Insight into the RssB-Mediated Recognition and Delivery of σs to the AAA+ Protease, ClpXP


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cloning
2.2. Protein Purification and Size Exclusion Chromatography
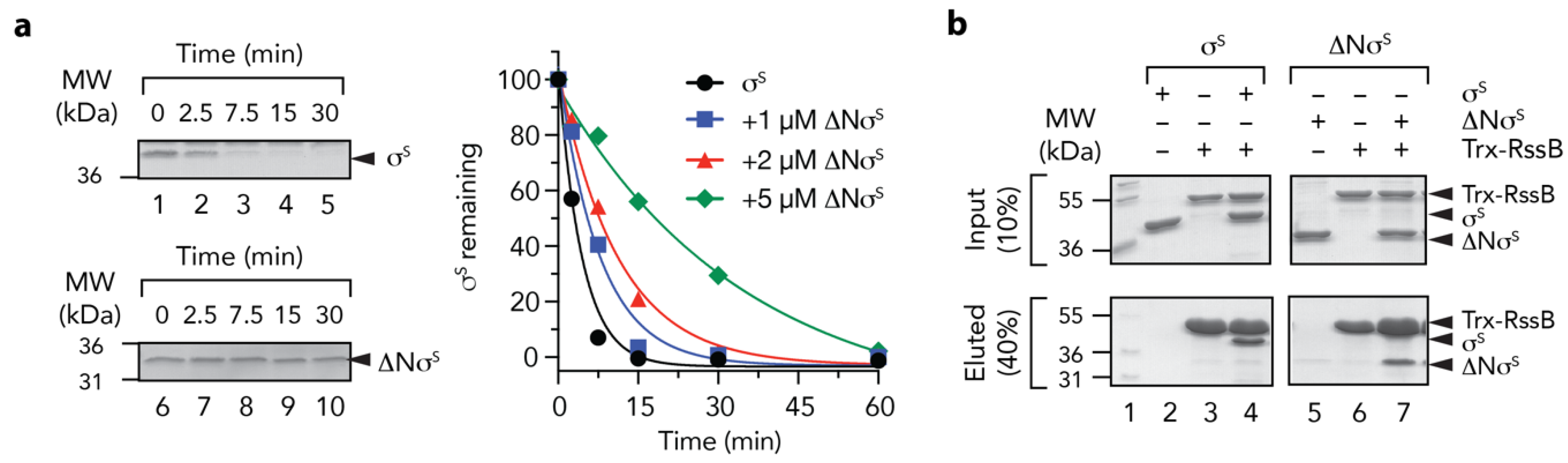
2.3. In Vitro Degradation Assays
2.4. Glutaraldehyde Cross-Linking
2.5. Peptide Library
2.6. In Vitro ‘Pull-Down’ Experiments
2.7. Co-Immunoprecipitation (Co-IP)
