Protein Backbone and Average Particle Dynamics in Reconstituted Discoidal and Spherical HDL Probed by Hydrogen Deuterium Exchange and Elastic Incoherent Neutron Scattering

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Studies
2.2. HDL Preparation
2.3. Compositional Analysis of Reconstituted HDL
2.4. Sample Preparation for Elastic Incoherent Neutron Scattering
2.5. Elastic Incoherent Neutron Scattering of Reconstituted HDL Particles
2.6. HDX-MS of Reconstituted Spherical HDL Particles
2.7. Analysis of HDX-MS Spectra
2.8. Kinetic Analysis of HDX-MS Data
3. Results and Discussion
3.1. Reconstituted HDL Particles Have Composition and Biological Activity Similar to Plasma HDL
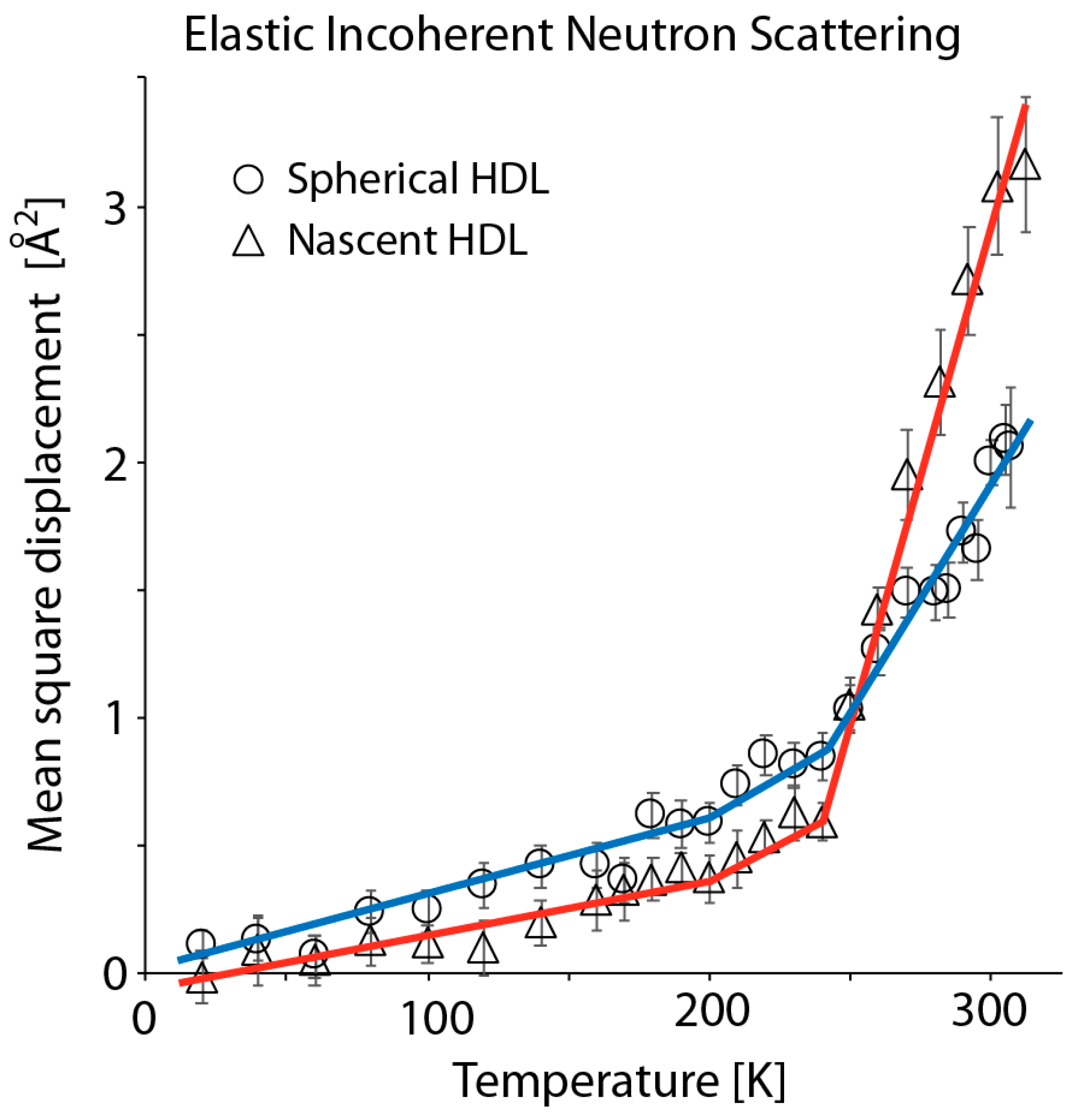
3.2. Elastic Incoherent Neutron Scattering of Reconstituted HDL Particles Reveals that Discoidal HDL is Softer and More Flexible than Spherical HDL at Ambient Temperature
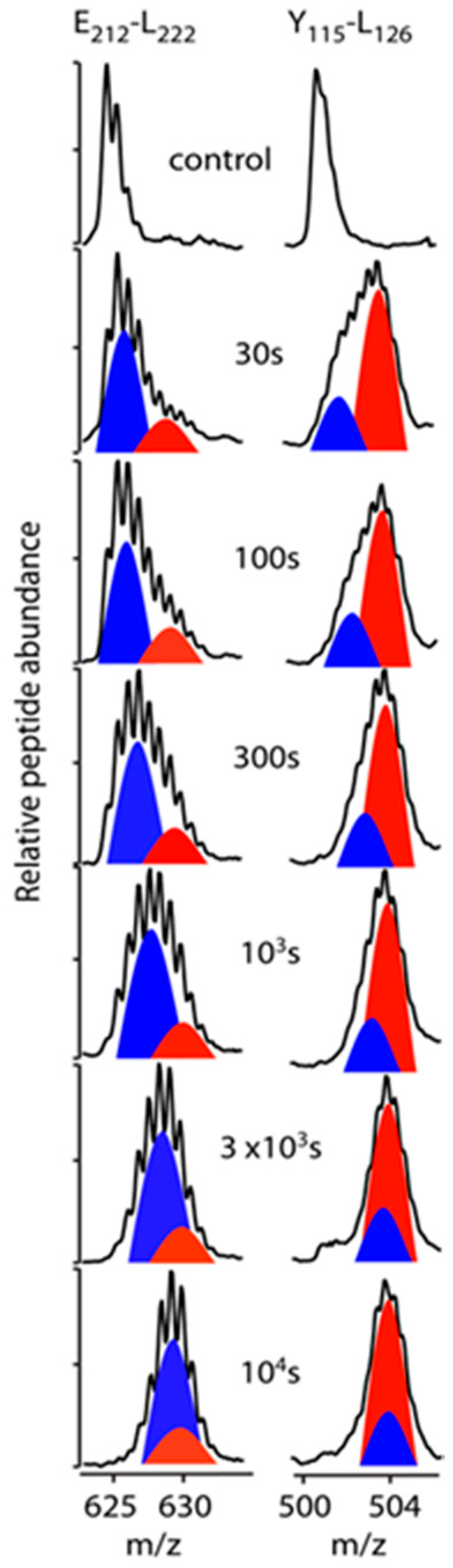
3.3. Bimodal HDX Kinetics in Reconstituted Spherical HDL Indicates Conformational Diversity and/or Distinct Molecular Environments for ApoA-I Chains
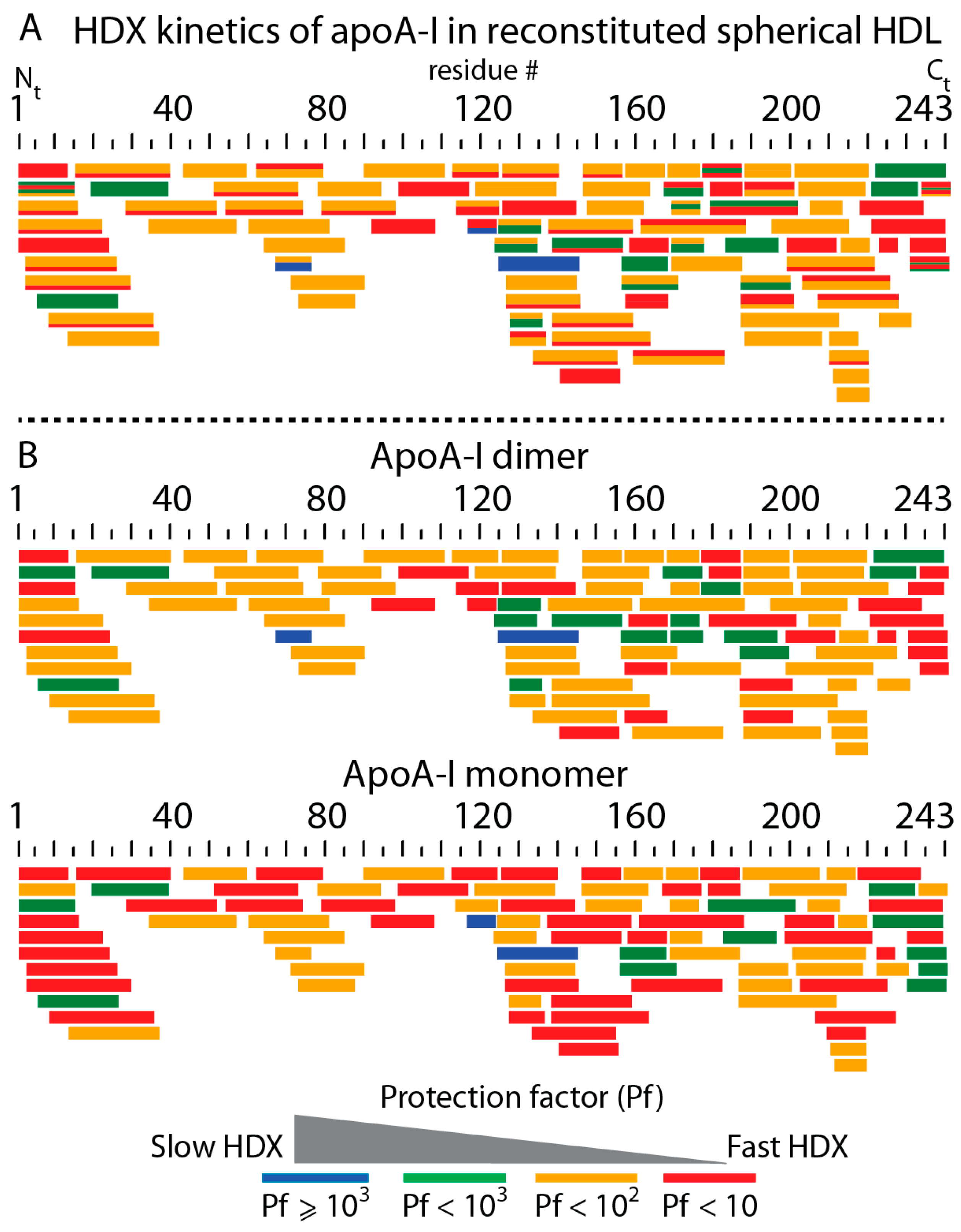
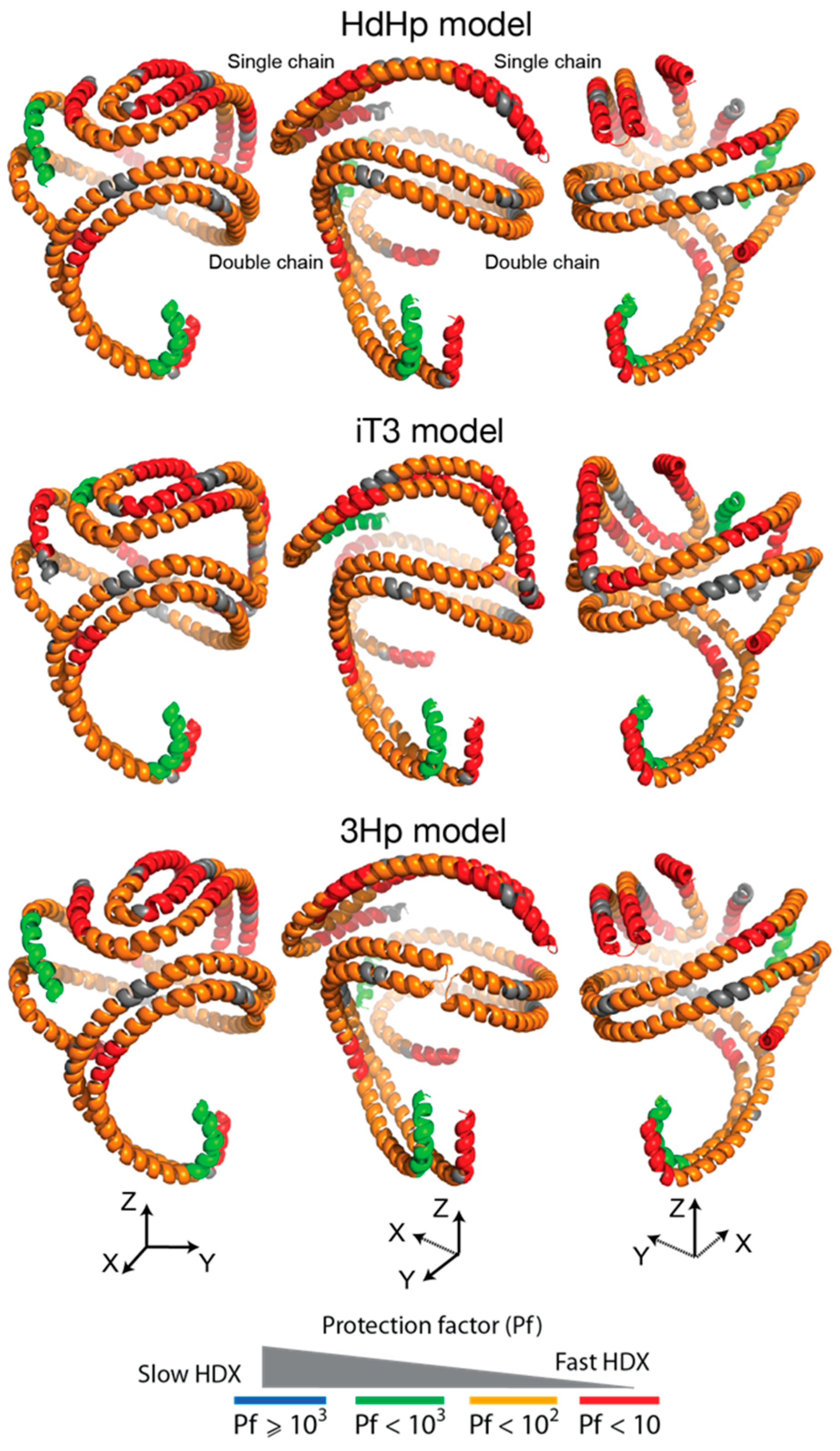
3.4. HDX-MS Kinetic Analyses Support the Model of Reconstituted Spherical HDL with the Three ApoA-I Chains in a Dimer/Monomer Configuration
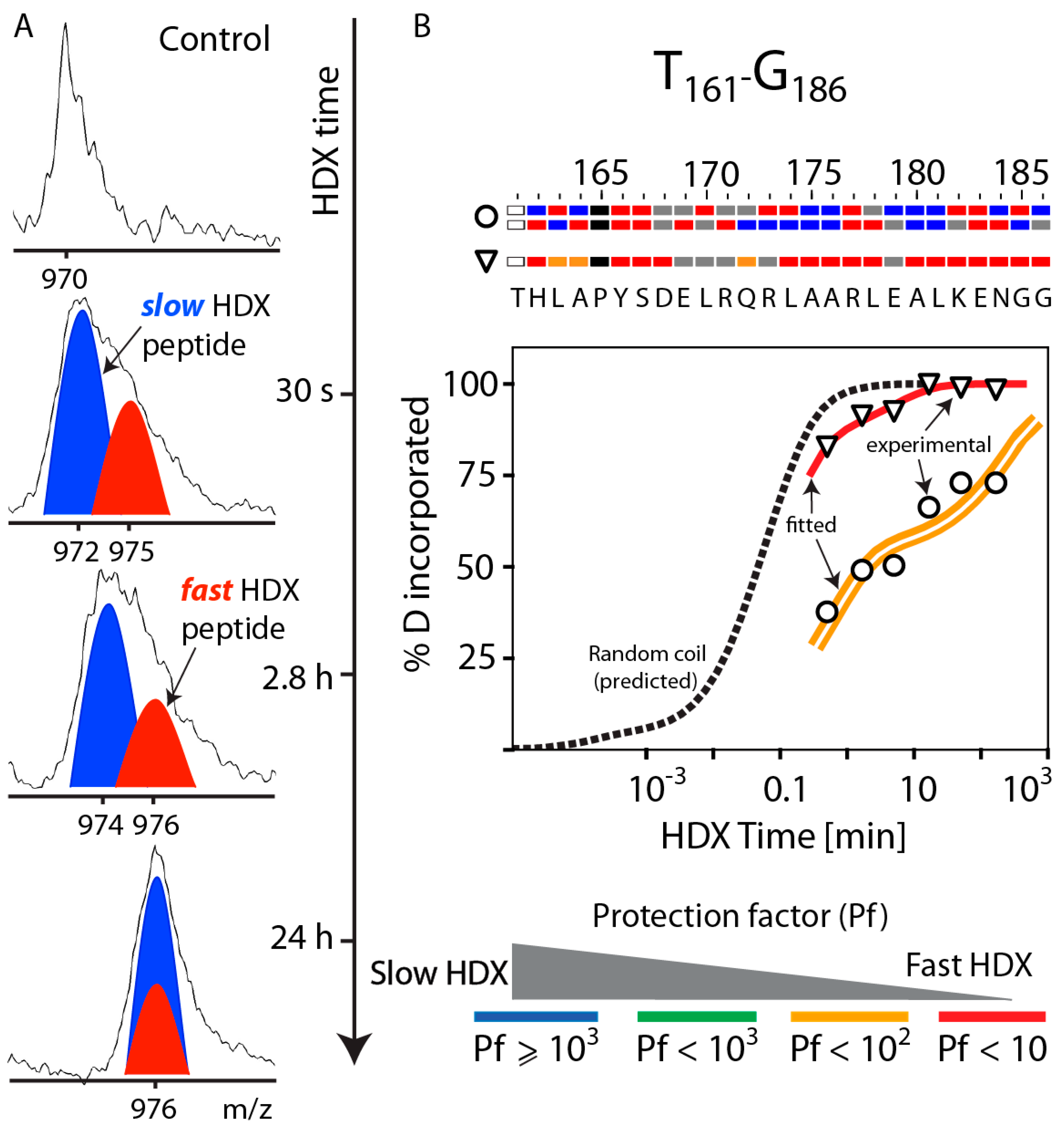
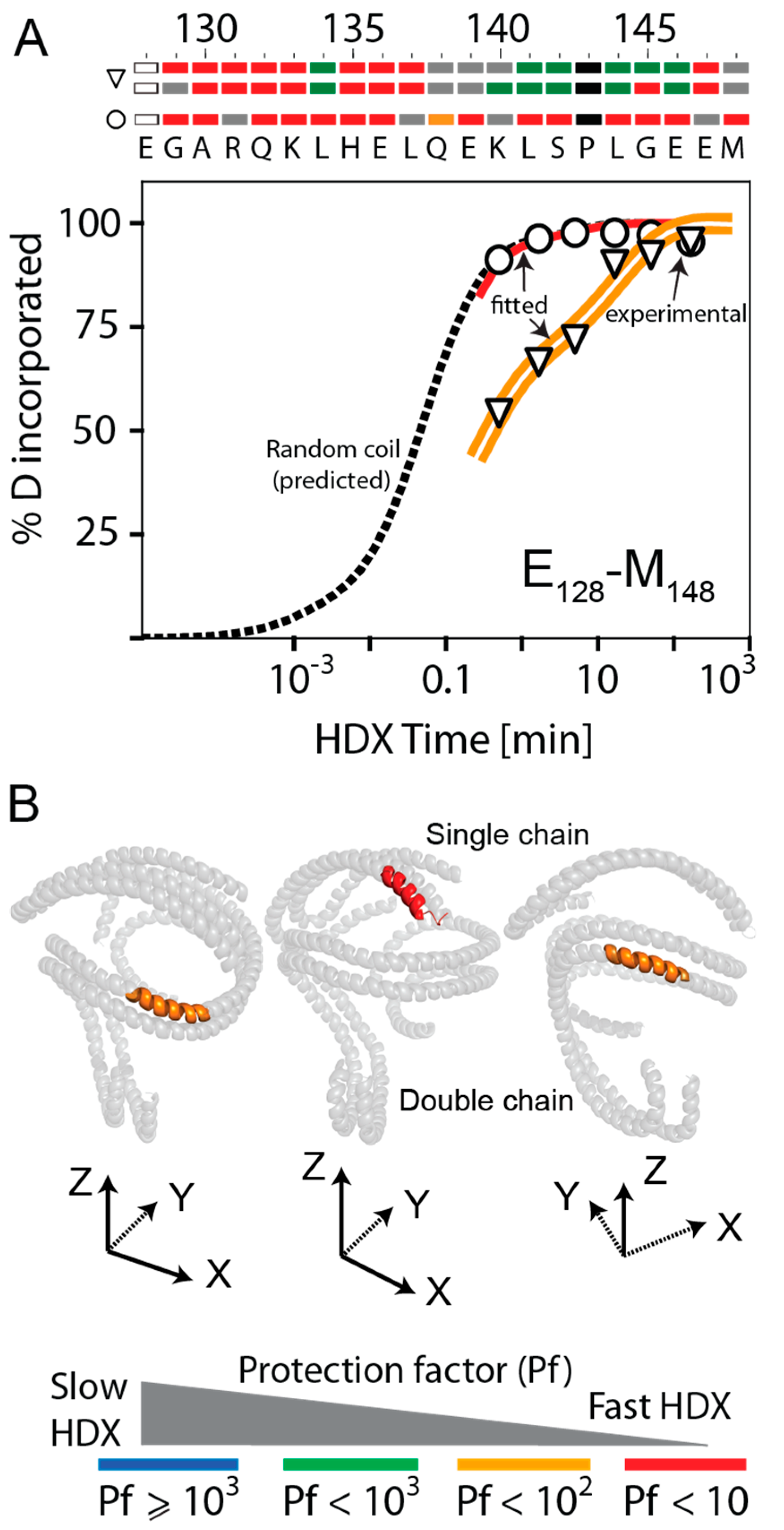
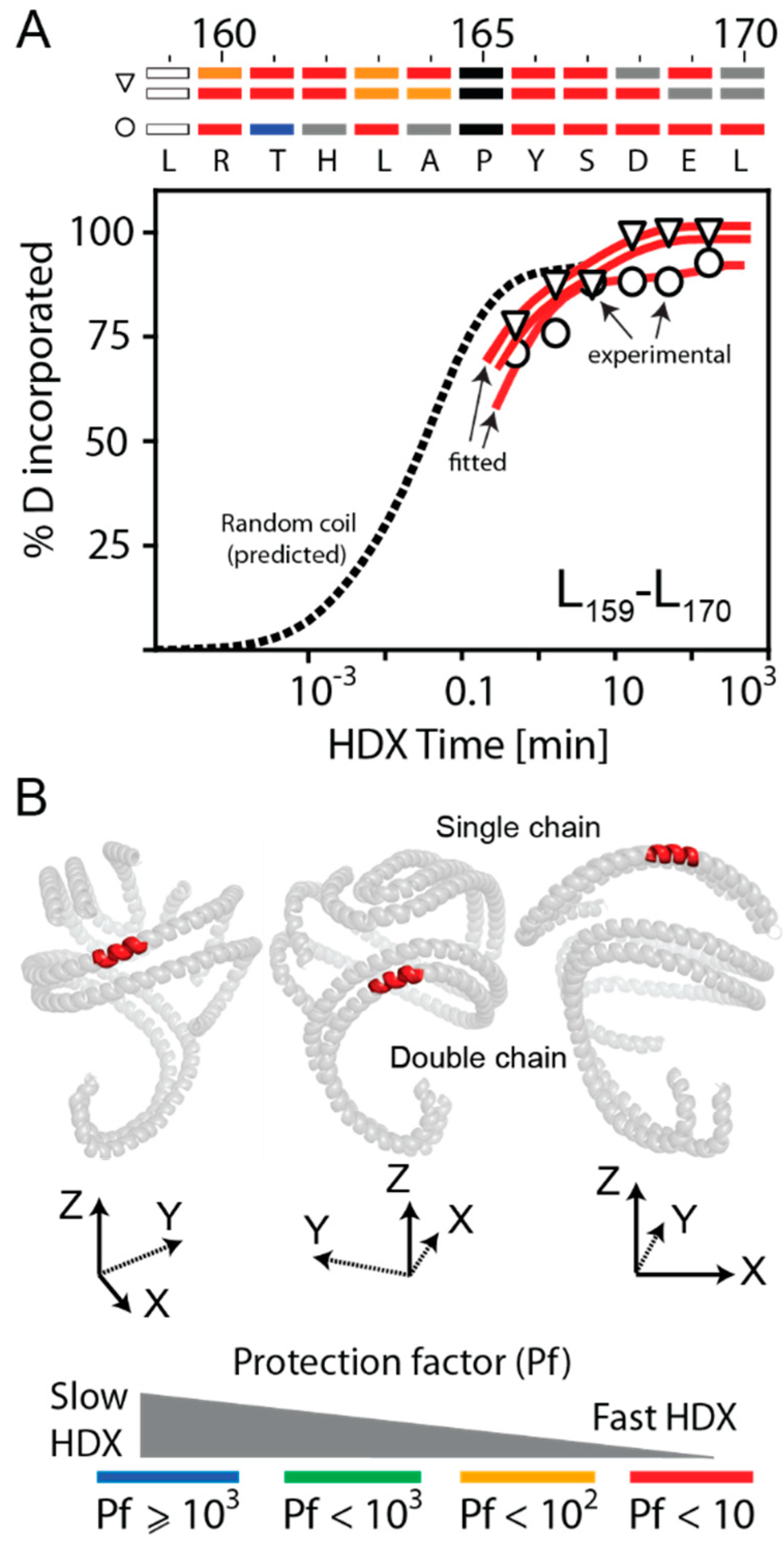
3.5. HDX Bimodal Kinetics Reveals a Complex Conformational Dynamics of ApoA-I in Reconstituted Spherical HDL
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zaccai, G. How soft is a protein? A protein dynamics force constant measured by neutron scattering. Science 2000, 288, 1604–1607. [Google Scholar] [CrossRef] [Green Version]
- Sears, V.F. Neutron scattering lengths and cross sections. Neutron News 1992, 3, 26–37. [Google Scholar] [CrossRef]
- Reat, V.; Zaccai, G.; Ferrand, C.; Pfister, C. Biological Macromolecular Dynamics, Proceedings of a Workshop on Inelastic and Quasielastic Neutron Scattering in Biology; Institut Laue-Langevin: Grenoble, France, 1996. [Google Scholar]
- Peters, J.; Marion, J.; Becher, F.J.; Trapp, M.; Gutberlet, T.; Bicout, D.J.; Heimburg, T. Thermodynamics of lipid multi-lamellar vesicles in presence of sterols at high hydrostatic pressure. Sci. Rep. 2017, 7, 15339. [Google Scholar] [CrossRef] [Green Version]
- Mikl, C.; Peters, J.; Trapp, M.; Kornmueller, K.; Schneider, W.J.; Prass, R. Softness of atherogenic lipoproteins: A comparison of very low density lipoprotein (VLDL) and low density lipoprotein (LDL) using elastic Incoherent neutron scattering (EINS). J. Am. Chem. Soc. 2011, 133, 13213–13215. [Google Scholar] [CrossRef]
