
Animal Models of Coenzyme Q Deficiency: Mechanistic and Translational Learnings

 ,
,  and
and
Abstract
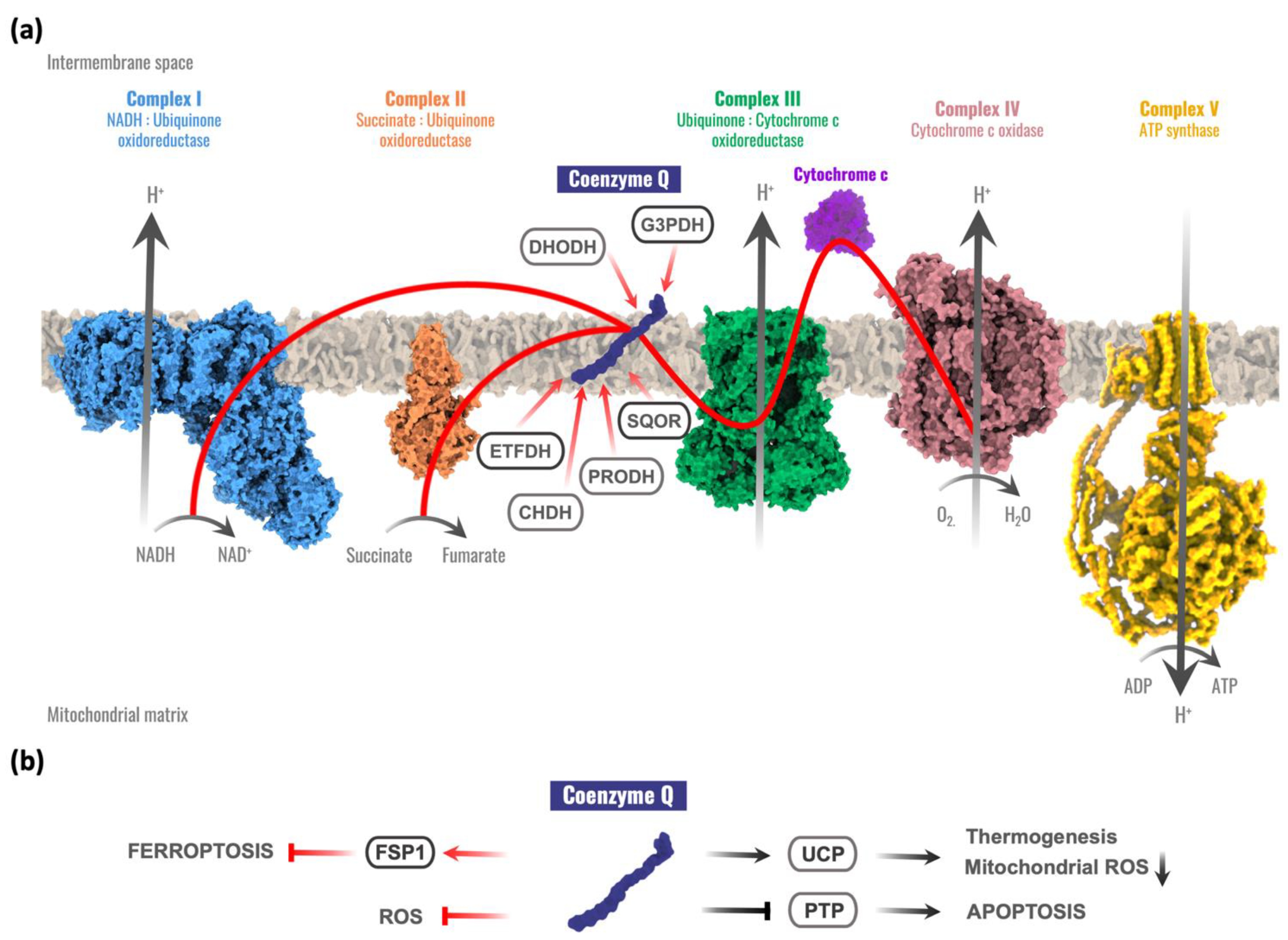
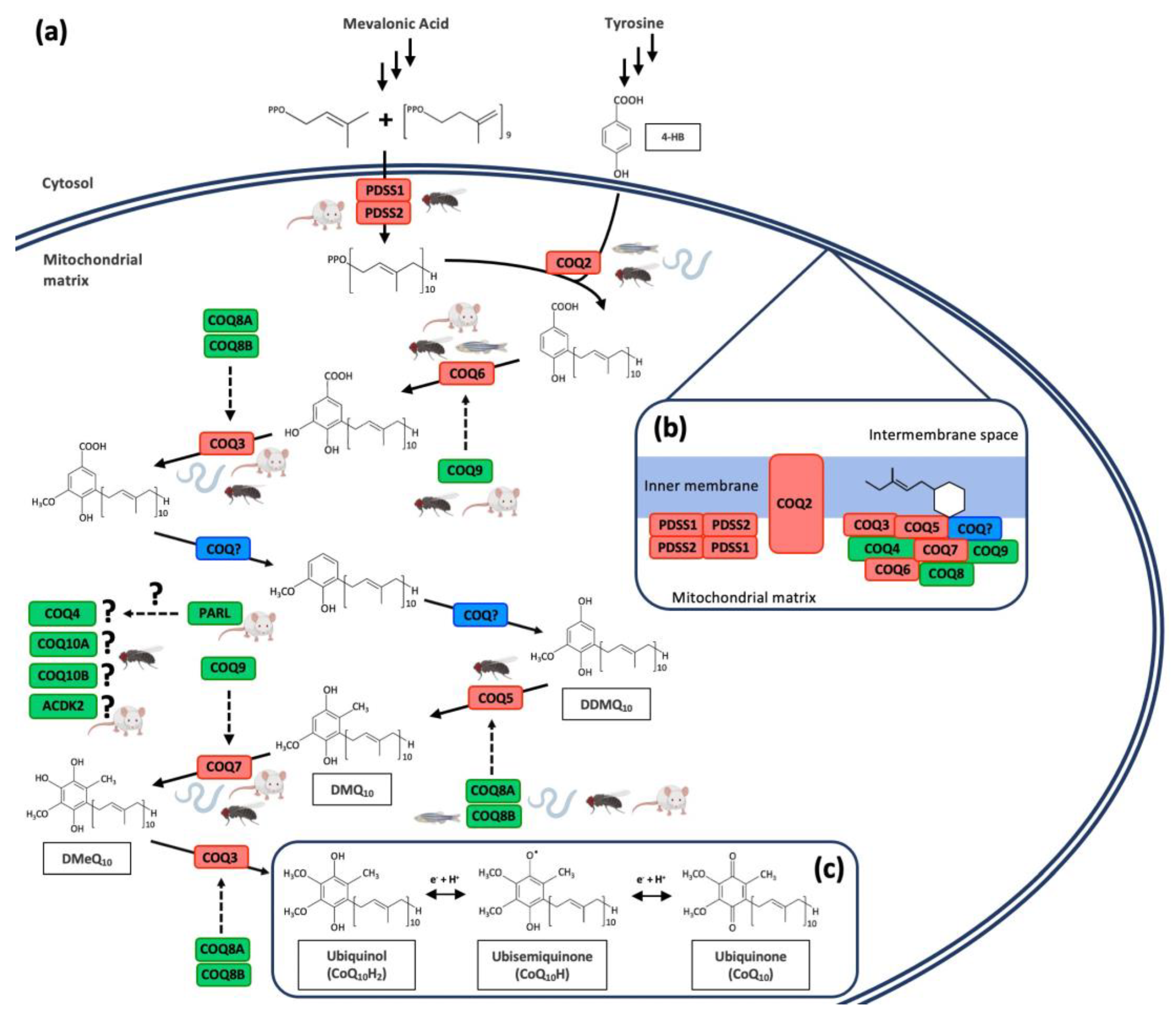
:1. Introduction
2. Invertebrate Models of CoQ Deficiency
2.1. Fruit Fly Models: Drosophila Melanogaster
2.2. Worm Models: Caenorhabditis Elegans
3. Vertebrate Models of CoQ Deficiency
3.1. Zebrafish Models of CoQ Deficiency
3.2. Mouse Models of CoQ Deficiency
3.2.1. Mouse Models with Spontaneous Mutation
3.2.2. Conditional Knockout Mouse Models
3.2.3. Constitutive Knockout and Knock-In Mouse Models
4. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Baschiera, E.; Sorrentino, U.; Calderan, C.; Desbats, M.A.; Salviati, L. The multiple roles of coenzyme Q in cellular homeostasis and their relevance for the pathogenesis of coenzyme Q deficiency. Free Radic. Biol. Med. 2021, 166, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Del-Rio, L.; Clarke, C.F. Coenzyme Q biosynthesis: An update on the origins of the benzenoid ring and discovery of new ring precursors. Metabolites 2021, 11, 385. [Google Scholar] [CrossRef] [PubMed]
- Kawamukai, M. Biosynthesis and bioproduction of coenzyme Q10 by yeasts and other organisms. Biotechnol. Appl. Biochem. 2009, 53, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Alcazar-Fabra, M.; Trevisson, E.; Brea-Calvo, G. Clinical syndromes associated with Coenzyme Q10 deficiency. Essays Biochem. 2018, 62, 377–398. [Google Scholar] [PubMed]
- Awad, A.M.; Bradley, M.C.; del Rio, L.F.; Nag, A.; Tsui, H.S.; Clarke, C.F. Coenzyme Q10 deficiencies: Pathways in yeast and humans. Essays Biochem. 2018, 62, 361–376. [Google Scholar] [PubMed] [Green Version]
- Hidalgo-Gutiérrez, A.; González-García, P.; Díaz-Casado, M.; Barriocanal-Casado, E.; López-Herrador, S.; Quinzii, C.; López, L. Metabolic targets of coenzyme Q10 in mitochondria. Antioxidants 2021, 10, 520. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.R.; Guy, H.I. Mammalian pyrimidine biosynthesis: Fresh insights into an ancient pathway. J. Biol. Chem. 2004, 279, 33035–33038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, X.; Luo, F.; Li, X.; Yi, S.; Yang, B.; Jiang, Z. Structural characteristics and catalytic cycle of dihydroorotate dehydrogenase-a review. Sheng Wu Gong Cheng Xue Bao. Chin. J. Biotechnol. 2020, 36, 2732–2740. [Google Scholar]
- Mracek, T.; Drahota, Z.; Houstek, J. The function and the role of the mitochondrial glycerol-3-phosphate dehydrogenase in mammalian tissues. Biochim. Biophys. Acta 2013, 1827, 401–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcazar-Fabra, M.; Navas, P.; Calvo, G.T.B. Coenzyme Q biosynthesis and its role in the respiratory chain structure. Biochim. Biophys. Acta 2016, 1857, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- Watmough, N.J.; Frerman, F.E. The electron transfer flavoprotein: Ubiquinone oxidoreductases. Biochim. Biophys. Acta 2010, 1797, 1910–1916. [Google Scholar] [CrossRef] [PubMed]
- Henriques, B.J.; Olsen, R.K.J.; Gomes, C.M.; Bross, P. Electron transfer flavoprotein and its role in mitochondrial energy metabolism in health and disease. Gene 2021, 776, 145407. [Google Scholar] [CrossRef] [PubMed]
- Blake, R.L.; Hall, J.G.; Russell, E.S. Mitochondrial proline dehydrogenase deficiency in hyperprolinemic PRO/Re mice: Genetic and enzymatic analyses. Biochem. Genet. 1976, 14, 739–757. [Google Scholar] [CrossRef]
- Salvi, F.; Gadda, G. Human choline dehydrogenase: Medical promises and biochemical challenges. Arch. Biochem. Biophys. 2013, 537, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Ziosi, M.; Di Meo, I.; Kleiner, G.; Gao, X.; Barca, E.; Sanchez-Quintero, M.J.; Tadesse, S.; Jiang, H.; Qiao, C.; Rodenburg, R.; et al. Coenzyme Q deficiency causes impairment of the sulfide oxidation pathway. EMBO Mol. Med. 2017, 9, 96–111. [Google Scholar] [CrossRef] [PubMed]
- Echtay, K.S.; Winkler, E.; Klingenberg, M. Coenzyme Q is an obligatory cofactor for uncoupling protein function. Nature 2000, 408, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Echtay, K.S.; Winkler, E.; Frischmuth, K.; Klingenberg, M. Uncoupling proteins 2 and 3 are highly active H (+) transporters and highly nucleotide sensitive when activated by coenzyme Q (ubiquinone). Proc. Natl. Acad. Sci. USA 2001, 98, 1416–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Echtay, K.S.; Roussel, D.; St-Pierre, J.; Jekabsons, M.B.; Cadenas, S.; Stuart, J.A.; Harper, J.A.; Roebuck, S.J.; Morrison, A.; Pickering, S.; et al. Superoxide activates mitochondrial uncoupling proteins. Nature 2002, 415, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Jaburek, M.; Garlid, K.D. Reconstitution of recombinant uncoupling proteins: UCP1, -2, and -3 have similar affinities for ATP and are unaffected by coenzyme Q10. J. Biol. Chem. 2003, 278, 25825–25831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontaine, E.; Ichas, F.; Bernardi, P. A ubiquinone-binding site regulates the mitochondrial permeability transition pore. J. Biol. Chem. 1998, 273, 25734–25740. [Google Scholar] [CrossRef] [Green Version]
- Walter, L.; Miyoshi, H.; Leverve, X.; Bernardi, P.; Fontaine, E. Regulation of the mitochondrial permeability transition pore by ubiquinone analogs. A progress report. Free Radic. Res. 2002, 36, 405–441. [Google Scholar] [CrossRef] [PubMed]
- Papucci, L.; Schiavone, N.; Witort, E.; Donnini, M.; Lapucci, A.; Tempestini, A.; Formigli, L.; Zecchi-Orlandini, S.; Orlandini, G.; Carella, G.; et al. Coenzyme q10 prevents apoptosis by inhibiting mitochondrial depolarization independently of its free radical scavenging property. J. Biol. Chem. 2003, 278, 28220–28228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Grocin, A.G.; da Silva, T.N.X.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Turunen, M.; Olsson, J.; Dallner, G. Metabolism and function of coenzyme Q. Biochim. Biophys. Acta 2004, 1660, 171–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gvozdjáková, A.; Low, H.; Sun, I.; Navas, P.; Crane, F. Plasma membrane coenzyme Q: Evidence for a role in autism. Biol. Targets Ther. 2014, 8, 199–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morre, D.J.; Morre, D.M. Non-mitochondrial coenzyme Q. Biofactors 2011, 37, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Cirilli, I.; Damiani, E.; Dludla, P.V.; Hargreaves, I.; Marcheggiani, F.; Millichap, L.E.; Orlando, P.; Silvestri, S.; Tiano, L. Role of coenzyme Q10 in health and disease: An update on the last 10 years (2010–2020). Antioxidants 2021, 10, 1325. [Google Scholar] [CrossRef]