2.8. Crystallization of RssBN and RssBC and Diffraction Data Collection
2.9. Structure Determination and Refinement
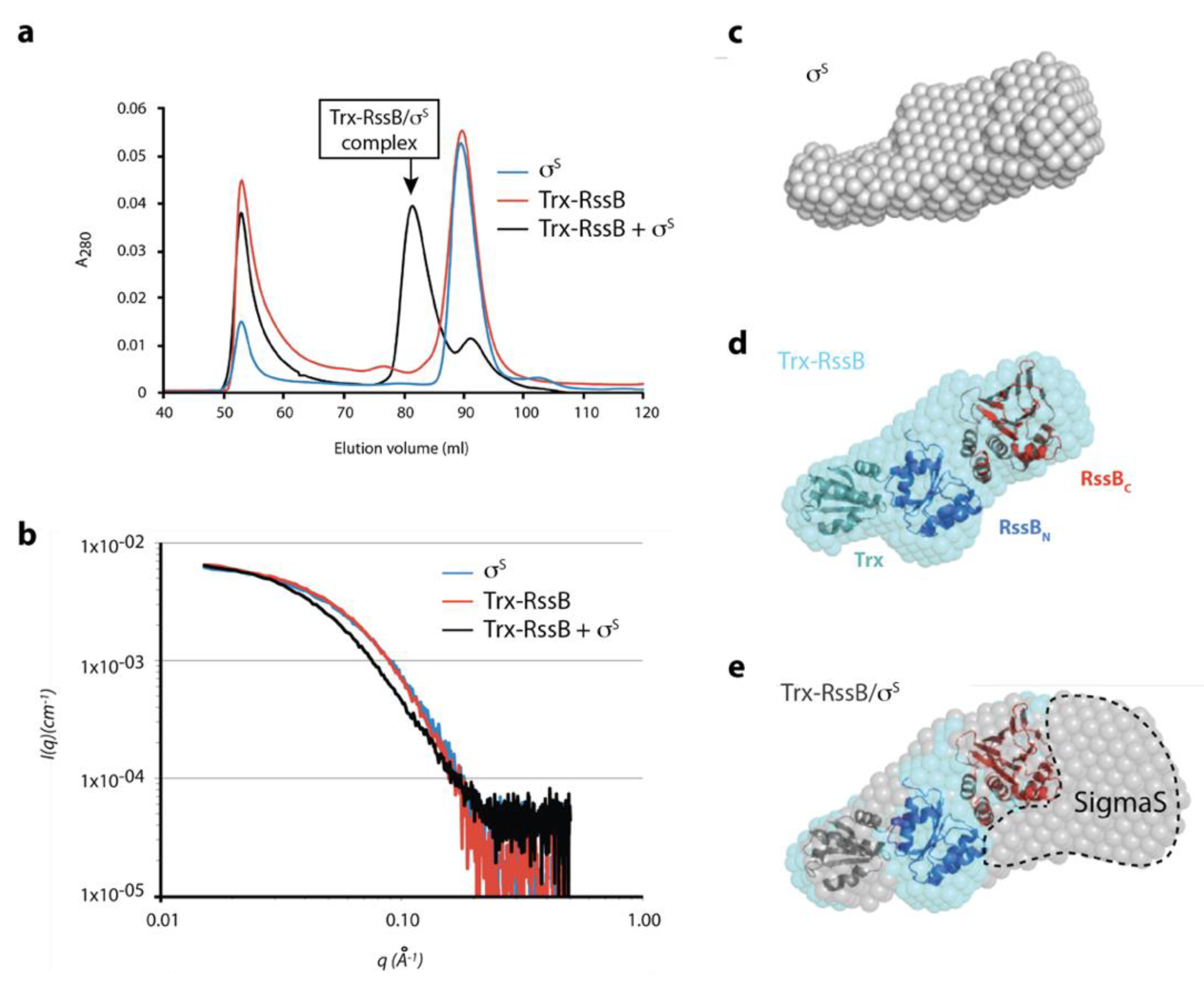
2.10. Small Angle X-ray Scattering (SAXS) of RssB, σS and the RssB/σS Complex
