
HO-1 Upregulation by Kaempferol via ROS-Dependent Nrf2-ARE Cascade Attenuates Lipopolysaccharide-Mediated Intercellular Cell Adhesion Molecule-1 Expression in Human Pulmonary Alveolar Epithelial Cells

 , ,
, ,  ,
,
Abstract
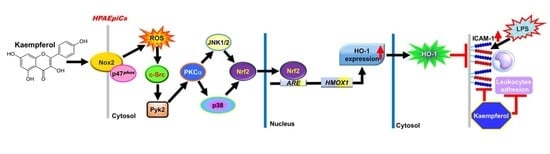
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Cell Culture and Treatment
2.3. Animal Care and Experimental Procedures
2.4. Immunohistochemical (IHC) Staining
2.5. Protein Preparation and Western Blot Analysis
2.6. Real-Time Quantitative PCR (RT-qPCR) Analysis
- ICAM-1 (NM_000201.3):
- Forward primer (5′ → 3′): GGCCTCAGTCAGTGTGA
- Reverse primer (5′ → 3′): AACCCCATTCAGCGTCA
- HO-1 (NM_002133.3):
- Forward primer (5′ → 3′): CTCCCAGGCTCCGCTTCT
- Reverse primer (5′ → 3′): GCATGCCTGCATTCACATG
- GAPDH (NM_001357943.2):
- Forward primer (5′ → 3′): GCCAGCCGAGCCACAT
- Reverse primer (5′ → 3′): CTTTACCAGAGTTAAAAGCAGCCC
2.7. Transient Transfection with siRNAs in HPAEpiCs
- Scrambled: 5′-UUCUCCGAACGUGUCACGU-3′,
- PKCα (NM_002737.3): 5′-AUAAGGAUCUGAAAGCCCGUUUGGA-3′,
- Pyk2 (NM_173174.3): 5′-CUGAUGACCUGGUGUACCU-3′,
- JNK1 (NM_001278547.2): 5′-GCAGAAGCAAGCGUGACAACA-3′,
- JNK2 (NM_001135044.2): 5′-AAUUGGUUUCAGCUGGUAACGU-3′,
- p38α (NM_139014.3):
- (1) 5′-AUGAAUGAUGGACUGAAAUGGUCUG-3′,
- (2) 5′-AAACAAUGUUCUUCCAGUCAACAGC-3′,
- (3) 5′-UUAGGUCCCUGUGAAUUAUGUCAGC-3′,
- c-Jun (NM_002228.4):
- 5′-AAGUUGCUGAGGUUUGCGUAGACCG-3′,
- 5′-AACUGCUGCGUUAGCAUGAGUUGGC-3′,
- 5′-AUAGAAGGUCGUUUCCAUCUUUGCA-3′.
2.8. Chromatin Immunoprecipitation (ChIP) Assay
2.9. Transfection and Promoter Activity Assay
2.10. NADPH Oxidase Activity Assay
2.11. Measurement of Intracellular ROS Accumulation
2.12. Adhesion Assay
2.13. Cell Viability Assay
2.14. Immunofluorescent Staining
2.15. Data and Statistical Analysis
3. Results
3.1. KPR Upregulates HO-1 and Reduces ICAM-1 Expression Induced by LPS
3.2. KPR Upregulates HO-1 to Reduce LPS-Induced ICAM-1 Expression In Vivo
3.3. KPR Activates Transcriptional and Translational Processes to Induce HO-1 Protein and mRNA Expression
3.4. KPR Stimulates NOX2 and Enhances ROS Generation to Induce HO-1 Expression
3.5. c-Src Is Needed for HO-1 Expression Induced by KPR
3.6. Pyk2 Is Involved in KPR-Induced HO-1 Expression
3.7. PKCα Is Needed for HO-1 Expression Induced by KPR
3.8. KPR Induces HO-1 Expression Mediated through p38 MAPK and JNK1/2
3.9. KPR Stimulates Phosphorylation of Nrf2 Involved in HO-1 Expression
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cho, R.L.; Yang, C.C.; Lee, I.T.; Lin, C.C.; Chi, P.L.; Hsiao, L.D.; Yang, C.M. Lipopolysaccharide induces ICAM-1 expression via a c-Src/NADPH oxidase/ROS-dependent NF-κB pathway in human pulmonary alveolar epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L639–L657. [Google Scholar] [CrossRef] [Green Version]
- Zandvoort, A.; van der Geld, Y.M.; Jonker, M.R.; Noordhoek, J.A.; Vos, J.T.; Wesseling, J.; Kauffman, H.F.; Timens, W.; Postma, D.S. High ICAM-1 gene expression in pulmonary fibroblasts of COPD patients: A reflection of an enhanced immunological function. Eur. Respir. J. 2006, 28, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Kotteas, E.A.; Boulas, P.; Gkiozos, I.; Tsagkouli, S.; Tsoukalas, G.; Syrigos, K.N. The intercellular cell adhesion molecule-1 (ICAM-1) in lung cancer: Implications for disease progression and prognosis. Anticancer Res. 2014, 34, 4665–4672. [Google Scholar]