- Peters, J.; Martinez, N.; Lehofer, B.; Prassl, R. Low-density lipoproteins investigated under high hydrostatic pressure by elastic incoherent neutron scattering. Eur. Phys. J. E 2017, 40, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golub, M.; Lehofer, B.; Martinez, N.; Ollivier, J.; Kohlbrecher, J.; Prassl, R.; Peters, J. High hydrostatic pressure specifically affects molecular dynamics and shape of low-density lipoprotein particles. Sci. Rep. 2017, 7, 46034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castelli, W.P.; Doyle, J.T.; Gordon, T.; Hames, C.G.; Hjortland, M.C.; Hulley, S.B.; Kagan, A.; Zukel, W.J. HDL cholesterol and other lipids in coronary heart disease. The cooperative lipoprotein phenotyping study. Circulation 1977, 55, 767–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durrington, P.N.; Ishola, M.; Hunt, L.; Arrol, S.; Bhatnagar, D. Apolipoproteins (a), AI, and B and parental history in men with early onset ischaemic heart disease. Lancet 1988, 1, 1070–1073. [Google Scholar] [CrossRef]
- Pekkanen, J.; Linn, S.; Heiss, G.; Suchindran, C.M.; Leon, A.; Rifkind, B.M.; Tyroler, H.A.J. Ten-year mortality from cardiovascular disease in relation to cholesterol level among men with and without preexisting cardiovascular disease. New Engl. J. Med. 1990, 322, 1700–1707. [Google Scholar] [CrossRef]
- Fielding, C.J.; Fielding, P.E. Evidence for a lipoprotein carrier in human plasma catalyzing sterol efflux from cultured fibroblasts and its relationship to lecithin:cholesterol acyltransferase. Proc. Natl. Acad. Sci. USA 1981, 78, 3911–3914. [Google Scholar] [CrossRef] [Green Version]
- Oram, J.F.; Lawn, R.M.; Garvin, M.R.; Wade, D.P. ABCA1 is the cAMP-inducible apolipoprotein receptor that mediates cholesterol secretion from macrophages. J. Biol. Chem. 2000, 275, 34508–34511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Zanotti, I.; Reilly, M.P.; Glick, J.M.; Rothblat, G.H.; Rader, D.J. Overexpression of apolipoprotein A-I promotes reverse transport of cholesterol from macrophages to feces in vivo. Circulation 2003, 108, 661–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, M.C.; Segrest, J.P.; Albers, J.J.; Cone, J.T.; Brouillette, C.G.; Chung, B.H.; Kashyap, M.; Glasscock, M.A.; Anantharamaiah, G.M. Characterization of high density lipoprotein subspecies: Structural studies by single vertical spin ultracentrifugation and immunoaffinity chromatography. J. Lipid Res. 1987, 28, 913–929. [Google Scholar] [PubMed]
- Lund-Katz, S.; Liu, L.; Thuahnai, S.T.; Phillips, M.C. High density lipoprotein structure. Front. Biosci. 2003, 8, d1044–d1054. [Google Scholar] [CrossRef]
