Nephroprotective Effect of Cilastatin against Gentamicin-Induced Renal Injury In Vitro and In Vivo without Altering Its Bactericidal Efficiency

 ,
,
Abstract
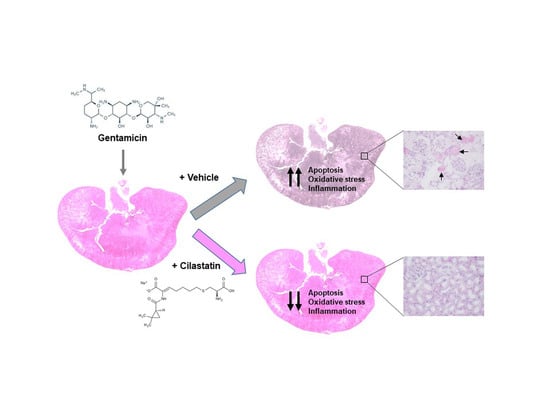
:
1. Introduction
2. Materials and Methods
2.1. Drugs
2.2. Experimental Animals
2.3. Renal Proximal Tubular Primary Cell Culture
2.4. Cell Death Study in RPTECs
2.4.1. Morphology Analysis
2.4.2. Quantification of Cell Detachment
2.4.3. Localization of Cleaved Caspase 3
2.4.4. Nucleosomal Quantification
2.5. In Vivo Model and Experimental Protocols
2.6. Renal Function Parameters
2.7. Renal Histology Studies
2.8. Western Blots and Immunohistochemistry
2.9. TUNEL Assay
2.10. Measurement of TNFα and Antioxidant Capacity
2.11. Determination of Gentamicin Levels
2.12. Megalin mRNA Expression
2.13. Bacterial Susceptibility Assays
2.14. Statistical Methods
3. Results
3.1. Cilastatin Prevents Apoptosis and Reduces the Accumulation of the Antibiotic in Gentamicin-Treated RPTECs
3.2. Cilastatin Ameliorates Body Weight Loss and Renal Function in Rats with Gentamicin-Induced AKI
3.3. Cilastatin Attenuates Gentamicin-Induced Renal Tubular Damage
3.4. Cilastatin Reduces Gentamicin-Induced Apoptosis
3.5. Cilastatin Reduces Gentamicin-Induced Oxidative Stress
3.6. Cilastatin Reduces Gentamicin-Induced Renal Inflammation
3.7. Effects of Cilastatin on Intracellular Gentamicin Accumulation and Megalin Expression
3.8. Cilastatin Has No Effect on the Bactericidal Actions of Gentamicin
4. Discussion
5. Conclusions
6. Patents
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nagai, J.; Takano, M. Molecular aspects of renal handling of aminoglycosides and strategies for preventing the nephrotoxicity. Drug Metab. Pharmacokinet. 2004, 19, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Novoa, J.M.; Quiros, Y.; Vicente, L.; Morales, A.I.; López-Hernandez, F.-J. New insights into the mechanism of aminoglycoside nephrotoxicity: An integrative point of view. Kidney Int. 2011, 79, 33–45. [Google Scholar] [CrossRef] [Green Version]
- Silan, C.; Uzun, O.; Comunoğlu, N.U.; Gokçen, S.; Bedirhan, S.; Cengiz, M. Gentamicin-induced nephrotoxicity in rats ameliorated and healing effects of resveratrol. Biol. Pharm. Bull. 2007, 30, 79–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leehey, D.J.; Braun, B.I.; Tholl, D.A.; Chung, L.S.; Gross, C.A.; Roback, J.A.; Lentino, J.R. Can pharmacokinetic dosing decrease nephrotoxicity associated with aminoglycoside therapy? J. Am. Soc. Nephrol. 1993, 4, 81–90. [Google Scholar] [PubMed]
- Watanabe, A.; Nagai, J.; Adachi, Y.; Katsube, T.; Kitahara, Y.; Murakami, T.; Takano, M. Targeted prevention of renal accumulation and toxicity of gentamicin by aminoglycoside binding receptor antagonists. J. Control Release 2004, 95, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Moestrup, S.K.; Cui, S.; Vorum, H.; Bregengård, C.; Bjørn, S.E.; Norris, K.; Gliemann, J.; Christensen, E.I. Evidence that epithelial glycoprotein 330/megalin mediates uptake of polybasic drugs. J. Clin. Investig. 1995, 96, 1404–1413. [Google Scholar] [CrossRef]