3. Results
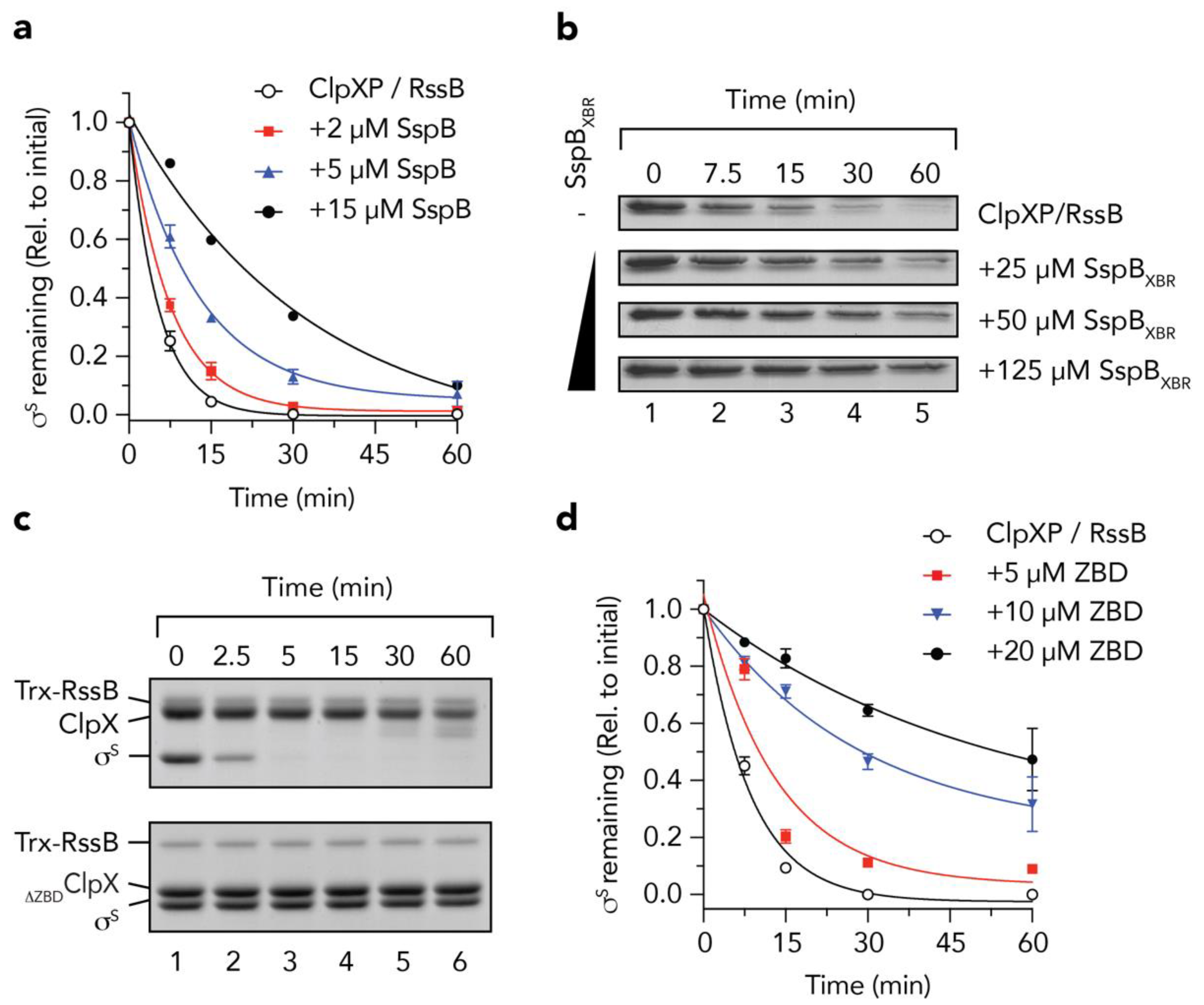
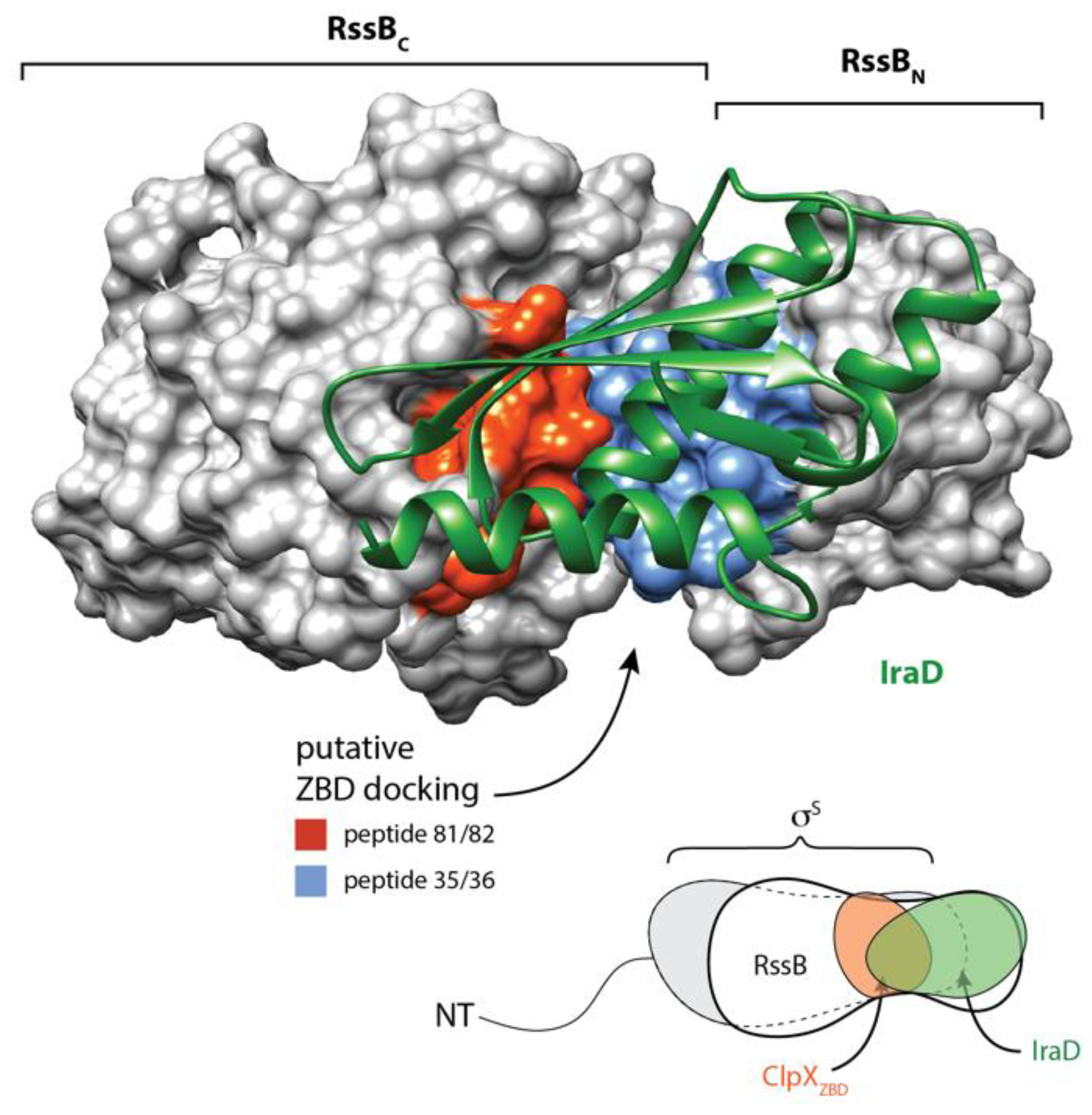
3.1. The RssB/σs Complex Docks to the ZBD of ClpX
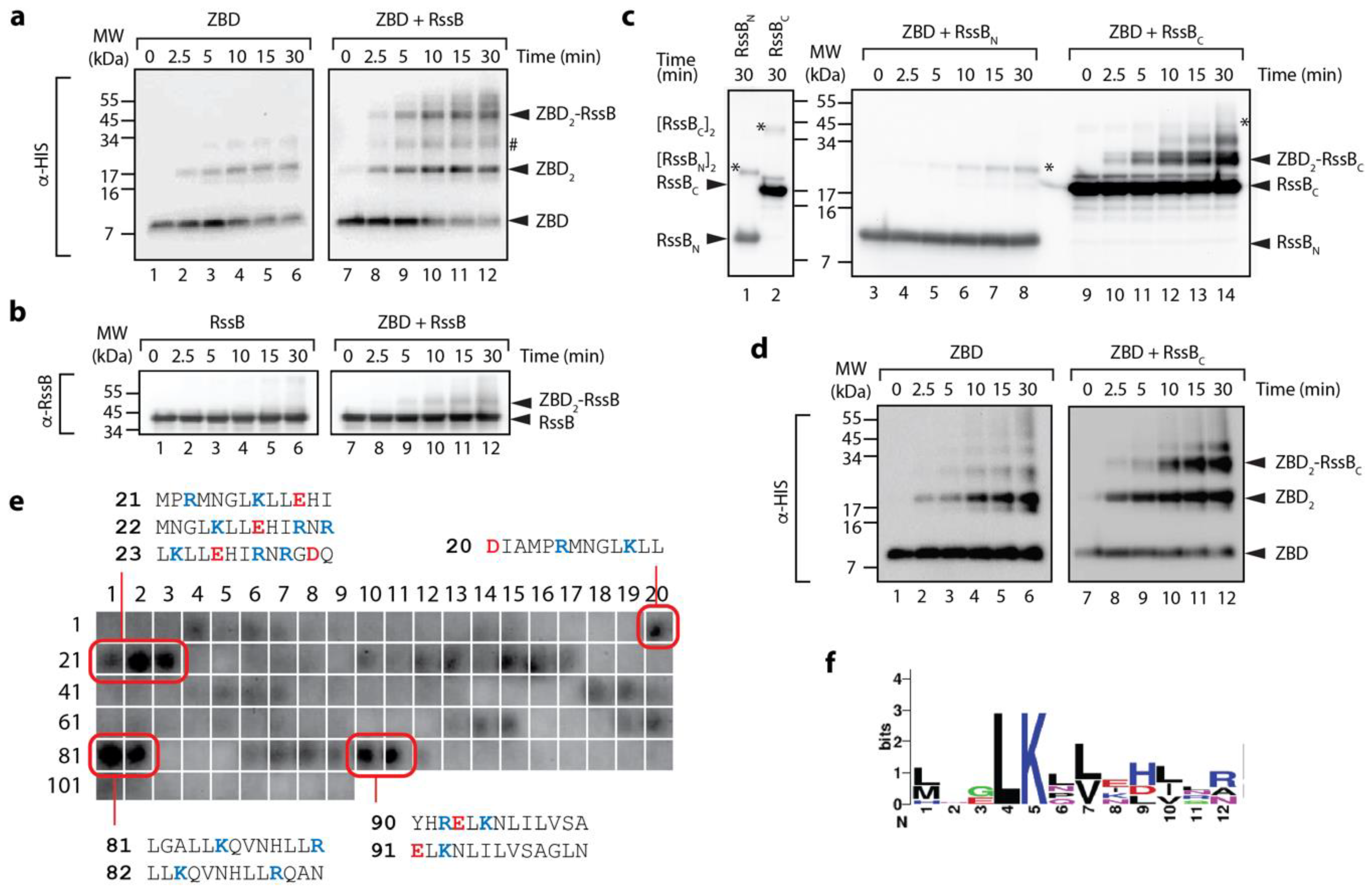
3.2. The C-Terminal Domain of RssB (RssBC) Docks Directly to the ZBD
