
Combined Activity of the Redox-Modulating Compound Setanaxib (GKT137831) with Cytotoxic Agents in the Killing of Acute Myeloid Leukemia Cells


 , , and
, , and
Abstract
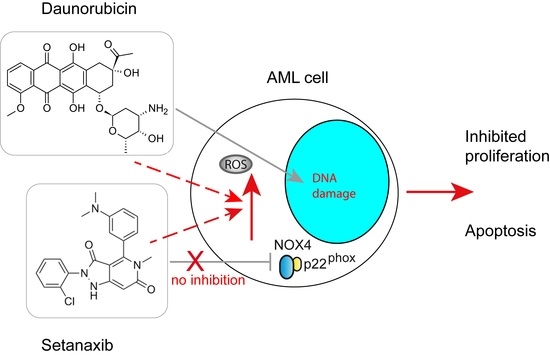
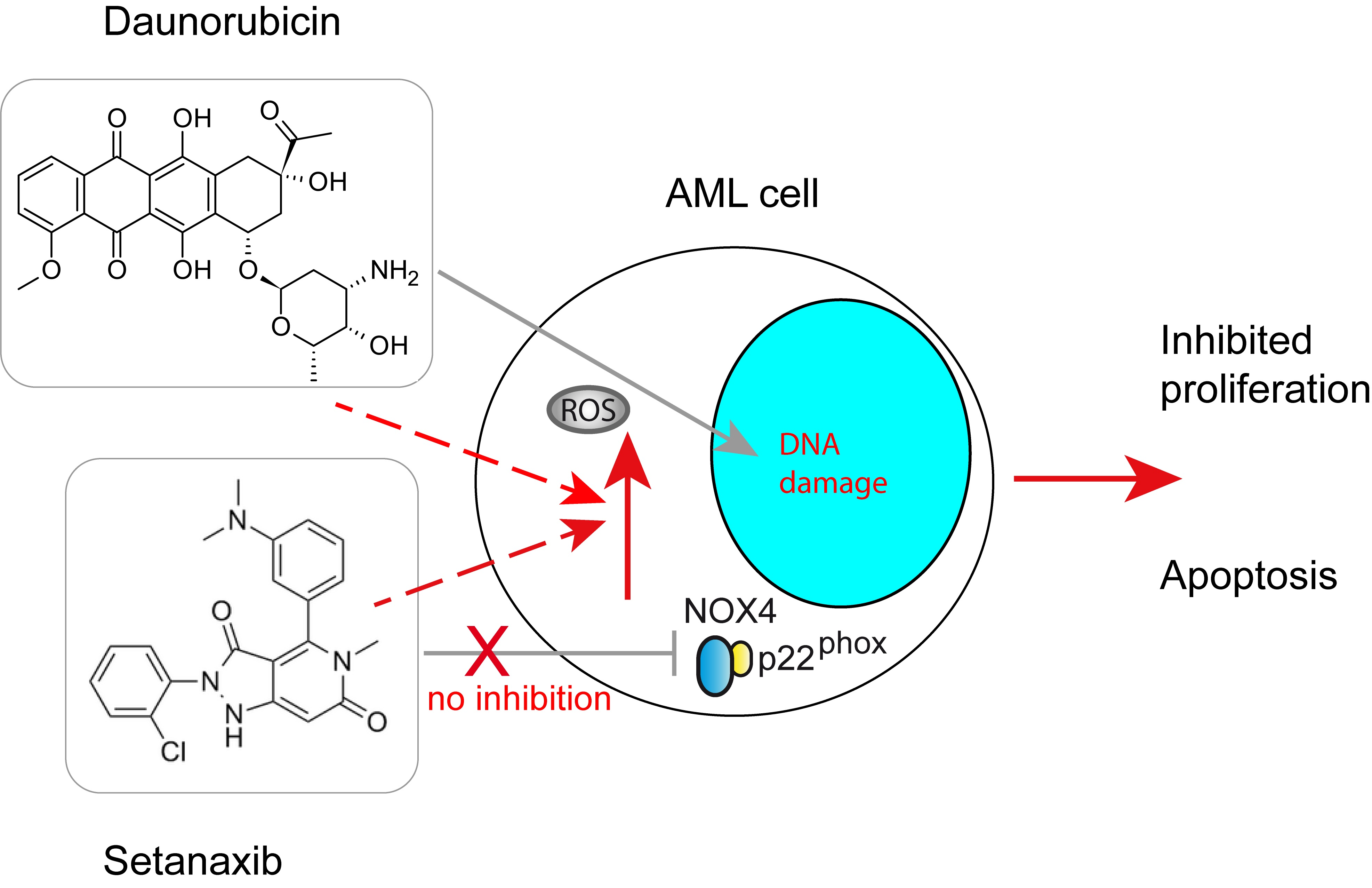
:
1. Introduction
2. Material and Methods
2.1. Cell Lines and Reagents
2.2. Generation of Genetically Modified Cell Lines
2.3. Proliferation and Apoptosis Assays
2.4. Animal Experiments
2.5. Immunoblotting and Reactive Oxygen Assays
2.6. Determination of Synergism and Statistics
3. Results
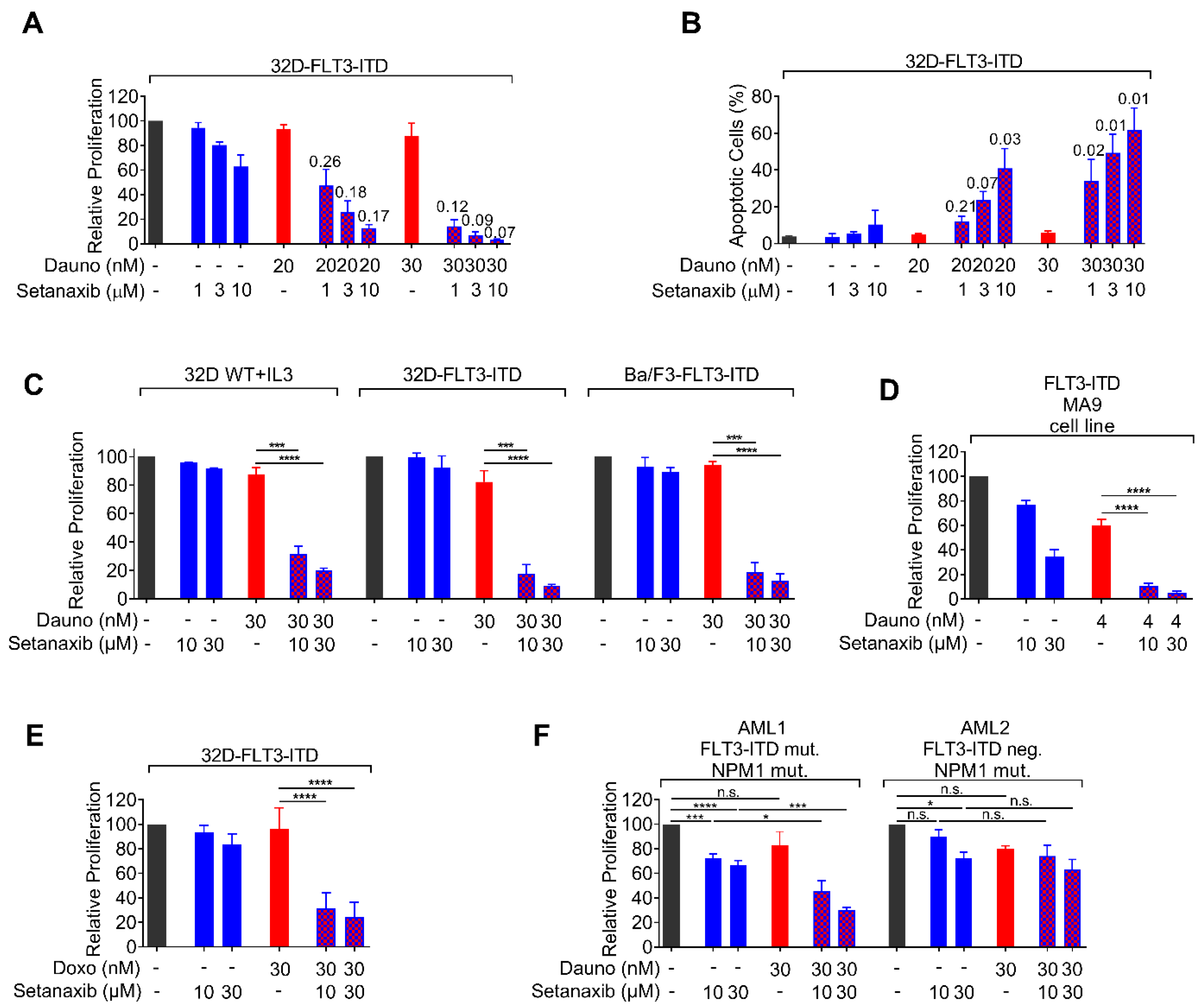
3.1. Setanaxib Has Inhibitory Activity on Growth of AML Cells In Vitro
3.2. Setanaxib Is Synergistic with Daunorubicin in FLT3-ITD-Positive Cells
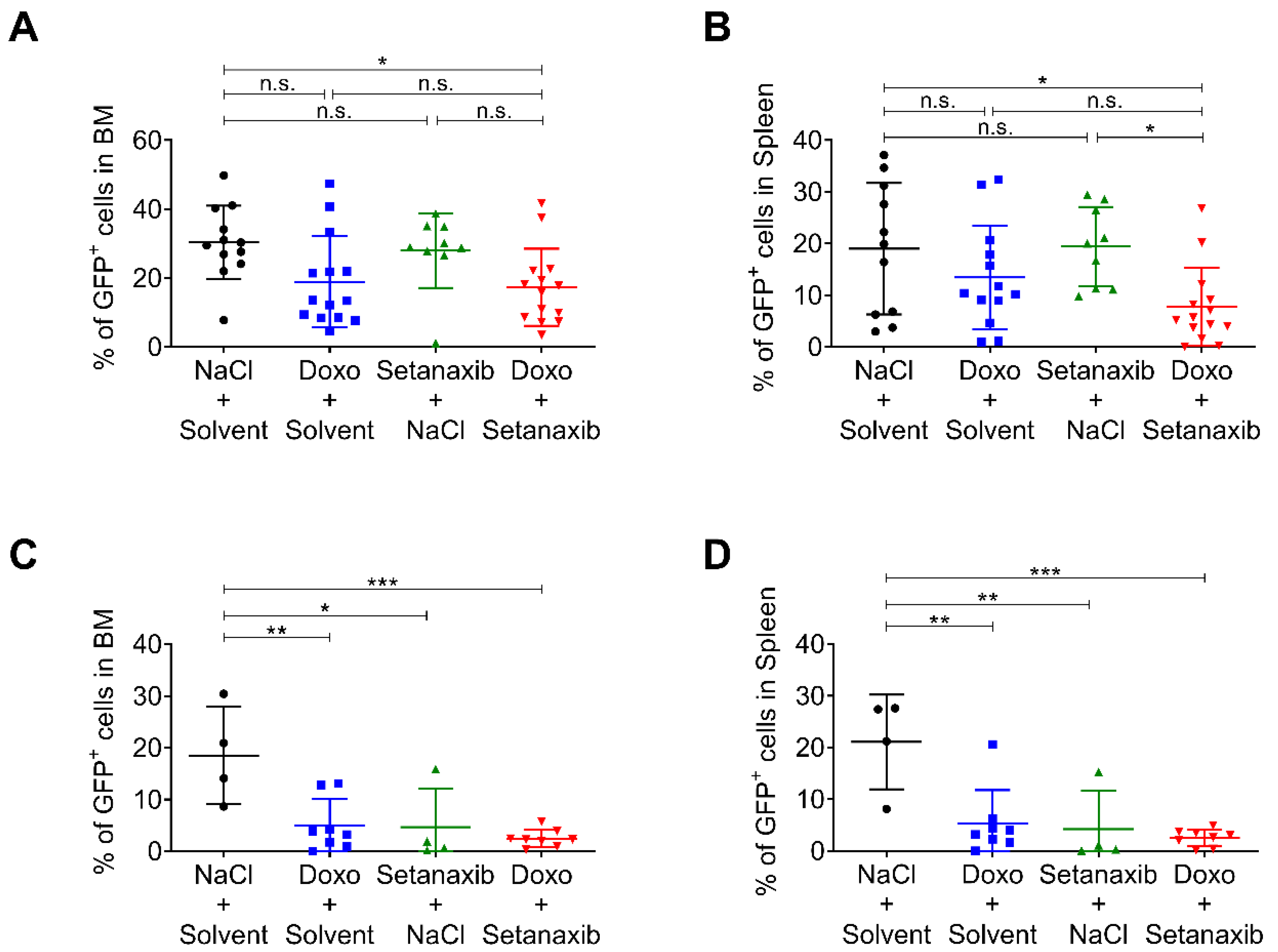
3.3. Setanaxib Has Efficiency In Vivo
3.4. Effects of Setanaxib Are Independent of FLT3-Signaling, Presence of NOX4, or ROS Quenching
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Shekhar, S.; Liu, Y.; Wang, S.; Zhang, H.; Fang, X.; Zhang, J.; Fan, L.; Zheng, B.; Roman, R.J.; Wang, Z.; et al. Novel mechanistic insights and potential therapeutic impact of TRPC6 in neurovascular coupling and ischemic stroke. Int. J. Mol. Sci. 2021, 22, 2074. [Google Scholar] [CrossRef] [PubMed]
- Tretter, V.; Hochreiter, B.; Zach, M.L.; Krenn, K.; Klein, K.U. Understanding cellular redox homeostasis: A challenge for precision medicine. Int. J. Mol. Sci. 2021, 23, 106. [Google Scholar] [CrossRef]
- Chen, K.; Lu, P.; Beeraka, N.M.; Sukocheva, O.A.; Madhunapantula, S.V.; Liu, J.; Sinelnikov, M.Y.; Nikolenko, V.N.; Bulygin, K.V.; Mikhaleva, L.M.; et al. Mitochondrial mutations and mitoepigenetics: Focus on regulation of oxidative stress-induced responses in breast cancers. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2020. [Google Scholar] [CrossRef]