- Schmitz, C.; Hilpert, J.; Jacobsen, C.; Boensch, C.; Christensen, E.I.; Luft, F.C.; Willnow, T.E. Megalin deficiency offers protection from renal aminoglycoside accumulation. J. Biol. Chem. 2002, 277, 618–622. [Google Scholar] [CrossRef] [Green Version]
- Quiros, Y.; Vicente-Vicente, L.; Morales, A.I.; López-Novoa, J.M.; López-Hernández, F.J. An integrative overview on the mechanisms underlying the renal tubular cytotoxicity of gentamicin. Toxicol. Sci. 2011, 119, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Karasawa, T.; Steyger, P.S. Intracellular mechanisms of aminoglycoside-induced cytotoxicity. Integr. Biol. 2011, 3, 879–886. [Google Scholar] [CrossRef] [Green Version]
- Hosohata, K. Role of Oxidative Stress in Drug-Induced Kidney Injury. Int. J. Mol. Sci. 2016, 17, 1826. [Google Scholar] [CrossRef] [Green Version]
- Morales, A.I.; Detaille, D.; Prieto, M.; Puente, A.; Briones, E.; Arévalo, M.; Leverve, X.; López-Novoa, J.M.; El-Mir, M.Y. Metformin prevents experimental gentamicin-induced nephropathy by a mitochondria-dependent pathway. Kidney Int. 2010, 77, 861–869. [Google Scholar] [CrossRef]
- Erdem, A.; Gündoğan, N.U.; Usubütün, A.; Kilinç, K.; Erdem, S.R.; Kara, A.; Bozkurt, A. The protective effect of taurine against gentamicin-induced acute tubular necrosis in rats. Nephrol. Dial. Transplant. 2000, 15, 1175–1182. [Google Scholar] [CrossRef] [PubMed]
- Pérez, M.; Castilla, M.; Torres, A.M.; Lázaro, J.A.; Sarmiento, E.; Tejedor, A. Inhibition of brush border dipeptidase with cilastatin reduces toxic accumulation of cyclosporin A in kidney proximal tubule epithelial cells. Nephrol. Dial. Transplant. 2004, 19, 2445–2455. [Google Scholar] [CrossRef] [Green Version]
- Camano, S.; Lazaro, A.; Moreno-Gordaliza, E.; Torres, A.M.; de Lucas, C.; Humanes, B.; Lazaro, J.A.; Gomez-Gomez, M.M.; Bosca, L.; Tejedor, A. Cilastatin attenuates cisplatin-induced proximal tubular cell damage. J. Pharmacol. Exp. Ther. 2010, 334, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Humanes, B.; Jado, J.C.; Camaño, S.; López-Parra, V.; Torres, A.M.; Álvarez-Sala, L.A.; Cercenado, E.; Tejedor, A.; Lázaro, A. Protective Effects of Cilastatin against Vancomycin-Induced Nephrotoxicity. Biomed. Res. Int. 2015, 2015, 704382. [Google Scholar] [CrossRef] [Green Version]
- Humanes, B.; Lazaro, A.; Camano, S.; Moreno-Gordaliza, E.; Lazaro, J.A.; Blanco-Codesido, M.; Lara, J.M.; Ortiz, A.; Gomez-Gomez, M.M.; Martín-Vasallo, P.; et al. Cilastatin protects against cisplatin-induced nephrotoxicity without compromising its anticancer efficiency in rats. Kidney Int. 2012, 82, 652–663. [Google Scholar] [CrossRef] [Green Version]
- Humanes, B.; Camaño, S.; Lara, J.M.; Sabbisetti, V.; González-Nicolás, M.A.; Bonventre, J.V.; Tejedor, A.; Lázaro, A. Cisplatin-induced renal inflammation is ameliorated by cilastatin nephroprotection. Nephrol. Dial. Transplant. 2017, 32, 1645–1655. [Google Scholar] [CrossRef] [Green Version]
- Lazaro, A.; Camaño, S.; Humanes, B.; Tejedor, A. Novel strategies in drug-induced acute kidney injury. In Pharmacology; Gallelli, L., Ed.; Intech: Rijeka, Croatia, 2012; pp. 381–396. [Google Scholar] [CrossRef] [Green Version]