- Thomas, M.J.; Bhat, S.; Sorci-Thomas, M.G. Three-dimensional models of HDL apoA-I: Implications for its assembly and function. J. Lipid. Res. 2008, 49, 1875–1883. [Google Scholar] [CrossRef] [Green Version]
- Segrest, J.P.; Jones, M.K.; Klon, A.E.; Sheldahl, C.J.; Hellinger, M.; De Loof, H.; Harvey, S.C. A detailed molecular belt model for apolipoprotein A-I in discoidal high density lipoprotein. J. Biol. Chem. 1999, 274, 31755–31758. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Wagner, M.A.; Zheng, L.; Parks, J.S.; Shy III, J.M.; Smith, J.D.; Gogonea, V.; Hazen, S.L. The refined structure of nascent HDL reveals a key functional domain for particle maturation and dysfunction. Nat. Struct. Mol. Biol. 2007, 14, 861–868. [Google Scholar] [CrossRef]
- Jones, M.K.; Zhang, L.; Catte, A.; Li, L.; Oda, M.N.; Ren, G.; Segrest, J.P. Assessment of the Validity of the Double Superhelix Model for Reconstituted High Density Lipoproteins. A COMBINED COMPUTATIONAL-EXPERIMENTAL APPROACH. J. Biol. Chem. 2010, 285, 41161–41171. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Song, J.; Cavigiolio, G.; Ishida, B.Y.; Zhang, S.; Kane, J.P.; Weisgraber, K.H.; Oda, M.N.; Rye, K.A.; Pownall, H.J.; et al. Morphology and structure of lipoproteins revealed by an optimized negative-staining protocol of electron microscopy. J. Lipid Res. 2011, 52, 175–184. [Google Scholar] [CrossRef] [Green Version]
- Gu, F.; Jones, M.K.; Chen, J.; Patterson, J.C.; Catte, A.; Jerome, W.G.; Li, L.; Segrest, J.P. Structures of Discoidal High Density Lipoproteins A COMBINED COMPUTATIONAL-EXPERIMENTAL APPROACH. J. Biol. Chem. 2010, 285, 4652–4665. [Google Scholar] [CrossRef] [Green Version]
- Lagerstedt, J.O.; Cavigiolio, G.; Budamagunta, M.S.; Pagani, I.; Voss, C.J.; Oda, M.N. Structure of apolipoprotein A-I N terminus on nascent high density lipoproteins. J. Biol. Chem. 2011, 286, 2966–2975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gogonea, V. Structural Insights into High Density Lipoprotein: Old Models and New Facts. Front. Pharmacol. 2015, 6, 311–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Gogonea, V.; Lee, X.; Wagner, M.A.; Li, X.-M.; Huang, Y.; Undurti, A.; May, R.P.; Haertlein, M.; Moulin, M.; et al. The double super helix model of high density lipoprotein. J. Biol. Chem. 2009, 284, 36605–36619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gogonea, V.; Wu, Z.; Lee, X.; Pipich, V.; Li, X.-M.; Ioffe, A.I.; DiDonato, J.A.; Hazen, S.L. Congruency between biophysical data from multiple platforms and molecular dynamics simulation of the double-super helix model of nascent high-density lipoprotein. Biochemistry 2010, 49, 7323–7343. [Google Scholar] [CrossRef] [Green Version]