- Casado, D.; Quiles, J.L.; Casado, B.; González-García, P.; Battino, M.; López, L.C.; Varela-López, A. The paradox of coenzyme Q10 in aging. Nutrients 2019, 11, 2221. [Google Scholar] [CrossRef] [Green Version]
- Teclebrhan, H.; Jakobsson-Borin, A.; Brunk, U.; Dallner, G. Relationship between the endoplasmic reticulum-Golgi membrane system and ubiquinone biosynthesis. Biochim. Biophys. Acta 1995, 1256, 157–165. [Google Scholar] [CrossRef]
- Mugoni, V.; Medana, C.; Santoro, M.M. 13C-isotope-based protocol for prenyl lipid metabolic analysis in zebrafish embryos. Nat. Protoc. 2013, 8, 2337–2347. [Google Scholar] [CrossRef]
- Eisenberg-Bord, M.; Tsui, H.S.; Antunes, D.; del Rio, L.F.; Bradley, M.C.; Dunn, C.; Nguyen, T.P.T.; Rapaport, D.; Clarke, C.F.; Schuldiner, M. The endoplasmic reticulum-mitochondria encounter structure complex coordinates coenzyme Q biosynthesis. Contact 2019, 2, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Hekimi, S. The complexity of making ubiquinone. Trends Endocrinol. Metab. 2019, 30, 929–943. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hekimi, S. Understanding ubiquinone. Trends Cell Biol. 2016, 26, 367–378. [Google Scholar] [CrossRef]
- Pierrel, F. Impact of chemical analogs of 4-hydroxybenzoic acid on coenzyme Q Biosynthesis: From inhibition to bypass of coenzyme q deficiency. Front. Physiol. 2017, 8, 436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombard, J.; Moreira, D. Origins and early evolution of the mevalonate pathway of isoprenoid biosynthesis in the three domains of life. Mol. Biol. Evol. 2011, 28, 87–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefely, J.A.; Pagliarini, D.J. Biochemistry of mitochondrial coenzyme Q biosynthesis. Trends Biochem. Sci. 2017, 42, 824–843. [Google Scholar] [CrossRef] [PubMed]
- Ozeir, M.; Mühlenhoff, U.; Webert, H.; Lill, R.; Fontecave, M.; Pierrel, F. Coenzyme Q biosynthesis: Coq6 is required for the C5-hydroxylation reaction and substrate analogs rescue Coq6 deficiency. Chem. Biol. 2011, 18, 1134–1142. [Google Scholar] [CrossRef] [PubMed]
- Jonassen, T.; Clarke, C.F. Isolation and functional expression of human COQ3, a gene encoding a methyltransferase required for ubiquinone biosynthesis. J. Biol. Chem. 2000, 275, 12381–12387. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.P.; Casarin, A.; Desbats, M.A.; Doimo, M.; Trevisson, E.; Santos-Ocaña, C.; Navas, P.; Clarke, C.F.; Salviati, L. Molecular characterization of the human COQ5 C-methyltransferase in coenzyme Q10 biosynthesis. Biochim. Biophys. Acta 2014, 1841, 1628–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marbois, B.N.; Clarke, C.F. The COQ7 gene encodes a protein in saccharomyces cerevisiae necessary for ubiquinone biosynthesis. J. Biol. Chem. 1996, 271, 2995–3004. [Google Scholar] [CrossRef] [Green Version]
- Reidenbach, A.G.; Kemmerer, Z.A.; Aydin, D.; Jochem, A.; McDevitt, M.T.; Hutchins, P.; Stark, J.L.; Stefely, J.; Reddy, T.; Hebert, A.S.; et al. Conserved lipid and small-molecule modulation of COQ8 reveals regulation of the ancient kinase-like UbiB family. Cell Chem. Biol. 2018, 25, 154–165.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohman, D.C.; Aydin, D.; Von Bank, H.; Smith, R.W.; Linke, V.; Weisenhorn, E.; McDevitt, M.T.; Hutchins, P.; Wilkerson, E.M.; Wancewicz, B.; et al. An isoprene lipid-binding protein promotes eukaryotic coenzyme Q biosynthesis. Mol. Cell 2019, 73, 763–774.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.H.; Black, D.S.; Nguyen, T.P.; Wang, C.; Srinivasan, C.; Clarke, C.F. Yeast Coq9 controls deamination of coenzyme Q intermediates that derive from para-aminobenzoic acid. Biochim. Biophys. Acta 2015, 1851, 1227–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Corzo, L.; Sánchez, M.L.; Doerrier, C.; García, J.A.; Guarás, A.; Acin-Perez, R.; Bullejos-Peregrín, J.; López, A.; Escames, G.; Enríquez, J.A.; et al. Dysfunctional Coq9 protein causes predominant encephalomyopathy associated with CoQ deficiency. Hum. Mol. Genet. 2013, 22, 1233–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsui, H.S.; Pham, N.V.B.; Amer, B.R.; Bradley, M.C.; Gosschalk, J.E.; Gallagher-Jones, M.; Ibarra, H.; Clubb, R.T.; Blaby, C.; Clarke, C.F. Human COQ10A and COQ10B are distinct lipid-binding START domain proteins required for coenzyme Q function. J. Lipid Res. 2019, 60, 1293–1310. [Google Scholar] [CrossRef]
- Marbois, B.; Gin, P.; Gulmezian, M.; Clarke, C.F. The yeast Coq4 polypeptide organizes a mitochondrial protein complex essential for coenzyme Q biosynthesis. Biochim. Biophys. Acta 2009, 1791, 69–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcázar-Fabra, M.; Rodríguez-Sánchez, F.; Trevisson, E.; Brea-Calvo, G. Primary coenzyme Q deficiencies: A literature review and online platform of clinical features to uncover genotype-phenotype correlations. Free Radic. Biol. Med. 2021, 167, 141–180. [Google Scholar] [CrossRef] [PubMed]
- Doimo, M.; Desbats, M.A.; Cerqua, C.; Cassina, M.; Trevisson, E.; Salviati, L. Genetics of coenzyme Q10 deficiency. Mol. Syndr. 2014, 5, 156–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navas, P.; Cascajo, M.V.; Alcázar-Fabra, M.; Hernández-Camacho, J.D.; Sánchez-Cuesta, A.; Rodríguez, A.B.C.; Ballesteros-Simarro, M.; Arroyo-Luque, A.; Rodríguez-Aguilera, J.C.; Fernández-Ayala, D.J.M.; et al. Secondary CoQ10 deficiency, bioenergetics unbalance in disease and aging. Biofactors 2021, 47, 551–569. [Google Scholar] [CrossRef] [PubMed]