- Dunyaporn, T.; Jerome, A.; Peng, H. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Reczek, C.; Chandel, N. The two faces of reactive oxygen species in cancer. Ann. Rev. Cancer Biol. 2017, 1, 79–98. [Google Scholar] [CrossRef]
- Azmanova, M.; Pitto-Barry, A. Oxidative stress in cancer therapy: Friend or enemy? Chembiochem 2022, e202100641. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [CrossRef] [Green Version]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Buchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [Green Version]
- Döhner, H.; Estey, E.H.; Amadori, S.; Appelbaum, F.R.; Buchner, T.; Burnett, A.K.; Dombret, H.; Fenaux, P.; Grimwade, D.; Larson, R.A.; et al. Diagnosis and management of acute myeloid leukemia in adults: Recommendations from an international expert panel, on behalf of the European leukemianet. Blood 2010, 115, 453–474. [Google Scholar] [CrossRef]
- Kazi, J.U.; Rönnstrand, L. FMS-like tyrosine kinase 3/FLT3: From basic science to clinical implications. Physiol. Rev. 2019, 99, 1433–1466. [Google Scholar] [CrossRef] [PubMed]
- Kiyoi, H.; Towatari, M.; Yokota, S.; Hamaguchi, M.; Ohno, R.; Saito, H.; Naoe, T. Internal tandem duplication of the FLT3 gene is a novel modality of elongation mutation which causes constitutive activation of the product. Leukemia 1998, 12, 1333–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Small, D. Targeting FLT3 for the treatment of leukemia. Semin. Hematol. 2008, 45, S17–S21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhary, C.; Olsen, J.V.; Brandts, C.; Cox, J.; Reddy, P.N.; Böhmer, F.D.; Gerke, V.; Schmidt-Arras, D.E.; Berdel, W.E.; Müller-Tidow, C.; et al. Mislocalized activation of oncogenic RTKs switches downstream signaling outcomes. Mol. Cell 2009, 36, 326–339. [Google Scholar] [CrossRef]