- Anderson, D.W.; Nichols, A.V.; Pan, S.S.; Lindgren, F.T. High density lipoprotein distribution. Resolution and determination of three major components in a normal population sample. Atherosclerosis 1978, 29, 161–179. [Google Scholar] [CrossRef]
- Blanche, P.J.; Gong, E.L.; Forte, T.M.; Nichols, A.V. Characterization of human high-density lipoproteins by gradient gel electrophoresis. Biochim. Biophys. Acta 1981, 665, 408–419. [Google Scholar] [CrossRef]
- Francone, O.L.; Gurakar, A.; Fielding, C. Distribution and functions of lecithin:cholesterol acyltransferase and cholesteryl ester transfer protein in plasma lipoproteins. Evidence for a functional unit containing these activities together with apolipoproteins A-I and D that catalyzes the esterification and transfer of cell-derived cholesterol. J. Biol. Chem. 1989, 264, 7066–7072. [Google Scholar]
- Silva, R.A.G.D.; Huang, R.; Morris, J.; Fang, J.; Gracheva, E.O.; Ren, G.; Kontush, A.; Jerome, W.G.; Rye, K.-A.; Davidson, W.S. Structure of apolipoprotein A-I in spherical high density lipoproteins of different sizes. Proc. Natl. Acad. Sci. USA 2008, 105, 12176–12181. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Gogonea, V.; Lee, X.; May, M.J.; Pipich, V.; Wagner, M.A.; Undurti, A.; Tallant, T.C.; Baleanu-Gogonea, C.; Charlton, F.; et al. The low resolution structure of ApoA1 in spherical high density lipoprotein revealed by small angle neutron scattering. J. Biol. Chem. 2011, 286, 12495–12508. [Google Scholar] [CrossRef] [Green Version]
- Chetty, P.S.; Nguyen, D.; Nickel, M.; Lund-Katz, S.; Mayne, L.; Englander, S.W.; Phillips, M.C. Comparison of apoA-I helical structure and stability in discoidal and spherical HDL particles by HX and mass spectrometry. J. Lipid Res. 2013, 54, 1589–1597. [Google Scholar] [CrossRef] [Green Version]
- Jonas, A.; Wald, J.H.; Toohill, K.L.; Krul, E.S.; Kezdy, K.E. Apolipoprotein A-I structure and lipid properties in homogeneous, reconstituted spherical and discoidal high density lipoproteins. J. Biol. Chem. 1990, 265, 22123–22129. [Google Scholar] [PubMed]
- Li, H.-H.; Lyles, D.S.; Pan, W.; Alexander, E.; Thomas, M.J.; Sorci-Thomas, M.G. ApoA-I Structure on discs and spheres. Variable helix registry and conformational states. J. Biol. Chem. 2002, 277, 39093–39101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, W.S.; Sparks, D.L.; Lund-Katz, S.; Phillips, M.C. The molecular basis for the difference in charge between pre- beta- and alpha-migrating high density lipoproteins. J. Biol. Chem. 1994, 269, 8959–8965. [Google Scholar] [PubMed]
- Dalton, M.B.; Swaney, J.B. Structural and functional domains of apolipoprotein A-I within high density lipoproteins. J. Biol. Chem. 1993, 268, 19274–19283. [Google Scholar]
