotoxicity in Rats. Oxid. Med. Cell. Longev. 2020, 2020. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.G.; Darley-Usmar, V.M.; Wu, M.; Jensen, P.B.; Rogers, G.W.; Ferrick, D.A. Bioenergetic profile experiment using C2C12 myoblast cells. J. Vis. Exp. 2010, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Misra, H.P.; Fridovich, I. The role of superoxide anion in the autoxidation of epinephrine and a simple assay for superoxide dismutase. J. Biol. Chem. 1972, 247, 3170–3175. [Google Scholar] [CrossRef]
- Cebrián-Silla, A.; Alfaro-Cervelló, C.; Herranz-Pérez, V.; Kaneko, N.; Park, D.H.; Sawamoto, K.; Alvarez-Buylla, A.; Lim, D.A.; García-Verdugo, J.M. Unique organization of the nuclear envelope in the post-natal quiescent neural stem cells. Stem Cell Rep. 2017, 9, 203–216. [Google Scholar] [CrossRef] [Green Version]
- López, L.C.; Akman, H.O.; García-Cazorla, Á.; Dorado, B.; Martí, R.; Nishino, I.; Tadesse, S.; Pizzorno, G.; Shungu, D.; Bonilla, E.; et al. Unbalanced deoxynucleotide pools cause mitochondrial DNA instability in thymidine phosphorylase-deficient mice. Hum. Mol. Genet. 2009, 18, 714–722. [Google Scholar] [CrossRef] [Green Version]
- Venegas, C.; García, J.A.; Doerrier, C.; Volt, H.; Escames, G.; Lõpez, L.C.; Reiter, R.J.; Acuña-Castroviejo, D. Analysis of the daily changes of melatonin receptors in the rat liver. J. Pineal. Res. 2013, 54, 313–321. [Google Scholar] [CrossRef]
- Lin, C.S.; Huang, Y.Y.; Pan, S.C.; Cheng, C.T.; Liu, C.C.; Shih, C.H.; Ho, H.L.; Yeh, Y.C.; Chou, T.Y.; Lee, M.Y.; et al. Involvement of increased p53 expression in the decrease of mitochondrial DNA copy number and increase of SUVmax of FDG-PET scan in esophageal squamous cell carcinoma. Mitochondrion 2019, 47, 54–63. [Google Scholar] [CrossRef]
- Hoppins, S.; Lackner, L.; Nunnari, J. The machines that divide and fuse mitochondria. Annu. Rev. Biochem. 2007, 76, 751–780. [Google Scholar] [CrossRef]
- Frazier, A.E.; Taylor, R.D.; Mick, D.U.; Warscheid, B.; Stoepel, N.; Meyer, H.E.; Ryan, M.T.; Guiard, B.; Rehling, P. Mdm38 interacts with ribosomes and is a component of the mitochondrial protein export machinery. J. Cell Biol. 2006, 172, 553–564. [Google Scholar] [CrossRef] [Green Version]
- Piao, L.; Li, Y.; Kim, S.J.; Sohn, K.C.; Yang, K.J.; Park, K.A.; Byun, H.S.; Won, M.; Hong, J.; Hur, G.M.; et al. Regulation of OPA1-mediated mitochondrial fusion by leucine zipper/EF-hand-containing transmembrane protein-1 plays a role in apoptosis. Cell. Signal. 2009, 21, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Maycotte, P.; Marín-Hernández, A.; Goyri-Aguirre, M.; Anaya-Ruiz, M.; Reyes-Leyva, J.; Cortés-Hernández, P. Mitochondrial dynamics and cancer. Tumor Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Wang, L.; Wang, Y.; Zhang, S.; Zhou, G.; Lieshout, R.; Ma, B.; Liu, J.; Qu, C.; Verstegen, M.M.A.; et al. Mitochondrial fusion via OPA1 and MFN1 supports liver tumor cell metabolism and growth. Cells 2020, 9, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guido, C.; Whitaker-Menezes, D.; Lin, Z.; Pestell, R.G.; Howell, A.; Zimmers, T.A.; Casimiro, M.C.; Aquila, S.; Ando’, S.; Martinez-Outschoorn, U.E.; et al. Mitochondrial fission induces glycolytic reprogramming in cancer-associated myofibroblasts, driving stromal lactate production, and early tumor growth. Oncotarget 2012, 3, 798–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberghina, L.; Mavelli, G.; Drovandi, G.; Palumbo, P.; Pessina, S.; Tripodi, F.; Coccetti, P.; Vanoni, M. Cell growth and cell cycle in Saccharomyces cerevisiae: Basic regulatory design and protein-protein interaction network. Biotechnol. Adv. 2012, 30, 52–72. [Google Scholar] [CrossRef]