- Mollet, J.; Giurgea, I.; Schlemmer, D.; Dallner, G.; Chretien, D.; Delahodde, A.; Bacq, D.; De Lonlay, P.; Munnich, A.; Rotig, A. Prenyldiphosphate synthase, subunit 1 (PDSS1) and OH-benzoate polyprenyltransferase (COQ2) mutations in ubiquinone deficiency and oxidative phosphorylation disorders. J. Clin. Investig. 2007, 117, 765–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López, L.C.; Schuelke, M.; Quinzii, C.M.; Kanki, T.; Rodenburg, R.J.; Naini, A.; DiMauro, S.; Hirano, M. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) Mutations. Am. J. Hum. Genet. 2006, 79, 1125–1129. [Google Scholar] [CrossRef] [Green Version]
- Quinzii, C.; Naini, A.; Salviati, L.; Trevisson, E.; Navas, P.; DiMauro, S.; Hirano, M. A mutation in para-hydroxybenzoate-polyprenyl transferase (COQ2) causes primary coenzyme Q10 deficiency. Am. J. Hum. Genet. 2006, 78, 345–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diomedi-Camassei, F.; Di Giandomenico, S.; Santorelli, F.M.; Caridi, G.; Piemonte, F.; Montini, G.; Ghiggeri, G.M.; Murer, L.; Barisoni, L.; Pastore, A.; et al. COQ2 Nephropathy: A newly described inherited mitochondriopathy with primary renal involvement. J. Am. Soc. Nephrol. 2007, 18, 2773–2780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salviati, L.; Trevisson, E.; Rodríguez-Hernández; Casarin, A.; Pertegato, V.; Doimo, M.; Cassina, M.; Agosto, C.; Desbats, M.A.; Sartori, G.; et al. Haploinsufficiency ofCOQ4causes coenzyme Q10deficiency. J. Med. Genet. 2012, 49, 187–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malicdan, M.C.V.; Vilboux, T.; Ben-Zeev, B.; Guo, J.; Eliyahu, A.; Pode-Shakked, B.; Dori, A.; Kakani, S.; Chandrasekharappa, S.C.; Ferreira, C.R.; et al. A novel inborn error of the coenzyme Q10 biosynthesis pathway: Cerebellar ataxia and static encephalomyopathy due to COQ5 C-methyltransferase deficiency. Hum. Mutat. 2017, 39, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Heeringa, S.F.; Chernin, G.; Chaki, M.; Zhou, W.; Sloan, A.J.; Ji, Z.; Vie, L.X.; Salviati, L.; Hurd, T.W.; Vega-Warner, V.; et al. COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. J. Clin. Investig. 2011, 121, 2013–2024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freyer, C.; Stranneheim, H.; Naess, K.; Mourier, A.; Felser, A.; Maffezzini, C.; Lesko, N.; Bruhn, H.; Engvall, M.; Wibom, R.; et al. Rescue of primary ubiquinone deficiency due to a novel COQ7 defect using 2,4-dihydroxybensoic acid. J. Med. Genet. 2015, 52, 779–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Smith, C.; Parboosingh, J.S.; Khan, A.; Innes, M.; Hekimi, S. Pathogenicity of two COQ7 mutations and responses to 2,4-dihydroxybenzoate bypass treatment. J. Cell. Mol. Med. 2017, 21, 2329–2343. [Google Scholar] [CrossRef]
- Lagier-Tourenne, C.; Tazir, M.; López, L.C.; Quinzii, C.M.; Assoum, M.; Drouot, N.; Busso, C.; Makri, S.; Ali-Pacha, L.; Benhassine, T.; et al. ADCK3, an ancestral kinase, is mutated in a form of recessive ataxia associated with coenzyme Q10 deficiency. Am. J. Hum. Genet. 2008, 82, 661–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mollet, J.; Delahodde, A.; Serre, V.; Chretien, D.; Schlemmer, D.; Lombes, A.; Boddaert, N.; Desguerre, I.; De Lonlay, P.; De Baulny, H.O.; et al. CABC1 gene mutations cause ubiquinone deficiency with cerebellar ataxia and seizures. Am. J. Hum. Genet. 2008, 82, 623–630. [Google Scholar] [CrossRef] [Green Version]
- Feng, C.; Wang, Q.; Wang, J.; Liu, F.; Shen, H.; Fu, H.; Mao, J. Coenzyme Q10 supplementation therapy for 2 children with proteinuria renal disease and ADCK4 mutation: Case reports and literature review. Medicine 2017, 96, e8880. [Google Scholar] [CrossRef]
- Ashraf, S.; Gee, H.Y.; Woerner, S.; Xie, L.X.; Vega-Warner, V.; Lovric, S.; Fang, H.; Song, X.; Cattran, D.C.; Avila-Casado, C.; et al. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J. Clin. Investig. 2013, 123, 5179–5189. [Google Scholar] [CrossRef] [Green Version]
- Duncan, A.J.; Bitner-Glindzicz, M.; Meunier, B.; Costello, H.; Hargreaves, I.P.; Lopez, L.C.; Hirano, M.; Quinzii, C.M.; Sadowski, M.; Hardy, J.; et al. A Nonsense Mutation in COQ9 causes autosomal-recessive neonatal-onset primary coenzyme Q10 deficiency: A potentially treatable form of mitochondrial disease. Am. J. Hum. Genet. 2009, 84, 558–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danhauser, K.; Herebian, D.; Haack, T.B.; Rodenburg, R.J.; Strom, T.M.; Meitinger, T.; Klee, D.; Mayatepek, E.; Prokisch, H.; Distelmaier, F. Fatal neonatal encephalopathy and lactic acidosis caused by a homozygous loss-of-function variant in COQ9. Eur. J. Hum. Genet. 2015, 24, 450–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musumeci, O.; Naini, A.; Slonim, A.E.; Skavin, N.; Hadjigeorgiou, G.L.; Krawiecki, N.; Weissman, B.M.; Tsao, C.-Y.; Mendell, J.R.; Shanske, S.; et al. Familial cerebellar ataxia with muscle coenzyme Q10 deficiency. Neurology 2001, 56, 849–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinzii, C.M.; Kattah, A.G.; Naini, A.; Akman, H.O.; Mootha, V.K.; DiMauro, S.; Hirano, M. Coenzyme Q deficiency and cerebellar ataxia associated with an aprataxin mutation. Neurology 2005, 64, 539–541. [Google Scholar] [CrossRef] [PubMed]
- Le Ber, I.; Dubourg, O.; Benoist, J.F.; Jardel, C.; Mochel, F.; Koenig, M.; Brice, A.; Lombea, A.; Durr, A. Muscle coenzyme Q10 deficiencies in ataxia with oculomotor apraxia 1. Neurology 2007, 68, 295–297. [Google Scholar] [CrossRef] [PubMed]
- Gempel, K.; Topaloglu, H.; Talim, B.; Schneiderat, P.; Schoser, B.; Hans, V.H.; Pálmafy, B.; Kale, G.; Tokatli, A.; Quinzii, C.; et al. The myopathic form of coenzyme Q10 deficiency is caused by mutations in the electron-transferring-flavoprotein dehydrogenase (ETFDH) gene. Brain 2007, 130, 2037–2044. [Google Scholar] [CrossRef] [PubMed]
- Aeby, A.; Sznajer, Y.; Cavé, H.; Rebuffat, E.; Van Coster, R.; Rigal, O.; Van Bogaert, P. Cardiofaciocutaneous (CFC) syndrome associated with muscular coenzyme Q10 deficiency. J. Inherit. Metab. Dis. 2007, 30, 827. [Google Scholar] [CrossRef] [PubMed]
- Yubero, D.; Montero, R.; Martín, M.; Montoya, J.; Ribes, A.; Grazina, M.; Trevisson, E.; Aguilera, J.C.R.; Hargreaves, I.P.; Salviati, L.; et al. Secondary coenzyme Q 10 deficiencies in oxidative phosphorylation (OXPHOS) and non-OXPHOS disorders. Mitochondrion 2016, 30, 51–58. [Google Scholar] [CrossRef]