- Verpooten, G.A.; Verbist, L.; Buntinx, A.P.; Entwistle, L.A.; Jones, K.H.; De Broe, M.E. The pharmacokinetics of imipenem (thienamycin-formamidine) and the renal dehydropeptidase inhibitor cilastatin sodium in normal subjects and patients with renal failure. Br. J. Clin. Pharmacol. 1984, 18, 183–193. [Google Scholar] [CrossRef] [Green Version]
- Norrby, S.R.; Alestig, K.; Björnegård, B.; Burman, L.A.; Ferber, F.; Huber, J.L.; Jones, K.H.; Kahan, F.M.; Kahan, J.S.; Kropp, H.; et al. Urinary recovery of N-formimidoyl thienamycin (MK0787) as affected by coadministration of N-formimidoyl thienamycin dehydropeptidase inhibitors. Antimicrob. Agents Chemother. 1983, 23, 300–307. [Google Scholar] [CrossRef] [Green Version]
- Bell, S.; Davey, P.; Nathwani, D.; Marwick, C.; Vadiveloo, T.; Sneddon, J.; Patton, A.; Bennie, M.; Fleming, S.; Donnan, P.T. Risk of AKI with gentamicin as surgical prophylaxis. J. Am. Soc. Nephrol. 2014, 25, 2625–2632. [Google Scholar] [CrossRef] [Green Version]
- Denamur, S.; Boland, L.; Beyaert, M.; Verstraeten, S.L.; Fillet, M.; Tulkens, P.M.; Bontemps, F.; Mingeot-Leclercq, M.P. Subcellular mechanisms involved in apoptosis induced by aminoglycoside antibiotics: Insights on p53, proteasome and endoplasmic reticulum. Toxicol. Appl. Pharmacol. 2016, 309, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Juan, S.H.; Chen, C.H.; Hsu, Y.H.; Hou, C.C.; Chen, T.H.; Lin, H.; Chu, Y.L.; Sue, Y.M. Tetramethylpyrazine protects rat renal tubular cell apoptosis induced by gentamicin. Nephrol. Dial. Transplant. 2007, 22, 732–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahu, B.D.; Tatireddy, S.; Koneru, M.; Borkar, R.M.; Kumar, J.M.; Kuncha, M.; Srinivas, R.; Shyam Sunder, R.; Sistla, R. Naringin ameliorates gentamicin-induced nephrotoxicity and associated mitochondrial dysfunction, apoptosis and inflammation in rats: Possible mechanism of nephroprotection. Toxicol. Appl. Pharmacol. 2014, 277, 8–20. [Google Scholar] [CrossRef] [PubMed]
- El Mouedden, M.; Laurent, G.; Mingeot-Leclercq, M.P.; Tulkens, P.M. Gentamicin-induced apoptosis in renal cell lines and embryonic rat fibroblasts. Toxicol. Sci. 2000, 56, 229–239. [Google Scholar] [CrossRef] [Green Version]
- Padanilam, B.J. Cell death induced by acute renal injury: A perspective on the contributions of apoptosis and necrosis. Am. J. Physiol. Renal Physiol. 2003, 284, F608–F627. [Google Scholar] [CrossRef] [Green Version]
- Abdelrahman, R.S. Protective effect of apocynin against gentamicin-induced nephrotoxicity in rats. Hum. Exp. Toxicol. 2018, 37, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Raheem, I.T.; Abdel-Ghany, A.A.; Mohamed, G.A. Protective effect of quercetin against gentamicin-induced nephrotoxicity in rats. Biol. Pharm. Bull. 2009, 32, 61–67. [Google Scholar] [CrossRef] [Green Version]
- Sayed-Ahmed, M.M.; Nagi, M.N. Thymoquinone supplementation prevents the development of gentamicin-induced acute renal toxicity in rats. Clin. Exp. Pharmacol. Physiol. 2007, 34, 399–405. [Google Scholar] [CrossRef]