- Carbajo-Pescador, S.; Martín-Renedo, J.; García-Palomo, A.; Tuñón, M.J.; Mauriz, J.L.; González-Gallego, J. Changes in the expression of melatonin receptors induced by melatonin treatment in hepatocarcinoma HepG2 cells. J. Pineal. Res. 2009, 47, 330–338. [Google Scholar] [CrossRef]
- Meng, F.; Wu, L.; Dong, L.; Mitchell, A.V.; Block, J.C.; Liu, J.; Zhang, H.; Lu, Q.; Song, W.M.; Zhang, B.; et al. EGFL9 promotes breast cancer metastasis by inducing cMET activation and metabolic reprogramming. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Pacold, M.E.; Brimacombe, K.R.; Chan, S.H.; Rohde, J.M.; Lewis, C.A.; Swier, L.J.; Possemato, R.; Chen, W.W.; Sullivan, L.B.; Fiske, B.P.; et al. A PHGDH inhibitor reveals coordination of serine synthesis and 1-carbon unit fate HHS public access author manuscript. Nat. Chem. Biol. 2016, 12, 452–458. [Google Scholar] [CrossRef] [Green Version]
- Locasale, J.W.; Grassian, A.R.; Melman, T.; Lyssiotis, C.A.; Mattaini, K.R.; Bass, A.J.; Heffron, G.; Metallo, C.M.; Muranen, T.; Sharfi, H.; et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 2011, 43, 869–874. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Shestov, A.A.; Swain, P.; Yang, C.; Parker, S.J.; Wang, Q.A.; Terada, L.S.; Adams, N.D.; McCabe, M.T.; Pietrak, B.; et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature 2016, 532, 255–258. [Google Scholar] [CrossRef]
- Zhang, Q.; Green, M.D.; Lang, X.; Lazarus, J.; Parsels, J.D.; Wei, S.; Parsels, L.A.; Shi, J.; Ramnath, N.; Wahl, D.R.; et al. Inhibition of ATM increases interferon signaling and sensitizes pancreatic cancer to immune checkpoint blockade therapy. Cancer Res. 2019, 79, 3940–3951. [Google Scholar] [CrossRef]
- Gui, D.Y.; Sullivan, L.B.; Luengo, A.; Hosios, A.M.; Bush, L.N.; Gitego, N.; Davidson, S.M.; Freinkman, E.; Thomas, C.J.; Heiden, M.G. Vander environment dictates dependence on mitochondrial complex I for NAD+ and aspartate production and determines cancer cell sensitivity to metformin. Cell Metab. 2016, 24, 716–727. [Google Scholar] [CrossRef] [Green Version]
- Weinber, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, K.; Klionsky, D.J. Participation of mitochondrial fission during mitophagy. Cell Cycle 2013, 12, 3131–3132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.K.; Gao, J.; Chen, Z.; Shi, H.; Yuan, J.; Cui, H.L.; Yeh, C.N.; Bränström, R.; Larsson, C.; Li, S.; et al. Heterogeneity of metabolic vulnerability in imatinib—Resistant gastrointestinal stromal tumor. Cells 2020, 9, 1333. [Google Scholar] [CrossRef]
- Eboli, M.L.; Paradies, G.; Galeotti, T.; Papa, S. Pyruvate transport in tumor-cell mitochondria. Biochim. Biophys. Acta 1977, 460, 183–187. [Google Scholar] [CrossRef]
- Hirschhaeuser, F.; Sattler, U.G.A.; Mueller-Klieser, W. Lactate: A metabolic key player in cancer. Cancer Res. 2011, 71, 6921–6925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, P.C.; Mao, M.; Abreu, D.A.L.P.; Ansenberger-Fricano, K.; Ekoue, D.N.; Ganini, D.; Kajdacsy-Balla, A.; Diamond, A.M.; Minshall, R.D.; Consolaro, M.E.L.; et al. MnSOD upregulation sustains the Warburg effect via mitochondrial ROS and AMPK-dependent signalling in cancer. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Pelicci, P.G.; Dalton, P.; Giorgio, M. The other face of ROS: A driver of stem cell expansion in colorectal cancer. Cell Stem Cell 2013, 12, 635–636. [Google Scholar] [CrossRef] [Green Version]