- Montero, R.; Sánchez-Alcázar, J.A.; Briones, P.; Navarro-Sastre, A.; Gallardo, E.; Bornstein, B.; Herrero-Martín, D.; Rivera, H.; Martín, M.; Marti, R.; et al. Coenzyme Q10 deficiency associated with a mitochondrial DNA depletion syndrome: A case report. Clin. Biochem. 2009, 42, 742–745. [Google Scholar] [CrossRef]
- Desbats, M.A.; Lunardi, G.; Doimo, M.; Trevisson, E.; Salviati, L. Genetic bases and clinical manifestations of coenzyme Q10 (CoQ10) deficiency. J. Inherit. Metab. Dis. 2014, 38, 145. [Google Scholar] [CrossRef] [PubMed]
- Kühl, I.; Miranda, M.; Atanassov, I.; Kuznetsova, I.; Hinze, Y.; Mourier, A.; Filipovska, A.; Larsson, N.-G. Transcriptomic and proteomic landscape of mitochondrial dysfunction reveals secondary coenzyme Q deficiency in mammals. Elife 2017, 6, e30952. [Google Scholar] [CrossRef] [PubMed]
- Fazakerley, D.J.; Chaudhuri, R.; Yang, P.; Maghzal, G.J.; Thomas, K.C.; Krycer, J.R.; Humphrey, S.J.; Parker, B.L.; Fisher-Wellman, K.H.; Meoli, C.C.; et al. Mitochondrial CoQ deficiency is a common driver of mitochondrial oxidants and insulin resistance. Elife 2018, 7, e32111. [Google Scholar] [CrossRef] [PubMed]
- Quinzii, C.M.; Hirano, M. Coenzyme Q and mitochondrial disease. Dev. Disabil. Res. Rev. 2010, 16, 183–188. [Google Scholar] [CrossRef]
- Hargreaves, I.; Heaton, R.A.; Mantle, D. Disorders of human coenzyme Q10 metabolism: An Overview. Int. J. Mol. Sci. 2020, 21, 6695. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.; Saldanha, J.W.; Gould, A.P. A Drosophila model for primary coenzyme Q deficiency and dietary rescue in the developing nervous system. Dis. Model. Mech. 2010, 3, 799–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Wu, Q.; He, D.; Ma, T.; Du, L.; Dui, W.; Guo, X.; Jiao, R. Drosophila sbo regulates lifespan through its function in the synthesis of coenzyme Q in vivo. J. Genet. Genom. 2011, 38, 225–234. [Google Scholar] [CrossRef]
- Cheng, W.; Song, C.; Anjum, K.M.; Chen, M.; Li, D.; Zhou, H.; Wang, W.; Chen, J. Coenzyme Q plays opposing roles on bacteria/fungi and viruses in Drosophila innate immunity. Int. J. Immunogenet. 2011, 38, 331–337. [Google Scholar] [CrossRef]
- Hermle, T.; Braun, D.A.; Helmstädter, M.; Huber, T.B.; Hildebrandt, F. Modeling monogenic human nephrotic syndrome in the drosophila garland cell nephrocyte. J. Am. Soc. Nephrol. 2016, 28, 1521–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Ayala, D.J.; Guerra, I.; Sanz, A.; Navas, P. coq7 (cg14437) interference courses a primary coenzyme Q deficiency in Drosophila. FEBS J. 2012, 279, 13–123. [Google Scholar]
- Guerra, I.; Fernandez-Ayala, D.; Daniel, J.M.; Navas, P. RNA interference (RNAi) of genes involved in Coenzyme Q biosynthesis in Drosophila melanogaster models Coenzyme Q deficiency in humans. In Proceedings of the 22nd IUBMB and 37th FEBS Congress, Seville, Spain, 4–9 September 2012. [Google Scholar]
- Vasta, V.; Sedensky, M.; Morgan, P.; Hahn, S. Altered redox status of coenzyme Q9 reflects mitochondrial electron transport chain deficiencies in Caenorhabditis elegans. Mitochondrion 2011, 11, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Ishii, N.; Takahashi, K.; Tomita, S.; Keino, T.; Honda, S.; Yoshino, K.; Suzuki, K. A methyl viologen-sensitive mutant of the nematode Caenorhabditis elegans. Mutat. Res. 1990, 237, 165–171. [Google Scholar] [CrossRef]
- Ishii, N.; Senoo-Matsuda, N.; Miyake, K.; Yasuda, K.; Ishii, T.; Hartman, P.S.; Furukawa, S. Coenzyme Q10 can prolong C. elegans lifespan by lowering oxidative stress. Mech. Ageing Dev. 2004, 125, 41–46. [Google Scholar] [CrossRef]
- Jonassen, T.; Larsen, P.L.; Clarke, C.F. A dietary source of coenzyme Q is essential for growth of long-lived Caenorhabditis elegans clk-1 mutants. Proc. Natl. Acad. Sci. USA 2001, 98, 421–426. [Google Scholar] [PubMed] [Green Version]
- Kayser, E.-B.; Sedensky, M.M.; Morgan, P.G.; Hoppel, C.L. Mitochondrial oxidative phosphorylation is defective in the long-lived mutant clk-1. J. Biol. Chem. 2004, 279, 54479–54486. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.-Y.; Gangoiti, J.A.; Sedensky, M.M.; Morgan, P.G. The effect of different ubiquinones on lifespan in Caenorhabditis elegans. Mech. Ageing Dev. 2009, 130, 370–376. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.-Y.; Vasta, V.; Hahn, S.; Gangoiti, J.A.; Opheim, E.; Sedensky, M.M.; Morgan, P.G. The role of DMQ9 in the long-lived mutant clk-1. Mech. Ageing Dev. 2011, 132, 331–339. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, M.; Ogawara, M.; Shimizu, T.; Shirasawa, T. Restoration of the behavioral rates and lifespan in clk-1 mutant nematodes in response to exogenous coenzyme Q10. Exp. Gerontol. 2012, 47, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Gomez, F.; Saiki, R.; Chin, R.; Srinivasan, C.; Clarke, C.F. Restoring de novo coenzyme Q biosynthesis in Caenorhabditis elegans coq-3 mutants yields profound rescue compared to exogenous coenzyme Q supplementation. Gene 2012, 506, 106–116. [Google Scholar] [CrossRef] [Green Version]
- Gavilan, A.; Asencio, C.; Cabello, J.; Rodriguez-Aguilera, J.C.; Schnabel, R.; Navas, P. C. elegans knockouts in ubiquinone biosynthesis genes result in different phenotypes during larval development. Biofactors 2005, 25, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Earls, L.R.; Hacker, M.L.; Watson, J.D.; Miller, D.M. Coenzyme Q protects Caenorhabditis elegans GABA neurons from calcium-dependent degeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 14460–14465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hihi, A.K.; Gao, Y.; Hekimi, S. Ubiquinone is necessary for caenorhabditis elegans development at mitochondrial and non-mitochondrial sites. J. Biol. Chem. 2002, 277, 2202–2206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asencio, C.; Navas, P.; Cabello, J.; Schnabel, R.; Cypser, J.R.; Johnson, T.E.; Rodríguez-Aguilera, J.C. Coenzyme Q supports distinct developmental processes in Caenorhabditis elegans. Mech. Ageing Dev. 2009, 130, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Mugoni, V.; Postel, R.; Catanzaro, V.; De Luca, E.; Turco, E.; Digilio, G.; Silengo, L.; Murphy, M.; Medana, C.; Stainier, D.; et al. Ubiad1 is an antioxidant enzyme that regulates eNOS activity by CoQ10 synthesis. Cell 2013, 152, 504–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinzii, C.M.; Garone, C.; Emmanuele, V.; Tadesse, S.; Krishna, S.; Dorado, B.; Hirano, M. Tissue-specific oxidative stress and loss of mitochondria in CoQ-deficient Pdss2 mutant mice. FASEB J. 2013, 27, 612–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinzii, C.M.; Luna-Sanchez, M.; Ziosi, M.; Gutiérrez, A.H.; Kleiner, G.; Lopez, L.C. The role of sulfide oxidation impairment in the pathogenesis of primary CoQ deficiency. Front. Physiol. 2017, 8, 525. [Google Scholar] [CrossRef] [PubMed]