- Muthuraman, A.; Singla, S.K.; Rana, A.; Singh, A.; Sood, S. Reno-protective role of flunarizine (mitochondrial permeability transition pore inactivator) against gentamicin induced nephrotoxicity in rats. Yakugaku Zasshi. 2011, 131, 437–443. [Google Scholar] [CrossRef] [Green Version]
- Shirwaikar, A.; Issac, D.; Malini, S. Effect of Aerva lanata on cisplatin and gentamicin models of acute renal failure. J. Ethnopharmacol. 2004, 90, 81–86. [Google Scholar] [CrossRef]
- Khan, S.A.; Priyamvada, S.; Farooq, N.; Khan, S.; Khan, M.W.; Yusufi, A.N. Protective effect of green tea extract on gentamicin-induced nephrotoxicity and oxidative damage in rat kidney. Pharmacol. Res. 2009, 59, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Garrido, C. Size matters: Of the small HSP27 and its large oligomers. Cell Death Differ. 2002, 9, 483–485. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Nakano, S.; Owari, K.; Fuziwara, K.; Ogawa, N.; Otaka, M.; Tamaki, K.; Watanabe, S.; Komatsuda, A.; Wakui, H.; et al. Gentamicin inhibits HSP70-assisted protein folding by interfering with substrate recognition. FEBS Lett. 2010, 584, 645–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cárdenas-González, M.; Jacobo Estrada, T.; Rodríguez-Muñoz, R.; Barrera-Chimal, J.; Bobadilla, N.A.; Barbier, O.C.; Del Razo, L.M. Sub-chronic exposure to fluoride impacts the response to a subsequent nephrotoxic treatment with gentamicin. J. Appl. Toxicol. 2016, 36, 309–319. [Google Scholar] [CrossRef]
- Hori, Y.; Aoki, N.; Kuwahara, S.; Hosojima, M.; Kaseda, R.; Goto, S.; Iida, T.; De, S.; Kabasawa, H.; Kaneko, R.; et al. Megalin Blockade with Cilastatin Suppresses Drug-Induced Nephrotoxicity. J. Am. Soc. Nephrol. 2017, 28, 1783–1791. [Google Scholar] [CrossRef] [Green Version]
- Nagai, J.; Takano, M. Molecular-targeted approaches to reduce renal accumulation of nephrotoxic drugs. Expert Opin. Drug Metab. Toxicol. 2010, 6, 1125–1138. [Google Scholar] [CrossRef]
- Hansen, G.H.; Dalskov, S.M.; Rasmussen, C.R.; Immerdal, L.; Niels-Christiansen, L.L.; Danielsen, E.M. Cholera toxin entry into pig enterocytes occurs via a lipid raft- and clathrin-dependent mechanism. Biochemistry 2005, 44, 873–882. [Google Scholar] [CrossRef]
- Marzolo, M.P.; Yuseff, M.I.; Retamal, C.; Donoso, M.; Ezquer, F.; Farfán, P.; Li, Y.; Bu, G. Differential distribution of low-density lipoprotein-receptor-related protein (LPR) and megalin in polarized epithelial cells is determined by their cytoplasmic domains. Traffic 2003, 4, 273–288. [Google Scholar] [CrossRef]
- Scheel-Toellner, D.; Wang, K.; Singh, R.; Majeed, S.; Raza, K.; Curnow, S.J.; Salmon, M.; Lord, J.M. The death-inducing signaling complex is recruited to lipid rafts in Fas-induced apoptosis. Biochem. Biophys. Res. Commun. 2002, 297, 876–879. [Google Scholar] [CrossRef]
- Gajate, C.; Mollinedo, F. Lipid rafts and raft-mediated supramolecular entities in the regulation of CD95 death receptor apoptotic signaling. Apoptosis 2015, 20, 584–606. [Google Scholar] [CrossRef]