- Schmidt-Arras, D.; Böhmer, F.D. Mislocalisation of activated receptor tyrosine kinases—challenges for cancer therapy. Trends Mol. Med. 2020, 26, 833–847. [Google Scholar] [CrossRef]
- Daver, N.; Schlenk, R.F.; Russell, N.H.; Levis, M.J. Targeting FLT3 mutations in AML: Review of current knowledge and evidence. Leukemia 2019, 33, 299–312. [Google Scholar] [CrossRef] [Green Version]
- Scholl, S.; Fleischmann, M.; Schnetzke, U.; Heidel, F.H. Molecular mechanisms of resistance to FLT3 inhibitors in acute myeloid leukemia: Ongoing challenges and future treatments. Cells 2020, 9, 2493. [Google Scholar] [CrossRef]
- Stone, R.M.; Manley, P.W.; Larson, R.A.; Capdeville, R. Midostaurin: Its odyssey from discovery to approval for treating acute myeloid leukemia and advanced systemic mastocytosis. Blood Adv. 2018, 2, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Sallmyr, A.; Fan, J.; Datta, K.; Kim, K.T.; Grosu, D.; Shapiro, P.; Small, D.; Rassool, F. Internal tandem duplication of FLT3 (FLT3/ITD) induces increased ROS production, DNA damage, and misrepair: Implications for poor prognosis in AML. Blood 2008, 111, 3173–3182. [Google Scholar] [CrossRef] [Green Version]
- Hole, P.S.; Zabkiewicz, J.; Munje, C.; Newton, Z.; Pearn, L.; White, P.; Marquez, N.; Hills, R.K.; Burnett, A.K.; Tonks, A.; et al. Overproduction of NOX-derived ROS in AML promotes proliferation and is associated with defective oxidative stress signaling. Blood 2013, 122, 3322–3330. [Google Scholar] [CrossRef]
- Godfrey, R.; Arora, D.; Bauer, R.; Stopp, S.; Müller, J.P.; Heinrich, T.; Böhmer, S.A.; Dagnell, M.; Schnetzke, U.; Scholl, S.; et al. Cell transformation by FLT3 ITD in acute myeloid leukemia involves oxidative inactivation of the tumor suppressor protein-tyrosine phosphatase DEP-1/ PTPRJ. Blood 2012, 119, 4499–4511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayavelu, A.K.; Moloney, J.N.; Böhmer, F.D.; Cotter, T.G. NOX-driven ROS formation in cell transformation of FLT3-ITD-positive AML. Exp. Hematol. 