- Saiki, R.; Lunceford, A.L.; Shi, Y.; Marbois, B.; King, R.; Pachuski, J.; Kawamukai, M.; Gasser, D.L.; Clarke, C.F. Coenzyme Q10 supplementation rescues renal disease in Pdss2kd/kd mice with mutations in prenyl diphosphate synthase subunit 2. Am. J. Physiol. Physiol. 2008, 295, F1535–F1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleiner, G.; Barca, E.; Ziosi, M.; Emmanuele, V.; Xu, Y.; Gutiérrez, A.H.; Qiao, C.; Tadesse, S.; Area-Gomez, E.; Lopez, L.C.; et al. CoQ10 supplementation rescues nephrotic syndrome through normalization of H2S oxidation pathway. Biochim. Biophys. Acta Mol. Basis. Dis. 2018, 1864, 3708–3722. [Google Scholar] [CrossRef]
- Falk, M.J.; Polyak, E.; Zhang, Z.; Peng, M.; King, R.; Maltzman, J.S.; Okwuego, E.; Horyn, O.; Nakamaru-Ogiso, E.; Ostrovsky, J.; et al. Probucol ameliorates renal and metabolic sequelae of primary CoQ deficiency in Pdss2 mutant mice. EMBO Mol. Med. 2011, 3, 410–427. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Ostrovsky, J.; Kwon, Y.J.; Polyak, E.; Licata, J.; Tsukikawa, M.; Marty, E.; Thomas, J.; Felix, C.A.; Xiao, R.; et al. Inhibiting cytosolic translation and autophagy improves health in mitochondrial disease. Hum. Mol. Genet. 2015, 24, 4829–4847. [Google Scholar] [CrossRef] [Green Version]
- Sidhom, E.H.; Kim, C.; Kost-Alimova, M.; Ting, M.T.; Keller, K.; Avila-Pacheco, J.; Watts, A.J.B.; Vernon, K.A.; Marshall, J.L.; Reyes-Bricio, E.; et al. Targeting a Braf/Mapk pathway rescues podocyte lipid peroxidation in CoQ-deficiency kidney disease. J. Clin. Investig. 2021, 131, e141380. [Google Scholar] [CrossRef]
- Peng, M.; Falk, M.J.; Haase, V.H.; King, R.; Polyak, E.; Selak, M.; Yudkoff, M.; Hancock, W.W.; Meade, R.; Saiki, R.; et al. Primary coenzyme Q deficiency in Pdss2 mutant mice causes isolated renal disease. PLoS Genet. 2008, 4, e1000061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; Lu, L.-Y.; Liu, M.-F.; Yuan, Q.-J.; Sham, M.-H.; Guan, X.-Y.; Huang, J.-D. Cerebellar defects in Pdss2 conditional knockout mice during embryonic development and in adulthood. Neurobiol. Dis. 2011, 45, 219–233. [Google Scholar] [CrossRef] [Green Version]
- Widmeier, E.; Airik, M.; Hugo, H.; Schapiro, D.; Wedel, J.; Ghosh, C.C.; Nakayama, M.; Schneider, R.; Awad, A.M.; Nag, A.; et al. Treatment with 2,4-Dihydroxybenzoic Acid Prevents FSGS Progression and Renal Fibrosis in Podocyte-Specific Coq6 Knockout Mice. J. Am. Soc. Nephrol. 2019, 30, 393–405. [Google Scholar] [CrossRef] [Green Version]
- Widmeier, E.; Yu, S.; Nag, A.; Chung, Y.W.; Nakayama, M.; Fernández-Del-Río, L.; Hugo, H.; Schapiro, D.; Buerger, F.; Choi, W.-I.; et al. ADCK4 deficiency destabilizes the coenzyme Q complex, which is rescued by 2,4-dihydroxybenzoic acid treatment. J. Am. Soc. Nephrol. 2020, 31, 1191–1211. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hekimi, S. Mitochondrial respiration without ubiquinone biosynthesis. Hum. Mol. Genet. 2013, 22, 4768–4783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Oxer, D.; Hekimi, S. Mitochondrial function and lifespan of mice with controlled ubiquinone biosynthesis. Nat. Commun. 2015, 6, 6393. [Google Scholar] [CrossRef] [Green Version]
- Lapointe, J.; Wang, Y.; Bigras, E.; Hekimi, S. The submitochondrial distribution of ubiquinone affects respiration in long-lived Mclk1+/− mice. J. Cell Biol. 2012, 199, 215–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levavasseur, F.; Miyadera, H.; Sirois, J.; Tremblay, M.L.; Kita, K.; Shoubridge, E.; Hekimi, S. Ubiquinone is necessary for mouse embryonic development but is not essential for mitochondrial respiration. J. Biol. Chem. 2001, 276, 46160–46164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakai, D.; Yuasa, S.; Takahashi, M.; Shimizu, T.; Asaumi, S.; Isono, K.; Tako, T.; Suzuki, Y.-S.; Kuroyanagi, K.; Hirokawa, K.; et al. Mouse homologue of coq7/clk-1, longevity gene in Caenorhabditis elegans, is essential for coenzyme Q synthesis, maintenance of mitochondrial integrity, and neurogenesis. Biochem. Biophys. Res. Commun. 2001, 289, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jiang, N.; Hughes, B.; Bigras, E.; Shoubridge, E.; Hekimi, S. Evolutionary conservation of the clk-1-dependent mechanism of longevity: Loss of mclk1 increases cellular fitness and lifespan in mice. Genes Dev. 2005, 19, 2424–2434. [Google Scholar] [CrossRef] [Green Version]
- Stefely, J.; Licitra, F.; Laredj, L.; Reidenbach, A.G.; Kemmerer, Z.A.; Grangeray, A.; Jaeg-Ehret, T.; Minogue, C.E.; Ulbrich, A.; Hutchins, P.; et al. Cerebellar ataxia and coenzyme Q deficiency through loss of unorthodox kinase activity. Mol. Cell 2016, 63, 608–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez, M.L.; Casado, M.E.D.; Barca, E.; Tejada, M.; Montilla-García, A.; Cobos, E.J.; Escames, G.; Acuña-Castroviejo, D.; Quinzii, C.M.; López, L.C. The clinical heterogeneity of coenzyme Q 10 deficiency results from genotypic differences in the Coq9 gene. EMBO Mol. Med. 2015, 7, 670–687. [Google Scholar] [CrossRef]
- Rodríguez-Hidalgo, M.; Luna-Sánchez, M.; Gutiérrez, A.H.; Barriocanal-Casado, E.; Mascaraque, C.; Acuña-Castroviejo, D.; Rivera, M.; Escames, G.; López, L.C. Reduction in the levels of CoQ biosynthetic proteins is related to an increase in lifespan without evidence of hepatic mitohormesis. Sci. Rep. 2018, 8, 14013. [Google Scholar] [CrossRef]