- Stanciu, L.A.; Djukanovic, R. The role of ICAM-1 on T-cells in the pathogenesis of asthma. Eur. Respir. J. 1998, 11, 949–957. [Google Scholar] [CrossRef] [Green Version]
- Hadzic, S.; Wu, C.Y.; Avdeev, S.; Weissmann, N.; Schermuly, R.T.; Kosanovic, D. Lung epithelium damage in COPD—An unstoppable pathological event? Cell Signal 2020, 68, 109540. [Google Scholar] [CrossRef]
- Potaczek, D.P.; Miethe, S.; Schindler, V.; Alhamdan, F.; Garn, H. Role of airway epithelial cells in the development of different asthma phenotypes. Cell Signal 2020, 69, 109523. [Google Scholar] [CrossRef]
- Suzuki, K.; Ohkuma, M.; Nagaoka, I. Bacterial lipopolysaccharide and antimicrobial LL-37 enhance ICAM-1 expression and NF-κB p65 phosphorylation in senescent endothelial cells. Int. J. Mol. Med. 2019, 44, 1187–1196. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.C.; Lin, W.N.; Cho, R.L.; Yang, C.C.; Yeh, Y.C.; Hsiao, L.D.; Tseng, H.C.; Yang, C.M. Induction of HO-1 by Mevastatin mediated via a Nox/ROS-dependent c-Src/PDGFRα/PI3K/Akt/Nrf2/ARE cascade suppresses TNF-α-induced lung inflammation. J. Clin. Med. 2020, 9, 226. [Google Scholar] [CrossRef] [Green Version]
- Liou, C.J.; Lai, Y.R.; Chen, Y.L.; Chang, Y.H.; Li, Z.Y.; Huang, W.C. Matrine attenuates COX-2 and ICAM-1 expressions in human lung epithelial cells and prevents acute lung injury in LPS-induced mice. Mediat. Inflamm. 2016, 2016, 3630485. [Google Scholar] [CrossRef] [Green Version]
- Woodfin, A.; Beyrau, M.; Voisin, M.B.; Ma, B.; Whiteford, J.R.; Hordijk, P.L.; Hogg, N.; Nourshargh, S. ICAM-1-expressing neutrophils exhibit enhanced effector functions in murine models of endotoxemia. Blood 2016, 127, 898–907. [Google Scholar] [CrossRef] [Green Version]
- Mehta, J.; Rayalam, S.; Wang, X. Cytoprotective effects of natural compounds against oxidative stress. Antioxidants 2018, 7, 147. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.M.; Lin, C.C.; Yang, C.C.; Cho, R.L.; Hsiao, L.D. Mevastatin-induced AP-1-dependent HO-1 expression suppresses vascular cell adhesion molecule-1 expression and monocyte adhesion on human pulmonary alveolar epithelial cells challenged with TNF-α. Biomolecules 2020, 10, 381. [Google Scholar] [CrossRef] [Green Version]
- Fredenburgh, L.E.; Perrella, M.A.; Mitsialis, S.A. The role of heme oxygenase-1 in pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2007, 36, 158–165. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Qian, J.-M. Cytoprotective role of heme oxygenase-1 in liver ischemia reperfusion injury. Int. J. Clin. Exp. Med. 2015, 8, 19867. [Google Scholar]
- Waza, A.A.; Hamid, Z.; Ali, S.; Bhat, S.A.; Bhat, M.A. A review on heme oxygenase-1 induction: Is it a necessary evil. Inflamm. Res. 2018, 67, 579–588. [Google Scholar] [CrossRef]
- Chen, A.Y.; Chen, Y.C. A review of the dietary flavonoid, kaempferol on human health and cancer chemoprevention. Food Chem. 2013, 138, 2099–2107. [Google Scholar] [CrossRef] [Green Version]