2016, 44, 1113–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sillar, J.R.; Germon, Z.P.; DeIuliis, G.N.; Dun, M.D. The role of reactive oxygen species in acute myeloid leukaemia. Int. J. Mol. Sci. 2019, 20, 6003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayavelu, A.K.; Müller, J.P.; Bauer, R.; Bohmer, S.A.; Lässig, J.; Cerny-Reiterer, S.; Sperr, W.R.; Valent, P.; Maurer, B.; Moriggl, R.; et al. NOX4-driven ROS formation mediates PTP inactivation and cell transformation in FLT3ITD-positive AML cells. Leukemia 2016, 30, 473–483. [Google Scholar] [CrossRef]
- Laleu, B.; Gaggini, F.; Orchard, M.; Fioraso-Cartier, L.; Cagnon, L.; Houngninou-Molango, S.; Gradia, A.; Duboux, G.; Merlot, C.; Heitz, F.; et al. First in class, potent, and orally bioavailable NADPH oxidase isoform 4 (Nox4) inhibitors for the treatment of idiopathic pulmonary fibrosis. J. Med. Chem. 2010, 53, 7715–7730. [Google Scholar] [CrossRef]
- Teixeira, G.; Szyndralewiez, C.; Molango, S.; Carnesecchi, S.; Heitz, F.; Wiesel, P.; Wood, J.M. Therapeutic potential of NADPH oxidase 1/4 inhibitors. Br. J. Pharmacol. 2017, 174, 1647–1669. [Google Scholar] [CrossRef] [Green Version]
- Arora, D.; Stopp, S.; Böhmer, S.A.; Schons, J.; Godfrey, R.; Masson, K.; Razumovskaya, E.; Rönnstrand, L.; Tänzer, S.; Bauer, R.; et al. Protein-tyrosine phosphatase DEP-1 controls receptor tyrosine kinase FLT3 signaling. J. Biol. Chem. 2011, 286, 10918–10929. [Google Scholar] [CrossRef] [Green Version]
- Mizuki, M.; Fenski, R.; Halfter, H.; Matsumura, I.; Schmidt, R.; Muller, C.; Gruning, W.; Kratz-Albers, K.; Serve, S.; Steur, C.; et al. Flt3 mutations from patients with acute myeloid leukemia induce transformation of 32D cells mediated by the ras and STAT5 pathways. Blood 2000, 96, 3907–3914. [Google Scholar] [CrossRef]