- Sparks, D.L.; Phillips, M.C.; Lund-Katz, S. The conformation of apolipoprotein A-I in discoidal and spherical recombinant high density lipoprotein particles. 13C NMR studies of lysine ionization behavior. J. Biol. Chem. 1992, 267, 25830–25838. [Google Scholar]
- Gogonea, V.; Gerstenecker, G.S.; Wu, Z.; Lee, X.; Topbas, C.; Wagner, M.A.; Tallant, T.C.; Smith, J.D.; Callow, P.; Pipich, V.; et al. The low resolution structures of nascent high density lipoprotein reconstituted with DMPC with and without cholesterol reveals a mechanism for particle expansion. J. Lipid Res. 2013, 54, 966–983. [Google Scholar] [CrossRef] [Green Version]
- Silva, R.A.G.D.; Hillard, G.M.; Li, L.; Segrest, J.P.; Davidson, W.S. A mass spectrometric determination of the conformation of dimeric apolipoprotein A-I in discoidal high density lipoproteins. Biochemistry 2005, 44, 8600–8607. [Google Scholar] [CrossRef]
- Huang, R.; Silva, R.A.G.D.; Jerome, G.M.; Kontush, A.; Chapman, M.J.; Curtiss, L.K.; Hodges, T.J.; Davidson, W.S. Apolipoprotein A-I structural organization in high density lipoproteins isolated from human plasma. Nat. Struct. Mol. Bio. 2011, 18, 416–422. [Google Scholar] [CrossRef] [Green Version]
- Chetty, P.S.; Mayne, L.; Lund-Katz, S.; Stranz, D.; Englander, S.W.; Phillips, M.C. Helical structure and stability in human apolipoprotein A-I by hydrogen exchange and mass spectrometry. Proc. Natl. Acad. Sci. USA 2009, 106, 19005–19010. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Smith, D.L. Determination of amide hydrogen exchange by mass spectrometry: A new tool for protein structure elucidation. Protein Sci. 1993, 2, 522–531. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.M.; Bou-Assaf, G.M.; Emmett, M.R.; Marshall, A.G. Fast reversed-phase liquid chromatography to reduce back exchange and increase throughput in H/D exchange monitored by FT-ICR mass spectrometry. J. Am. Soc. Mass. Spectrom. 2009, 20, 520–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcoux, J.; Thierry, E.; Vives, C.; Signor, L.; Fieschi, F.; Forest, E. Investigating alternative acidic proteases for H/D exchange coupled to mass spectrometry: Plasmepsin 2 but not plasmepsin 4 is active under quenching conditions. J. Am. Soc. Mass. Spectrom. 2010, 21, 76–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayne, L.; Englander, S.W. Two-state vs. multistate protein unfolding studied by optical melting and hydrogen exchange. Prot. Sci. 2000, 9, 1873–1877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayne, L.; Kan, Z.Y.; Chetty, P.S.; Ricciuti, A.; Walters, B.T.; Englander, S.W. Many overlapping peptides for protein hydrogen exchange experiments by the fragment separation-mass spectrometry method. J. Am. Soc. Mass. Spectrom. 2011, 22, 1898–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matz, C.E.; Jonas, A. Micellar complexes of human apolipoprotein A-I with phosphatidylcholines and cholesterol prepared from cholate-lipid dispersions. J. Biol. Chem. 1982, 257, 4535–4540. [Google Scholar]