- Mori, K.; Shibanuma, M.; Nose, K. Invasive potential induced under long-term oxidative stress in mammary epithelial cells. Cancer Res. 2004, 64, 7464–7472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.L.; Uthaya Kumar, D.B.; Punj, V.; Xu, J.; Sher, L.; Tahara, S.M.; Hess, S.; Machida, K. NANOG Metabolically reprograms tumor-initiating stem-like cells through tumorigenic changes in oxidative phosphorylation and fatty acid metabolism. Cell Metab. 2016, 23, 206–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guido, C.; Whitaker-Menezes, D.; Capparelli, C.; Balliet, R.; Lin, Z.; Pestell, R.G.; Howell, A.; Aquila, S.; Ando, S.; Martinez-Outschoorn, U.; et al. Metabolic reprogramming of cancer-associated fibroblasts by TGF-β drives tumor growth: Connecting TGF-β signaling with “Warburg-like” cancer metabolism and L-lactate production. Cell Cycle 2012, 11, 3019–3035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Huang, Q.; Long, X.; Guo, X.; Sun, X.; Jin, X.; Li, Z.; Ren, T.; Yuan, P.; Huang, X.; et al. Mitochondrial elongation-mediated glucose metabolism reprogramming is essential fortumor cell survival during energy stress. Oncogene 2017, 36, 4901–4912. [Google Scholar] [CrossRef]
- Cogliati, S.; Frezza, C.; Soriano, M.E.; Varanita, T.; Quintana-Cabrera, R.; Corrado, M.; Cipolat, S.; Costa, V.; Casarin, A.; Gomes, L.C.; et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 2013, 155, 160–171. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, K.; Tsuno, N.H.; Sunami, E.; Tsurita, G.; Kawai, K.; Okaji, Y.; Nishikawa, T.; Shuno, Y.; Hongo, K.; Hiyoshi, M.; et al. Chloroquine potentiates the anti-cancer effect of 5-fluorouracil on colon cancer cells. BMC Cancer 2010, 10, 370. [Google Scholar] [CrossRef] [Green Version]
- Mencalha, A.; Victorino, V.J.; Cecchini, R.; Panis, C. Mapping oxidative changes in breast cancer: Understanding the basic to reach the clinics. Anticancer Res. 2014, 34, 1127–1140. [Google Scholar]
- Ma, T.; Li, J.; Xu, Y.; Yu, C.; Xu, T.; Wang, H.; Liu, K.; Cao, N.; Nie, B.M.; Zhu, S.Y.; et al. Atg5-independent autophagy regulates mitochondrial clearance and is essential for iPSC reprogramming. Nat. Cell Biol. 2015, 17, 1379–1387. [Google Scholar] [CrossRef]
- Ippolito, L.; Morandi, A.; Taddei, M.L.; Parri, M.; Comito, G.; Iscaro, A.; Raspollini, M.R.; Magherini, F.; Rapizzi, E.; Masquelier, J.; et al. Cancer-associated fibroblasts promote prostate cancer malignancy via metabolic rewiring and mitochondrial transfer. Oncogene 2019, 38, 5339–5355. [Google Scholar] [CrossRef] [PubMed]
- Wernig, M.; Lengner, C.J.; Hanna, J.; Lodato, M.A.; Foreman, R.; Staerk, J.; Markoulaki, S.; Jaenisch, R. A drug-inducible transgenic system for direct reprogramming of multiple somatic cell types. Nat. Biotechnol. 2008, 26, 916–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guerra-Librero, A.; Fernandez-Gil, B.I.; Florido, J.; Martinez-Ruiz, L.; Rodríguez-Santana, C.; Shen, Y.-Q.; García-Verdugo, J.M.; López-Rodríguez, A.; Rusanova, I.; Quiñones-Hinojosa, A.; et al. Melatonin Targets Metabolism in Head and Neck Cancer Cells by Regulating Mitochondrial Structure and Function. Antioxidants 2021, 10, 603. https://doi.org/10.3390/antiox10040603
Guerra-Librero A, Fernandez-Gil BI, Florido J, Martinez-Ruiz L, Rodríguez-Santana C, Shen Y-Q, García-Verdugo JM, López-Rodríguez A, Rusanova I, Quiñones-Hinojosa A, et al. Melatonin Targets Metabolism in Head and Neck Cancer Cells by Regulating Mitochondrial Structure and Function. Antioxidants. 2021; 10(4):603. https://doi.org/10.3390/antiox10040603
Chicago/Turabian StyleGuerra-Librero, Ana, Beatriz I. Fernandez-Gil, Javier Florido, Laura Martinez-Ruiz, César Rodríguez-Santana, Ying-Qiang Shen, José M. García-Verdugo, Alba López-Rodríguez, Iryna Rusanova, Alfredo Quiñones-Hinojosa, and et al. 2021. "Melatonin Targets Metabolism in Head and Neck Cancer Cells by Regulating Mitochondrial Structure and Function" Antioxidants 10, no. 4: 603. https://doi.org/10.3390/antiox10040603