- Arreba-Tutusaus, P.; Mack, T.S.; Bullinger, L.; Schnöder, T.M.; Polanetzki, A.; Weinert, S.; Ballaschk, A.; Wang, Z.; Deshpande, A.J.; Armstrong, S.A.; et al. Impact of FLT3-ITD location on sensitivity to TKI-therapy in vitro and in vivo. Leukemia 2016, 30, 1220–1225. [Google Scholar] [CrossRef]
- Serrander, L.; Cartier, L.; Bedard, K.; Banfi, B.; Lardy, B.; Plastre, O.; Sienkiewicz, A.; Forro, L.; Schlegel, W.; Krause, K.H. NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem. J. 2007, 406, 105–114. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.H.; Tothova, Z.; Levine, R.L.; Anderson, K.; Buza-Vidas, N.; Cullen, D.E.; McDowell, E.P.; Adelsperger, J.; Fröhling, S.; Huntly, B.J.; et al. FLT3 mutations confer enhanced proliferation and survival properties to multipotent progenitors in a murine model of chronic myelomonocytic leukemia. Cancer Cell 2007, 12, 367–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, T.C. Drug combination studies and their synergy quantification using the chou-talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuber, J.; Radtke, I.; Pardee, T.S.; Zhao, Z.; Rappaport, A.R.; Luo, W.; McCurrach, M.E.; Yang, M.M.; Dolan, M.E.; Kogan, S.C.; et al. Mouse models of human AML accurately predict chemotherapy response. Genes Dev. 2009, 23, 877–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augsburger, F.; Filippova, A.; Rasti, D.; Seredenina, T.; Lam, M.; Maghzal, G.; Mahiout, Z.; Jansen-Dürr, P.; Knaus, U.G.; Doroshow, J.; et al. Pharmacological characterization of the seven human NOX isoforms and their inhibitors. Redox Biol. 2019, 26, 101272. [Google Scholar] [CrossRef]
- Dao, V.T.; Elbatreek, M.H.; Altenhofer, S.; Casas, A.I.; Pachado, M.P.; Neullens, C.T.; Knaus, U.G.; Schmidt, H. Isoform-selective NADPH oxidase inhibitor panel for pharmacological target validation. Free Radic. Biol. Med. 2020, 148, 60–69. [Google Scholar] [CrossRef]