- Gong, J.H.; Shin, D.; Han, S.Y.; Kim, J.L.; Kang, Y.H. Kaempferol suppresses eosionphil infiltration and airway inflammation in airway epithelial cells and in mice with allergic asthma. J. Nutr. 2012, 142, 47–56. [Google Scholar] [CrossRef]
- Molitorisova, M.; Sutovska, M.; Kazimierova, I.; Barborikova, J.; Joskova, M.; Novakova, E.; Franova, S. The anti-asthmatic potential of flavonol kaempferol in an experimental model of allergic airway inflammation. Eur. J. Pharmacol. 2021, 891, 173698. [Google Scholar] [CrossRef]
- Yang, C.; Yang, W.; He, Z.; Guo, J.; Yang, X.; Wang, R.; Li, H. Kaempferol alleviates oxidative stress and apoptosis through mitochondria-dependent pathway during lung ischemia-reperfusion injury. Front Pharmacol. 2021, 12, 624402. [Google Scholar] [CrossRef]
- Motterlini, R.; Foresti, R. Heme oxygenase-1 as a target for drug discovery. Antioxid. Redox. Signal 2014, 20, 1810–1826. [Google Scholar] [CrossRef]
- Velagapudi, R.; Jamshaid, F.; Lepiarz, I.; Katola, F.O.; Hemming, K.; Olajide, O.A. The tiliroside derivative, 3-O-[(E)-(2-oxo-4-(p-tolyl) but-3-en-1-yl] kaempferol produced inhibition of neuroinflammation and activation of AMPK and Nrf2/HO-1 pathways in BV-2 microglia. Int. Immunopharmacol. 2019, 77, 105951. [Google Scholar] [CrossRef]
- Hong, J.T.; Yen, J.H.; Wang, L.; Lo, Y.H.; Chen, Z.T.; Wu, M.J. Regulation of heme oxygenase-1 expression and MAPK pathways in response to kaempferol and rhamnocitrin in PC12 cells. Toxicol. Appl. Pharmacol. 2009, 237, 59–68. [Google Scholar] [CrossRef]
- Cho, R.L.; Lin, W.N.; Wang, C.Y.; Yang, C.C.; Hsiao, L.D.; Lin, C.C.; Yang, C.M. Heme oxygenase-1 induction by rosiglitazone via PKCα/AMPKα/p38 MAPKα/SIRT1/PPARγ pathway suppresses lipopolysaccharide-mediated pulmonary inflammation. Biochem. Pharmacol. 2018, 148, 222–237. [Google Scholar] [CrossRef]
- Cho, R.L.; Yang, C.C.; Tseng, H.C.; Hsiao, L.D.; Lin, C.C.; Yang, C.M. Haem oxygenase-1 up-regulation by rosiglitazone via ROS-dependent Nrf2-antioxidant response elements axis or PPARγ attenuates LPS-mediated lung inflammation. Br. J. Pharmacol. 2018, 175, 3928–3946. [Google Scholar] [CrossRef] [Green Version]
- Percie du Sert, N.; Hurst, V.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. J. Cereb. Blood Flow Metab. 2020, 40, 1769–1777. [Google Scholar] [CrossRef]
- Curtis, M.J.; Alexander, S.; Cirino, G.; Docherty, J.R.; George, C.H.; Giembycz, M.A.; Hoyer, D.; Insel, P.A.; Izzo, A.A.; Ji, Y.; et al. Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. Br. J. Pharmacol. 2018, 175, 987–993. [Google Scholar] [CrossRef] [Green Version]
- Ryter, S.W.; Choi, A.M. Heme oxygenase-1: Molecular mechanisms of gene expression in oxygen-related stress. Antioxid. Redox. Signal 2002, 4, 625–632. [Google Scholar] [CrossRef]
- Cheng, S.E.; Lee, I.T.; Lin, C.C.; Kou, Y.R.; Yang, C.M. Cigarette smoke particle-phase extract induces HO-1 expression in human tracheal smooth muscle cells: Role of the c-Src/NADPH oxidase/MAPK/Nrf2 signaling pathway. Free Radic. Biol. Med. 2010, 48, 1410–1422. [Google Scholar] [CrossRef]
- Lee, I.T.; Wang, S.W.; Lee, C.W.; Chang, C.C.; Lin, C.C.; Luo, S.F.; Yang, C.M. Lipoteichoic acid induces HO-1 expression via the TLR2/MyD88/c-Src/NADPH oxidase pathway and Nrf2 in human tracheal smooth muscle cells. J. Immunol. 2008, 181, 5098–5110. [Google Scholar] [CrossRef] [Green Version]