- Rye, K.-A.; Barter, P.J. The Influence of Apolipoproteins on the Structure and Function of Spheroidal, Reconstituted High Density Lipoproteins. J. Biol. Chem. 1994, 269, 10298–10303. [Google Scholar]
- Perticaroli, S.; Ehlers, G.; Stanley, C.B.; Mamontov, E.; O’Neill, H.; Zhang, Q.; Cheng, X.; Myles, D.A.; Katsaras, J.; Nickels, J.D. Description of hydration water in protein (green fluorescent protein) solution. J. Am. Chem. Soc. 2017, 139, 1098–1105. [Google Scholar] [CrossRef]
- Natali, F.; Peters, J.; Russo, D.; Barbieri, S.; Chiapponi, C.; Cupane, A.; Deriu, A.; Di Bari, M.T.; Farhi, E.; Gerelli, Y.; et al. IN13 Backscattering spectrometer at ILL: Looking for motions in biological macromolecules and organisms. Neutron News 2008, 19, 14–18. [Google Scholar]
- Frick, B.; Gonzalez, M. Five years operation of the second generation backscattering spectrometer IN16—A retrospective, recent developments and plans. Physica B Cond. Matt. 2001, 301, 8–19. [Google Scholar] [CrossRef]
- Tehei, M.; Franzetti, B.; Wood, K.; Gabel, F.; Fabiani, E.; Jasnin, M.; Zamponi, M.; Oesterhelt, D.; Zaccai, G.; Ginzburg, M.; et al. Neutron scattering reveals extremely slow cell water in a Dead Sea organism. Proc. Natl. Acad. Sci. USA 2007, 104, 766–771. [Google Scholar] [CrossRef] [Green Version]
- Rahman, A.; Singwi, K.S.; Sjölander, A. Theory of slow neutron scattering by liquids. I. Phys. Rev. 1962, 126, 986–996. [Google Scholar] [CrossRef]
- Bee, M. Quasielastic Neutron Scattering; Adam Hilger: Bristol, UK, 1988. [Google Scholar]
- Richard, D.; Ferrand, M.; Kearley, G.J. Analysis and visualisation of neutron scattering data. J. Neutron Res. 1996, 4, 33–39. [Google Scholar] [CrossRef]
- Paalman, H.H.; Pings, C.J. Numerical evaluation of x-ray absorption factors for cylindrical samples and annular sample cells. J. Appl. Phys. 1962, 33, 2635–2639. [Google Scholar] [CrossRef]
- Chetty, P.S.; Mayne, L.; Kan, Z.-Y.; Lund-Katz, S.; Englander, S.W.; Phillips, M.C. Apolipoprotein A-I helical structure and stability in discoidal high-density lipoprotein (HDL) particles by hydrogen exchange and mass spectrometry. Proc. Natl. Acad. Sci. USA 2012, 109, 11687–11692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, Y.; Milne, J.S.; Mayne, L.; Englander, S.W. Primary structure effects on peptide group hydrogen exchange. Proteins: Struc. Funct. Genet. 1993, 17, 75–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazen, S.L. HDL Structure, Function, Therapeutics, and Imaging. Arterioscler Thromb Vasc Biol. 2010, 30, 138. [Google Scholar] [CrossRef] [Green Version]