- Brandes, R.P.; Weissmann, N.; Schröder, K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic. Biol. Med. 2014, 76, 208–226. [Google Scholar] [CrossRef]
- Zarrinkar, P.P.; Gunawardane, R.N.; Cramer, M.D.; Gardner, M.F.; Brigham, D.; Belli, B.; Karaman, M.W.; Pratz, K.W.; Pallares, G.; Chao, Q.; et al. AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML). Blood 2009, 114, 2984–2992. [Google Scholar] [CrossRef]
- Kalyanaraman, B. Teaching the basics of the mechanism of doxorubicin-induced cardiotoxicity: Have we been barking up the wrong tree? Redox Biol. 2020, 29, 101394. [Google Scholar] [CrossRef]
- Doroshow, J.H. Mechanisms of anthracycline-enhanced reactive oxygen metabolism in tumor cells. Oxidative Med. Cell. Longev. 2019, 2019, 9474823. [Google Scholar] [CrossRef]
- Reddy, M.M.; Fernandes, M.S.; Salgia, R.; Levine, R.L.; Griffin, J.D.; Sattler, M. NADPH oxidases regulate cell growth and migration in myeloid cells transformed by oncogenic tyrosine kinases. Leukemia 2011, 25, 281–289. [Google Scholar] [CrossRef] [Green Version]
- Owada, S.; Endo, H.; Okada, C.; Yoshida, K.; Shida, Y.; Tatemichi, M. Setanaxib as a potent hypoxia-specific therapeutic agent against liver cancer. Anticancer Res. 2020, 40, 5071–5079. [Google Scholar] [CrossRef] [PubMed]
- Reutens, A.T.; Jandeleit-Dahm, K.; Thomas, M.; Salim, A.; De Livera, A.M.; Bach, L.A.; Colman, P.G.; Davis, T.M.E.; Ekinci, E.I.; Fulcher, G.; et al. A physician-initiated double-blind, randomised, placebo-controlled, phase 2 study evaluating the efficacy and safety of inhibition of NADPH oxidase with the first-in-class Nox-1/4 inhibitor, GKT137831, in adults with type 1 diabetes and persistently elevated urinary albumin excretion: Protocol and statistical considerations. Contemp. Clin. Trials 2020, 90, 105892. [Google Scholar] [CrossRef] [PubMed]