- Rosadini, C.V.; Kagan, J.C. Early innate immune responses to bacterial LPS. Curr. Opin. Immunol. 2017, 44, 14–19. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Qu, C.; Dong, P. The ICAM-1 K469E polymorphism is associated with the risk of coronary artery disease: A meta-analysis. Coron Artery Dis. 2014, 25, 665–670. [Google Scholar] [CrossRef]
- Lee, T.-S.; Chau, L.-Y. Heme oxygenase-1 mediates the anti-inflammatory effect of interleukin-10 in mice. Nat. Med. 2002, 8, 240–246. [Google Scholar] [CrossRef]
- Park, E.J.; Kim, Y.M.; Park, S.W.; Kim, H.J.; Lee, J.H.; Lee, D.U.; Chang, K.C. Induction of HO-1 through p38 MAPK/Nrf2 signaling pathway by ethanol extract of Inula helenium L. reduces inflammation in LPS-activated RAW 264.7 cells and CLP-induced septic mice. Food Chem. Toxicol. 2013, 55, 386–395. [Google Scholar] [CrossRef]
- Li, C.; Hossieny, P.; Wu, B.J.; Qawasmeh, A.; Beck, K.; Stocker, R. Pharmacologic induction of heme oxygenase-1. Antioxid. Redox. Signal 2007, 9, 2227–2239. [Google Scholar] [CrossRef] [Green Version]
- Gates, M.A.; Tworoger, S.S.; Hecht, J.L.; De Vivo, I.; Rosner, B.; Hankinson, S.E. A prospective study of dietary flavonoid intake and incidence of epithelial ovarian cancer. Int. J. Cancer 2007, 121, 2225–2232. [Google Scholar] [CrossRef]
- Suchal, K.; Malik, S.; Gamad, N.; Malhotra, R.K.; Goyal, S.N.; Bhatia, J.; Arya, D.S. Kampeferol protects against oxidative stress and apoptotic damage in experimental model of isoproterenol-induced cardiac toxicity in rats. Phytomedicine 2016, 23, 1401–1408. [Google Scholar] [CrossRef]
- Zang, Y.; Zhang, L.; Igarashi, K.; Yu, C. The anti-obesity and anti-diabetic effects of kaempferol glycosides from unripe soybean leaves in high-fat-diet mice. Food Funct. 2015, 6, 834–841. [Google Scholar] [CrossRef]
- Lin, F.; Luo, X.; Tsun, A.; Li, Z.; Li, D.; Li, B. Kaempferol enhances the suppressive function of Treg cells by inhibiting FOXP3 phosphorylation. Int. Immunopharmacol. 2015, 28, 859–865. [Google Scholar] [CrossRef]
- Li, S.; Pu, X.P. Neuroprotective effect of kaempferol against a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced mouse model of Parkinson’s disease. Biol. Pharm. Bull. 2011, 34, 1291–1296. [Google Scholar] [CrossRef] [Green Version]
- Shin, D.; Park, S.H.; Choi, Y.J.; Kim, Y.H.; Antika, L.D.; Habibah, N.U.; Kang, M.K.; Kang, Y.H. Dietary compound Kaempferol inhibits airway thickening induced by allergic reaction in a bovine serum albumin-induced model of asthma. Int. J. Mol. Sci. 2015, 16, 29980–29995. [Google Scholar] [CrossRef] [Green Version]