- Gajate, C.; Mollinedo, F. Isolation of Lipid Rafts Through Discontinuous Sucrose Gradient Centrifugation and Fas/CD95 Death Receptor Localization in Raft Fractions. Methods Mol. Biol. 2017, 1557, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Orlandi, P.A.; Fishman, P.H. Filipin-dependent inhibition of cholera toxin: Evidence for toxin internalization and activation through caveolae-like domains. J. Cell Biol. 1998, 141, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Chung, H.; Komada, T.; Platnich, J.M.; Sandall, C.F.; Choudhury, S.R.; Chun, J.; Naumenko, V.; Surewaard, B.G.; Nelson, M.C.; et al. Renal immune surveillance and dipeptidase-1 contribute to contrast-induced acute kidney injury. J. Clin. Investig. 2018, 128, 2894–2913. [Google Scholar] [CrossRef] [PubMed] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Initial Weight (g) | Final Weight (g) | Δ Weight (g) |
|---|---|---|---|
| Control | 296 ± 12 | 312 ± 10 | 16 ± 3 |
| Control + Cil | 287 ± 8 | 299 ± 8 | 12 ± 2 |
| Gentamicin | 268 ± 9 | 265 ± 9 | −3 ± 2 a |
| Gentamicin + Cil | 283 ± 9 | 288 ± 5 | 5 ± 3 b, c |
| Groups | SCreat (mg/dL) | BUN (mg/dL) | GFR (mL/min/100 g) | UProt (mg/24 h) | FENa+ | FEH2O |
|---|---|---|---|---|---|---|
| Control | 0.24 ± 0.03 | 30.00 ± 3.48 | 0.91 ± 0.08 | 9.12 ± 0.57 | 0.27 ± 0.02 | 0.24 ± 0.03 |
| Control + Cil | 0.27 ± 0.02 | 30.00 ± 1.32 | 0.72 ± 0.05 | 8.65 ± 0.66 | 0.27 ± 0.03 | 0.28 ± 0.04 |
| Gentamicin | 0.82 ± 0.12 a | 55.75 ± 7.10 e | 0.26 ± 0.04 b | 26.54 ± 4.06 f | 1.22 ± 0.17 a | 3.27 ± 0.51 g |
| Gentamicin + Cil | 0.50 ± 0.07 a,c | 38.67 ± 3.61 | 0.46 ± 0.09 b,d | 16.67 ± 2.09 | 0.76 ± 0.10 a,c | 1.02 ± 0.22 |
| Strain 1 | Strain 2 | Strain 3 | Strain 4 | |||||
|---|---|---|---|---|---|---|---|---|
| Staphylococcus aureus | Cil | Vehicle | Cil | Vehicle | Cil | Vehicle | Cil | Vehicle |
| MIC | 0.5 | 0.5 | 0.5 | 0.25 | 0.5 | 0.5 | 0.5 | 0.5 |
| MBC | 0.5 | 0.5 | 0.5 | 0.5 | 1 | 1 | 0.5 | 0.5 |
| Escherichia coli | Cil | Vehicle | Cil | Vehicle | Cil | Vehicle | Cil | Vehicle |
| MIC | 1 | 0.5 | 1 | 0.5 | 0.5 | 0.5 | 0.5 | 1 |
| MBC | 1 | 0.5 | 1 | 0.5 | 0.5 | 0.5 | 0.5 | 1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jado, J.C.; Humanes, B.; González-Nicolás, M.Á.; Camaño, S.; Lara, J.M.; López, B.; Cercenado, E.; García-Bordas, J.; Tejedor, A.; Lázaro, A. Nephroprotective Effect of Cilastatin against Gentamicin-Induced Renal Injury In Vitro and In Vivo without Altering Its Bactericidal Efficiency. Antioxidants 2020, 9, 821. https://doi.org/10.3390/antiox9090821
Jado JC, Humanes B, González-Nicolás MÁ, Camaño S, Lara JM, López B, Cercenado E, García-Bordas J, Tejedor A, Lázaro A. Nephroprotective Effect of Cilastatin against Gentamicin-Induced Renal Injury In Vitro and In Vivo without Altering Its Bactericidal Efficiency. Antioxidants. 2020; 9(9):821. https://doi.org/10.3390/antiox9090821
Chicago/Turabian StyleJado, Juan Carlos, Blanca Humanes, María Ángeles González-Nicolás, Sonia Camaño, José Manuel Lara, Beatriz López, Emilia Cercenado, Julio García-Bordas, Alberto Tejedor, and Alberto Lázaro. 2020. "Nephroprotective Effect of Cilastatin against Gentamicin-Induced Renal Injury In Vitro and In Vivo without Altering Its Bactericidal Efficiency" Antioxidants 9, no. 9: 821. https://doi.org/10.3390/antiox9090821