- Luna-Sánchez, M.; Hidalgo-Gutiérrez, A.; Hildebrandt, T.M.; Chaves-Serrano, J.; Barriocanal-Casado, E.; Santos-Fandila, Á.; Romero, M.; Sayed, R.K.; Duarte, J.; Prokisch, H.; et al. CoQ deficiency causes disruption of mitochondrial sulfide oxidation, a new pathomechanism associated with this syndrome. EMBO Mol. Med. 2016, 9, 78–95. [Google Scholar] [CrossRef] [PubMed]
- González-García, P.; Hidalgo-Gutiérrez, A.; Mascaraque, C.; Barriocanal-Casado, E.; Bakkali, M.; Ziosi, M.; Abdihankyzy, U.B.; Sánchez-Hernández, S.; Escames, G.; Prokisch, H.; et al. Coenzyme Q10 modulates sulfide metabolism and links the mitochondrial respiratory chain to pathways associated to one carbon metabolism. Hum. Mol. Genet. 2020, 29, 3296–3311. [Google Scholar] [CrossRef]
- García-Corzo, L.; Luna-Sánchez, M.; Doerrier, C.; Ortiz, F.; Escames, G.; Acuña-Castroviejo, D.; López, L.C. Ubiquinol-10 ameliorates mitochondrial encephalopathy associated with CoQ deficiency. Biochim. Biophys. Acta 2014, 1842, 893–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hidalgo-Gutierrez, A.; Barriocanal-Casado, E.; Bakkali, M.; Diaz-Casado, M.E.; Sanchez-Maldonado, L.; Romero, M.; Sayed, R.K.; Prehn, C.; Escames, G.; Duarte, J.; et al. beta-RA reduces DMQ/CoQ ratio and rescues the encephalopathic phenotype in Coq9 (R239X) mice. EMBO Mol. Med. 2019, 11, e9466. [Google Scholar] [CrossRef] [PubMed]
- Barriocanal-Casado, E.; Cueto-Ureña, C.; Benabdellah, K.; Gutiérrez-Guerrero, A.; Cobo, M.; Gutiérrez, A.H.; Rodríguez-Sevilla, J.J.; Martín, F.; Lopez, L.C. Gene therapy corrects mitochondrial dysfunction in hematopoietic progenitor cells and fibroblasts from Coq9R239X mice. PLoS ONE 2016, 11, e0158344. [Google Scholar] [CrossRef]
- Vázquez-Fonseca, L.; Schäefer, J.; Navas-Enamorado, I.; Santos-Ocaña, C.; Hernández-Camacho, J.D.; Guerra, I.; Cascajo, M.V.; Sánchez-Cuesta, A.; Horvath, Z.; Siendones, E.; et al. ADCK2 haploinsufficiency reduces mitochondrial lipid oxidation and causes myopathy associated with CoQ Deficiency. J. Clin. Med. 2019, 8, 1374. [Google Scholar] [CrossRef] [Green Version]
- Spinazzi, M.; Radaelli, E.; Horré, K.; Arranz, A.M.; Gounko, N.V.; Agostinis, P.; Maia, T.M.; Impens, F.; Morais, V.A.; Lopez-Lluch, G.; et al. PARL deficiency in mouse causes Complex III defects, coenzyme Q depletion, and Leigh-like syndrome. Proc. Natl. Acad. Sci. USA 2018, 116, 277–286. [Google Scholar] [CrossRef] [Green Version]
- Licitra, F.; Puccio, H. An Overview of Current Mouse Models Recapitulating Coenzyme Q10 Deficiency Syndrome. Mol. Syndr. 2014, 5, 180–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Ayala, D.J.M.; Gancedo, S.J.; Guerra, I.; Navas, P. Invertebrate Models for Coenzyme Q10 Deficiency. Mol. Syndr. 2014, 5, 170–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeibmann, A.; Paulus, W. Drosophila melanogaster as a Model Organism of Brain Diseases. Int. J. Mol. Sci. 2009, 10, 407–440. [Google Scholar] [CrossRef] [PubMed]
- Doimo, M.; Trevisson, E.; Airik, R.; Bergdoll, M.; Santos-Ocaña, C.; Hildebrandt, F.; Navas, P.; Pierrel, F.; Salviati, L. Effect of vanillic acid on COQ6 mutants identified in patients with coenzyme Q10 deficiency. Biochim. Biophys. Acta 2014, 1842, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamath, R.S.; Fraser, A.G.; Dong, Y.; Poulin, G.; Durbin, R.; Gotta, M.; Kanapin, A.; Le Bot, N.; Moreno, S.; Sohrmann, M.; et al. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 2003, 421, 231–237. [Google Scholar] [CrossRef]
- Wong, A.; Boutis, P.; Hekimi, S. Mutations in the clk-1 gene of Caenorhabditis elegans affect developmental and behavioral timing. Genetics 1995, 139, 1247–1259. [Google Scholar] [CrossRef] [PubMed]
- Cristina, D.; Cary, M.; Lunceford, A.; Clarke, C.; Kenyon, C. A regulated response to impaired respiration slows behavioral rates and increases lifespan in Caenorhabditis elegans. PLoS Genet. 2009, 5, e1000450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arroyo, A.; Santos-Ocaña, C.; Ruiz-Ferrer, M.; Padilla, S.; Gavilán, A.; Rodríguez-Aguilera, J.C.; Navas, P. Coenzyme Q is irreplaceable by demethoxy-coenzyme Q in plasma membrane of Caenorhabditis elegans. FEBS Lett. 2006, 580, 1740–1746. [Google Scholar] [CrossRef] [PubMed]
- Lieschke, G.J.; Currie, P. Animal models of human disease: Zebrafish swim into view. Nat. Rev. Genet. 2007, 8, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, K.; Sawada, N.; Hirota, Y.; Uchino, Y.; Suhara, Y.; Hasegawa, T.; Amizuka, N.; Okamoto, T.; Tsugawa, N.; Kamao, M.; et al. Vitamin K2 biosynthetic enzyme, UBIAD1 is essential for embryonic development of mice. PLoS ONE 2014, 9, e104078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegarty, J.M.; Yang, H.; Chi, N.C. UBIAD1-mediated vitamin K2 synthesis is required for vascular endothelial cell survival and development. Development 2013, 140, 1713–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, U.C.; Clarke, C.F. Endogenous synthesis of coenzyme Q in eukaryotes. Mitochondrion 2007, 7, S62–S71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyon, M.F.; Hulse, E.V. An inherited kidney disease of mice resembling human nephronophthisis. J. Med. Genet. 1971, 8, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Neilson, E.G.; McCafferty, E.; Feldman, A.; Clayman, M.D.; Zakheim, B.; Korngold, R. Spontaneous interstitial nephritis in kdkd mice. I. An experimental model of autoimmune renal disease. J. Immunol. 1984, 133, 2560–2565. [Google Scholar] [PubMed]
- Kelly, C.J.; Korngold, R.; Mann, R.; Clayman, M.; Haverty, T.; Neilson, E.G. Spontaneous interstitial nephritis in kdkd mice. II. Characterization of a tubular antigen-specific, H-2K-restricted Lyt-2+ effector T cell that mediates destructive tubulointerstitial injury. J Immunol. 1986, 136, 526–531. [Google Scholar] [PubMed]
- Hancock, W.W.; Tsai, T.-L.; Madaio, M.P.; Gasser, D.L. Cutting Edge: Multiple autoimmune pathways in kd/kd mice. J. Immunol. 2003, 171, 2778–2781. [Google Scholar] [CrossRef] [Green Version]