- Gong, J.-H.; Shin, D.; Han, S.-Y.; Park, S.-H.; Kang, M.-K.; Kim, J.-L.; Kang, Y.-H. Blockade of airway inflammation by kaempferol via disturbing Tyk-STAT signaling in airway epithelial cells and in asthmatic mice. Evid. Based Complement. Alternat. Med. 2013, 2013, 250725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.H.; Park, S.E.; Kim, S.J.; Kim, S. Kaempferol inhibits thrombosis and platelet activation. Biochimie 2015, 115, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, R.; Kumar, S.; Kumar, A.; Siddiqui, J.A.; Swarnkar, G.; Gupta, V.; Kendurker, A.; Dwivedi, A.K.; Romero, J.R.; Chattopadhyay, N. Kaempferol has osteogenic effect in ovariectomized adult Sprague-Dawley rats. Mol. Cell. Endocrinol. 2008, 289, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Imran, M.; Rauf, A.; Shah, Z.A.; Saeed, F.; Imran, A.; Arshad, M.U.; Ahmad, B.; Bawazeer, S.; Atif, M.; Peters, D.G.; et al. Chemo-preventive and therapeutic effect of the dietary flavonoid kaempferol: A comprehensive review. Phytother. Res. 2019, 33, 263–275. [Google Scholar] [CrossRef]
- Kamata, H.; Hirata, H. Redox regulation of cellular signalling. Cell Signal 1999, 11, 1–14. [Google Scholar] [CrossRef]
- Mulier, B.; Rahman, I.; Watchorn, T.; Donaldson, K.; MacNee, W.; Jeffery, P.K. Hydrogen peroxide-induced epithelial injury: The protective role of intracellular nonprotein thiols (NPSH). Eur. Respir. J. 1998, 11, 384–391. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.B.; Jang, J.Y.; Chae, Y.H.; Min, J.H.; Baek, J.Y.; Kim, M.; Park, Y.; Hwang, G.S.; Ryu, J.S.; Chang, T.S. Kaempferol suppresses collagen-induced platelet activation by inhibiting NADPH oxidase and protecting SHP-2 from oxidative inactivation. Free Radic. Biol. Med. 2015, 83, 41–53. [Google Scholar] [CrossRef]
- Sekiguchi, A.; Motegi, S.I.; Fujiwara, C.; Yamazaki, S.; Inoue, Y.; Uchiyama, A.; Akai, R.; Iwawaki, T.; Ishikawa, O. Inhibitory effect of kaempferol on skin fibrosis in systemic sclerosis by the suppression of oxidative stress. J. Dermatol. Sci. 2019, 96, 8–17. [Google Scholar] [CrossRef] [Green Version]
- Jang, Y.J.; Kim, J.; Shim, J.; Kim, J.; Byun, S.; Oak, M.H.; Lee, K.W.; Lee, H.J. Kaempferol attenuates 4-hydroxynonenal-induced apoptosis in PC12 cells by directly inhibiting NADPH oxidase. J. Pharmacol. Exp. Ther. 2011, 337, 747–754. [Google Scholar] [CrossRef] [Green Version]
- Holmström, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef]
- Newton, A.C. Protein kinase C: Perfectly balanced. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 208–230. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.M.; Heo, D.R.; Kim, Y.A.; Lee, J.; Kim, N.S.; Bang, O.S. Coniferaldehyde inhibits LPS-induced apoptosis through the PKC α/β II/Nrf-2/HO-1 dependent pathway in RAW264.7 macrophage cells. Environ. Toxicol. Pharmacol. 2016, 48, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Martel-Gallegos, G.; Casas-Pruneda, G.; Ortega-Ortega, F.; Sánchez-Armass, S.; Olivares-Reyes, J.A.; Diebold, B.; Pérez-Cornejo, P.; Arreola, J. Oxidative stress induced by P2X7 receptor stimulation in murine macrophages is mediated by c-Src/Pyk2 and ERK1/2. Biochim. Biophys. Acta 2013, 1830, 4650–4659. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.E.; Jeong, S.I.; Yang, H.; Park, C.S.; Jin, Y.H.; Park, Y.S. Fisetin induces Nrf2-mediated HO-1 expression through PKC-δ and p38 in human umbilical vein endothelial cells. J. Cell Biochem. 2011, 112, 2352–2360. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.S.; Kim, K.S.; Ko, W.; Li, B.; Jeong, G.S.; Jang, J.H.; Oh, H.; Kim, Y.C. The cytoprotective effect of sulfuretin against tert-butyl hydroperoxide-induced hepatotoxicity through Nrf2/ARE and JNK/ERK MAPK-mediated heme oxygenase-1 expression. Int. J. Mol. Sci. 2014, 15, 8863–8877. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.C.; Hsiao, L.D.; Cho, R.L.; Yang, C.M. Carbon monoxide releasing molecule-2-upregulated ROS-dependent heme oxygenase-1 axis suppresses lipopolysaccharide-induced airway inflammation. Int. J. Mol. Sci. 2019, 20, 3157. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.C.; Yang, C.C.; Chen, Y.W.; Hsiao, L.D.; Yang, C.M. Arachidonic acid induces ARE/Nrf2-dependent heme oxygenase-1 transcription in rat brain astrocytes. Mol. Neurobiol. 2018, 55, 3328–3343. [Google Scholar] [CrossRef]
- Lin, C.C.; Chiang, Y.C.; Cho, R.L.; Lin, W.N.; Yang, C.C.; Hsiao, L.D.; Yang, C.M. Up-regulation of PYK2/PKCα-dependent haem oxygenase-1 by CO-releasing molecule-2 attenuates TNF-α-induced lung inflammation. Br. J. Pharmacol. 2018, 175, 456–468. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.H.; Zhao, C.; Peng, Q.; Xie, P.; Liu, Q.H. Kaempferol inhibited VEGF and PGF expression and in vitro angiogenesis of HRECs under diabetic-like environment. Braz. J. Med. Biol. Res. 2017, 50, e5396. [Google Scholar] [CrossRef] [Green Version]
- Podder, B.; Song, K.S.; Song, H.Y.; Kim, Y.S. Cytoprotective effect of kaempferol on paraquat-exposed BEAS-2B cells via modulating expression of MUC5AC. Biol. Pharm. Bull. 2014, 37, 1486–1494. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.S.; Lee, E.G.; Sung, M.S.; Yoo, W.H. Kaempferol inhibits IL-1β-stimulated, RANKL-mediated osteoclastogenesis via downregulation of MAPKs, c-Fos, and NFATc1. Inflammation 2014, 37, 1221–1230. [Google Scholar] [CrossRef] [PubMed]
- Levonen, A.L. Activation of stress signaling pathways by oxidized and nitrated lipids. Free Radic. Biol. Med. 2014, 75 (Suppl. 1), S8. [Google Scholar] [CrossRef] [PubMed]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef] [PubMed] [Green Version]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, C.-C.; Hsiao, L.-D.; Wang, C.-Y.; Lin, W.-N.; Shih, Y.-F.; Chen, Y.-W.; Cho, R.-L.; Tseng, H.-C.; Yang, C.-M. HO-1 Upregulation by Kaempferol via ROS-Dependent Nrf2-ARE Cascade Attenuates Lipopolysaccharide-Mediated Intercellular Cell Adhesion Molecule-1 Expression in Human Pulmonary Alveolar Epithelial Cells. Antioxidants 2022, 11, 782. https://doi.org/10.3390/antiox11040782
Yang C-C, Hsiao L-D, Wang C-Y, Lin W-N, Shih Y-F, Chen Y-W, Cho R-L, Tseng H-C, Yang C-M. HO-1 Upregulation by Kaempferol via ROS-Dependent Nrf2-ARE Cascade Attenuates Lipopolysaccharide-Mediated Intercellular Cell Adhesion Molecule-1 Expression in Human Pulmonary Alveolar Epithelial Cells. Antioxidants. 2022; 11(4):782. https://doi.org/10.3390/antiox11040782
Chicago/Turabian StyleYang, Chien-Chung, Li-Der Hsiao, Chen-Yu Wang, Wei-Ning Lin, Ya-Fang Shih, Yi-Wen Chen, Rou-Ling Cho, Hui-Ching Tseng, and Chuen-Mao Yang. 2022. "HO-1 Upregulation by Kaempferol via ROS-Dependent Nrf2-ARE Cascade Attenuates Lipopolysaccharide-Mediated Intercellular Cell Adhesion Molecule-1 Expression in Human Pulmonary Alveolar Epithelial Cells" Antioxidants 11, no. 4: 782. https://doi.org/10.3390/antiox11040782