- Englander, S.W. Hydrogen exchange and mass spectrometry: A historical perspective. J. Am. Mass. Spectrom. 2006, 17, 1481–1489. [Google Scholar] [CrossRef] [Green Version]
- Murphy, A.J.; Chin-Dusting, J.; Sviridov, D. Reconstituted HDL: A Therapy for Atherosclerosis and Beyond. Clin. Lipidology 2009, 4, 731–739. [Google Scholar] [CrossRef]
- Javaheri, A.; Kolansky, D.M.; Cuchel, M. Reconstituted High-Density Lipoprotein Therapies. Arterioscl. Throm. Vas. 2014, 34, 1800–1802. [Google Scholar] [CrossRef] [Green Version]
- Darabi, M.; Guillas-Baudouin, I.; Le Goff, W.; Chapman, M.J.; Kontush, A. Therapeutic applications of reconstituted HDL: When structure meets function. Pharmacol. Therapeut. 2016, 157, 28–42. [Google Scholar] [CrossRef]
- Stadler, A.M.; Digel, I.; Embs, J.P.; Unruh, T.; Tehei, M.; Zaccai, G.; Büldt, G.; Artmann, G.M. From powder to solution: Hydration dependence of human hemoglobin dynamics correlated to body temperature. Biophys. J. 2009, 96, 5073–5081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, J.; Zanotti, J.-M.; Durand, D. Evolution of the Internal Dynamics of Two Globular Proteins from Dry Powder to Solution. Biophys. J. 1999, 77, 454–469. [Google Scholar] [CrossRef] [Green Version]
- Doster, W.; Cusack, S.; Petry, W. Dynamical transition of myoglobin revealed by inelastic neutron scattering. Nature 1989, 337, 754–756. [Google Scholar] [CrossRef]
- Trapp, M.; Marion, J.; Tehei, M.; Demé, B.; Gutberlet, T.; Peters, J. High hydrostatic pressure effects in-vestigated by neutron scattering on lipid multilamellar vesicles. Phys. Chem. Chem. Phys. 2013, 15, 20951–20956. [Google Scholar] [CrossRef] [PubMed]
- Frölich, A.; Gabel, F.; Jasnin, M.; Lehnert, U.; Oesterhelt, D.; Stadler, A.M.; Tehei, M.; Weik, M.; Wood, K.; Zaccai, G. From Shell to cell: Neutron scattering studies of biological water dynamics and coupling to activity. Faraday Discuss. 2009, 141, 117. [Google Scholar] [CrossRef] [Green Version]
- Frauenfelder, H.; Parak, F.; Young, R.D. Conformational sub-states in proteins. Ann. Rev. Biophys. Biophys. Chem. 1988, 17, 451–479. [Google Scholar] [CrossRef] [PubMed]
- Kido, T.; Kurata, H.; Kondo, K.; Itakura, H.; Okazaki, M.; Urata, T.; Yokoyama, S. Bioinformatic analysis of plasma apolipoproteins A-I and A-II revealed unique features of A-I/A-II HDL particles in human plasma. Sci. Rep. 2016, 6, 31532. [Google Scholar] [CrossRef] [Green Version]