- Madaio, M.P.; Ahima, R.S.; Meade, R.; Rader, D.J.; Mendoza, A.; Peng, M.; Tomaszewski, J.E.; Hancock, W.W.; Gasser, D.L. Glomerular and tubular epithelial defects in kd/kd mice lead to progressive renal failure. Am. J. Nephrol. 2005, 25, 604–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, M.; Jarett, L.; Meade, R.; Madaio, M.P.; Hancock, W.W.; George, A.L.; Neilson, E.G.; Gasser, D.L. Mutant prenyltransferase-like mitochondrial protein (PLMP) and mitochondrial abnormalities in kd/kd mice. Kidney Int. 2004, 66, 20–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rötig, A.; Mollet, J.; Rio, M.; Munnich, A. Infantile and pediatric quinone deficiency diseases. Mitochondrion 2007, 7, S112–S121. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-M.; Ng, A.H.-L.; Tanner, J.A.; Wu, W.-T.; Copeland, N.G.; Jenkins, N.A.; Huang, J.-D. Highly restricted expression of Cre recombinase in cerebellar Purkinje cells. Genesis 2004, 40, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Hsu, A.Y.; Poon, W.; Shepherd, J.A.; Myles, D.C.; Clarke, C.F. Complementation of coq3 mutant yeast by mitochondrial targeting of the escherichia coli UbiG polypeptide: Evidence that UbiG catalyzes both O-methylation steps in ubiquinone biosynthesis. Biochemistry 1996, 35, 9797–9806. [Google Scholar] [CrossRef]
- Lapointe, J.; Hekimi, S. Early mitochondrial dysfunction in long-lived Mclk1+/− mice. J. Biol. Chem. 2008, 283, 26217–26227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapointe, J.; Stepanyan, Z.; Bigras, E.; Hekimi, S. Reversal of the mitochondrial phenotype and slow development of oxidative biomarkers of aging in long-lived Mclk1+/− mice. J. Biol. Chem. 2009, 284, 20364–20374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Malo, D.; Hekimi, S. Elevated mitochondrial reactive oxygen species generation affects the immune response via hypoxia-inducible factor-1alpha in long-lived Mclk1+/− mouse mutants. J. Immunol. 2010, 184, 582–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, H.; Lapointe, J.; Hekimi, S. Lifelong protection from global cerebral ischemia and reperfusion in long-lived Mclk1(+/) (−) mutants. Exp. Neurol. 2010, 223, 557–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, R.; Zhang, F.; Chen, G.; Han, C.; Liu, J.; Ren, Z.; Zhu, Y.; Waddington, J.L.; Zheng, L.T.; Zhen, X. Clk1 deficiency promotes neuroinflammation and subsequent dopaminergic cell death through regulation of microglial metabolic reprogramming. Brain Behav. Immun. 2017, 60, 206–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakowski, B.; Hekimi, S. Determination of Life-Span in Caenorhabditis elegans by Four Clock Genes. Science 1996, 272, 1010–1013. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo-Gutiérrez, A.; Barriocanal-Casado, E.; Díaz-Casado, M.E.; González-García, P.; Chiozzi, R.Z.; Acuña-Castroviejo, D.; López, L.C. β-RA Targets mitochondrial metabolism and adipogenesis, leading to therapeutic benefits against CoQ deficiency and age-related overweight. Biomedicines 2021, 9, 1457. [Google Scholar] [CrossRef] [PubMed]
- Barriocanal-Casado, E.; Hidalgo-Gutiérrez, A.; Raimundo, N.; González-García, P.; Acuña-Castroviejo, D.; Escames, G.; López, L.C. Rapamycin administration is not a valid therapeutic strategy for every case of mitochondrial disease. EBioMedicine 2019, 42, 511–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, S.C.; Yanos, M.E.; Kayser, E.-B.; Quintana, A.; Sangesland, M.; Castanza, A.; Uhde, L.; Hui, J.; Wall, V.Z.; Gagnidze, A.; et al. mTOR inhibition alleviates mitochondrial disease in a mouse model of leigh syndrome. Science 2013, 342, 1524–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef] [Green Version]
- Düsterhöft, S.; Künzel, U.; Freeman, M. Rhomboid proteases in human disease: Mechanisms and future prospects. Biochim. Biophys. Acta Bioenerg. 2017, 1864, 2200–2209. [Google Scholar] [CrossRef]
- Herebian, D.; Seibt, A.; Smits, S.H.J.; Rodenburg, R.J.; Mayatepek, E.; Distelmaier, F. 4-Hydroxybenzoic acid restores CoQ10 biosynthesis in human COQ2 deficiency. Ann. Clin. Transl. Neurol. 2017, 4, 902–908. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Model | Strain | Phenotype | Coenzyme Q | CoQ Deficiencies | Ref | ||
|---|---|---|---|---|---|---|---|
| Biosynthesis | Functions | Pathological Mechanisms | Treatments | ||||
| Drosophila melanogaster | qless mutant | Nervous system failure | Caspase-dependent apoptosis in neurons | CoQ4, CoQ9 or CoQ10 rescue the phenotype. | [78] | ||
| sbo mutant | Small larvae phenotype Controversial extended lifespan | CoQ is important in the early stages of development and fertility. | More susceptible to bacterial and fungal infections | CoQ10 partially rescue the phenotype. | [79,80] | ||
| Coq2 mutant | Renal failure Larval lethality | CoQ is important in the early stages of development. | ROS accumulation | Glutathione and vanillic acid rescued the phenotype. | [81,82] | ||
| Coq3 mutant | Larval lethality | Existence of complex Q | CoQ is important in the early stages of development. | DMQ accumulation | [82,83] | ||
| Coq5 mutant | Larval lethality | CoQ is important in the early stages of development. | [82,83] | ||||
| Coq6 mutant | Existence of complex Q | DMQ accumulation | [82,83] | ||||
| Coq7 mutant | Larval lethality | Existence of complex Q | CoQ is important in the early stages of development. | DMQ accumulation | [82,83] | ||
| Coq8 mutant | Larval lethality | CoQ is important in the early stages of development. | [82,83] | ||||
| Coq9 mutant | Larval lethality | Existence of complex Q | CoQ is important in the early stages of development. | DMQ accumulation | [82,83] | ||
| Coq10 mutant | Larval lethality | CoQ is important in the early stages of development. | [82,83] | ||||
| Caernorhabditis elegans | Mev-1 mutant | Shortened lifespan | CoQ is an important link between lifespan and oxidative stress. | ROS accumulation. Altered reduced and oxidized forms of CoQ | CoQ10 rescued the phenotype. | [84,85,86] | |