- Clinical Trial News Release. Available online: https://www.prnewswire.com/news-releases/positive-phase-1-results-in-high-dose-setanaxib-trial-301209899 (accessed on 1 January 2022).
- Elbatreek, M.H.; Mucke, H.; Schmidt, H. NOX Inhibitors: From bench to naxibs to bedside. Handb. Exp. Pharmacol. 2021, 264, 145–168. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proliferation/Viability (Cell Titer Blue) | ||||
|---|---|---|---|---|
| Chemotherapy drugs | 32D FLT3-ITD | MV4-11 | MOLM13 | |
| Setanaxib | Cytarabine | +++ | ± | ± |
| Daunorubicin | +++++ | +++ | +++ | |
| Midostaurin | ++++ | +++ | ++ | |
| Apoptosis (Annexin V + 7-AAD) | ||||
| Chemotherapy drugs | 32D FLT3-ITD | MV4-11 | ||
| Setanaxib | Cytarabine | +++ | ± | |
| Daunorubicin | +++++ | + | ||
| Midostaurin | ++++ | ++ | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Demircan, M.B.; Mgbecheta, P.C.; Kresinsky, A.; Schnoeder, T.M.; Schröder, K.; Heidel, F.H.; Böhmer, F.D. Combined Activity of the Redox-Modulating Compound Setanaxib (GKT137831) with Cytotoxic Agents in the Killing of Acute Myeloid Leukemia Cells. Antioxidants 2022, 11, 513. https://doi.org/10.3390/antiox11030513
Demircan MB, Mgbecheta PC, Kresinsky A, Schnoeder TM, Schröder K, Heidel FH, Böhmer FD. Combined Activity of the Redox-Modulating Compound Setanaxib (GKT137831) with Cytotoxic Agents in the Killing of Acute Myeloid Leukemia Cells. Antioxidants. 2022; 11(3):513. https://doi.org/10.3390/antiox11030513
Chicago/Turabian StyleDemircan, Muhammed Burak, Peter C. Mgbecheta, Anne Kresinsky, Tina M. Schnoeder, Katrin Schröder, Florian H. Heidel, and Frank D. Böhmer. 2022. "Combined Activity of the Redox-Modulating Compound Setanaxib (GKT137831) with Cytotoxic Agents in the Killing of Acute Myeloid Leukemia Cells" Antioxidants 11, no. 3: 513. https://doi.org/10.3390/antiox11030513