- Flenner, E.; Das, J.; Rheinstadter, M.C.; Kosztin, I. Subdiffusion and lateral diffusion coefficient of lipid atoms and molecules in phospholipid bilayers. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2009, 79, 011907. [Google Scholar] [CrossRef] [Green Version]
- Trapp, M.; Juranyi, F.; Tehei, M.; van Eijck, L.; Deme, B.; Gutberlet, T.; Peters, J. Elastic scattering studies of aligned DMPC multilayers on different hydrations. Spectroscopy 2010, 24, 461–466. [Google Scholar] [CrossRef]
- Peters, J.; Giudici-Orticoni, M.T.; Zaccai, G.; Guiral, M. Dynamics measured by neutron scattering cor-relates with the organization of bioenergetics complexes in natural membranes from hyperthermophile and mesophile bacteria. Eur. Phys. J. E Soft Matter. 2013, 36, 78. [Google Scholar] [CrossRef]
- Johs, A.; Hammel, M.; Waldner, I.; May, R.P.; Laggner, P.; Prassl, R. Modular structure of solubilized human apolipoprotein B-100: Low resolution model revealed by small angle neutron scattering. J. Biol. Chem. 2006, 281, 19732–19739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prassl, R. Human low density lipoprotein: The mystery of core lipid packing. J. Lipid Res. 2011, 52, 187–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lipoprotein | Instrument a | Force Constant b | ||
| 20–200 K c | 200–250 K | 250–310 K | ||
| rsHDL d | IN13 | 1.00 ± 0.13 | 0.25 ± 0.03 | 0.15 ± 0.03 |
| rHDL e | 1.13 ± 0.11 | 0.49 ± 0.11 | 0.067 ± 0.002 | |
| rsHDL | IN16 | 0.88 ± 0.03 | 0.17 ± 0.01 | 0.12 ± 0.02 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gogonea, V.; Peters, J.; Gerstenecker, G.S.; Topbas, C.; Hou, L.; Combet, J.; DiDonato, J.A.; Smith, J.D.; Rye, K.-A.; Hazen, S.L. Protein Backbone and Average Particle Dynamics in Reconstituted Discoidal and Spherical HDL Probed by Hydrogen Deuterium Exchange and Elastic Incoherent Neutron Scattering. Biomolecules 2020, 10, 121. https://doi.org/10.3390/biom10010121
Gogonea V, Peters J, Gerstenecker GS, Topbas C, Hou L, Combet J, DiDonato JA, Smith JD, Rye K-A, Hazen SL. Protein Backbone and Average Particle Dynamics in Reconstituted Discoidal and Spherical HDL Probed by Hydrogen Deuterium Exchange and Elastic Incoherent Neutron Scattering. Biomolecules. 2020; 10(1):121. https://doi.org/10.3390/biom10010121
Chicago/Turabian StyleGogonea, Valentin, Judith Peters, Gary S. Gerstenecker, Celalettin Topbas, Liming Hou, Jérôme Combet, Joseph A. DiDonato, Jonathan D. Smith, Kerry-Anne Rye, and Stanley L. Hazen. 2020. "Protein Backbone and Average Particle Dynamics in Reconstituted Discoidal and Spherical HDL Probed by Hydrogen Deuterium Exchange and Elastic Incoherent Neutron Scattering" Biomolecules 10, no. 1: 121. https://doi.org/10.3390/biom10010121