| Clk-1 mutant | Extended lifespan | COQ7 has catalytic activity. | CoQ10 has the highest antioxidant properties. | Accumulation of DMQ9. Lower production of ROS. Inhibition of complex I by DMQ9 | CoQ10 restored the phenotype. | [87,88,89,90,91,92] | |
| Coq-1 mutant | Larval lethality Muscle failure Nervous system failure | CoQ is important in the early stages of development and fertility. | Degeneration of GABA neurons | CoQ10 or feeding worms with bacteria containing its own CoQ8 did not improved the phenotype | [92,93,94] | ||
| Coq-2 mutant | Larval lethality Muscle failure Nervous system failure | CoQ is important in the early stages of development and fertility. | Degeneration of GABA neurons | CoQ10 or feeding worms with bacteria containing its own CoQ8 did not improved the phenotype | [92,93,94] | ||
| Coq-3 mutant | Larval lethality Nervous system failure | CoQ is important in the early stages of development and fertility. | Degeneration of GABA neurons | Extra-chromosomal array containing the own C. elegans coq-3 gene rescue the phenotype. | [92,94,95] | ||
| Coq8 mutant | Larval lethality | CoQ is important in the early stages of development and fertility. | CoQ-rich diet increased its lifespan | [93,96] | |||
| Danio rerio (zebrafish) | ubiad 1 (bar) mutant | Cardiovascular failure | UBIAD1 participates in non-mitochondrial CoQ biosynthesis. | UBIAD1-mediated CoQ10 production is important for mem-brane redox signaling and protection from lipid peroxidation. | ROS accumulation. | [97] | |
| Coq2 mutant | Normal | COQ2-mediated CoQ10 production is for mitochondrial respiratory chain function and energy production. | ROS accumulation. | [97] | |||
| Coq6 mutant | Apoptosis increase. | [57] | |||||
| Coq8b mutant | Renal failure | [63] | |||||
| Mus Musculus | Pdss2kd/kd | Renal failure | Pdss2 participates in the biosynthesis of CoQ. | Disruption of sulfide oxidation pathway. Inhibition of short-chain acyl-CoA dehy-drogenase. Glutathione depletion. | CoQ10 rescued proteinuria and intersti-tial nephritis. Probucol had a more powerful health improvement than high-dose CoQ10. Rapamycin reduced the proteinuria. Treatment with GDC0879 ameliorated kidney disease. | [98,99,100,101,102,103,104] | |
| Podocin/cre, Pdss2loxP/loxP | Nephrotic syndrome | Renal glomerular podocytes display the greatest sensitivity to Pdss2 impairment | [105] | ||||
| Pdss2f/-; Pax2-cre | Cerebellar ataxia | Defect in cell migration, cell proliferation, and increased apoptosis in cerebellum | [106] | ||||
| Pdss2f/−; Pcp2-cre | Cerebellar ataxia | Ataxia at old age. Progressive decreased of Purkinje cells and neuron death by apoptosis | [106] | ||||
| Coq6podKO | Nephrotic syndrome | 2,4-diHB rescued the phenotype. | [107] | ||||
| Adck4ΔPodocyte | Nephrotic syndrome | ADCK4 is a protein component in CoQ biosynthesis with no catalytic activity. | 2,4-diHB rescued the phenotype. | [108] | |||
| Coq7liver-KO | Hepatic failure | A small amount of CoQ is required in the respiratory chain function in liver. | Accumulation of DMQ9. DMQ seems not to interfere with CoQ-mediated mitochondrial electron transport in the liver | CoQ10 partially rescued the phenotype. | [109] | ||
| aogCoq7 (Coq7 KO by TM) | Early death, but unclear phenotype | Energy-demanding tissues are not highly susceptible to CoQ deficit and defective mitochondrial energy metabolism. | Accumulation of DMQ9 | CoQ10 was ineffective. 2,4-diHB partially rescued the phenotype. | [110] | ||
| Pdss2−/− | Embryonic lethality | CoQ is important in the early stages of development. | [105] | ||||
| Coq3−/− | Embryonic lethality | CoQ is important in the early stages of development. | [111] | ||||
| Coq7−/− | Embryonic lethality | CoQ is important in the early stages of development. | Accumulation of DMQ | [112,113] | |||
| Coq7+/− | Extended lifespan | Decreased CoQ levels in the inner mitochondrial membrane coupled with higher CoQ levels in the outer mitochondrial membrane. Early hepatic mitochondrial dysfunction, which induces a protective physiological response (mitohormesis) | CoQ10 normalized the CoQ levels. | [111,114] | |||
| Coq8a−/− | Cerebellar ataxia | COQ8a participates in the stability of complex Q. | Purkinje cells displayed Golgi morphology defects, but normal mitochondria. Model to study CoQ production across different organelles, cells, and tissues | [115] | |||
| Coq9Q95X | Extended lifespan | COQ9 is needed for the stability and activity of Coq7. | Disruption of mitochondrial sulfide oxidation pathway | [116,117] | |||
| Coq9R239X | Encephalopathy | COQ9 is needed for the stability and activity of COQ7. | Accumulation of DMQ. Disruption of mitochondrial sulfide oxidation pathway | Ubiquinone-10 had limited efficacy. Ubiquinol-10 provided better therapeutic outcomes. 2,4-diHB full rescued the phenotype. Rapamycin has no efficacy. | [45,116,118,119,120,121,122] | ||
| Adck2+/− | Myopathy | ADCK2 participates in the control of CoQ levels. | Impairment in lipid metabolism | CoQ partially recovered the phenotype. | [123] | ||
| Parl−/− | Encephalopathy | Parl is required for efficient CoQ biosynthesis in the brain by stabilization of COQ4 expression. | Downregulation of the CIII-regulating protein TTC19 | [124] | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-García, P.; Barriocanal-Casado, E.; Díaz-Casado, M.E.; López-Herrador, S.; Hidalgo-Gutiérrez, A.; López, L.C. Animal Models of Coenzyme Q Deficiency: Mechanistic and Translational Learnings. Antioxidants 2021, 10, 1687. https://doi.org/10.3390/antiox10111687
González-García P, Barriocanal-Casado E, Díaz-Casado ME, López-Herrador S, Hidalgo-Gutiérrez A, López LC. Animal Models of Coenzyme Q Deficiency: Mechanistic and Translational Learnings. Antioxidants. 2021; 10(11):1687. https://doi.org/10.3390/antiox10111687
Chicago/Turabian StyleGonzález-García, Pilar, Eliana Barriocanal-Casado, María Elena Díaz-Casado, Sergio López-Herrador, Agustín Hidalgo-Gutiérrez, and Luis C. López. 2021. "Animal Models of Coenzyme Q Deficiency: Mechanistic and Translational Learnings" Antioxidants 10, no. 11: 1687. https://doi.org/10.3390/antiox10111687