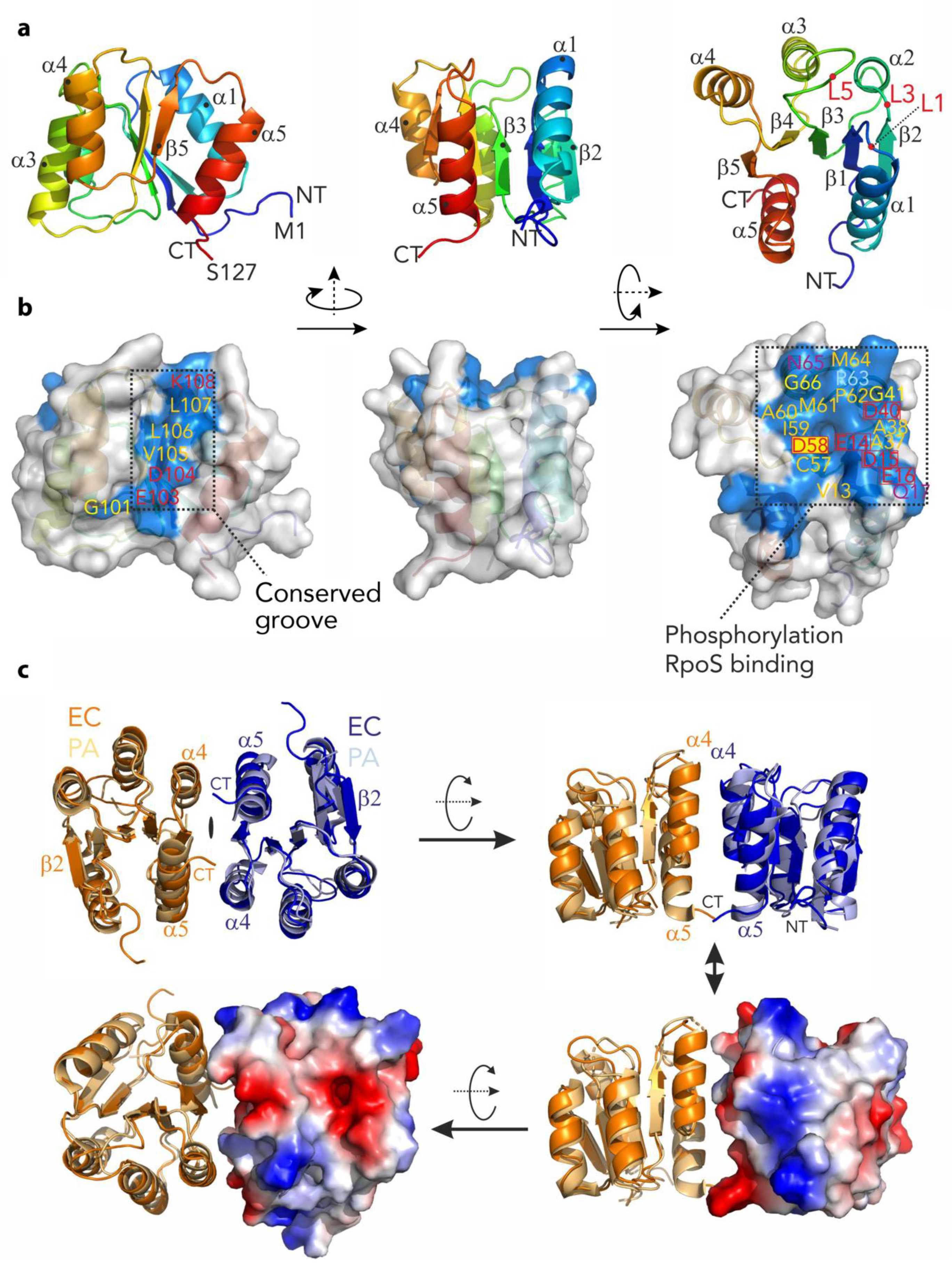
3.3. Structure of the C-Terminal Phosphatase Domain of RssB
3.4. The N-Terminal Domain of RssB Adopts the Fold of a Two Component System Regulator
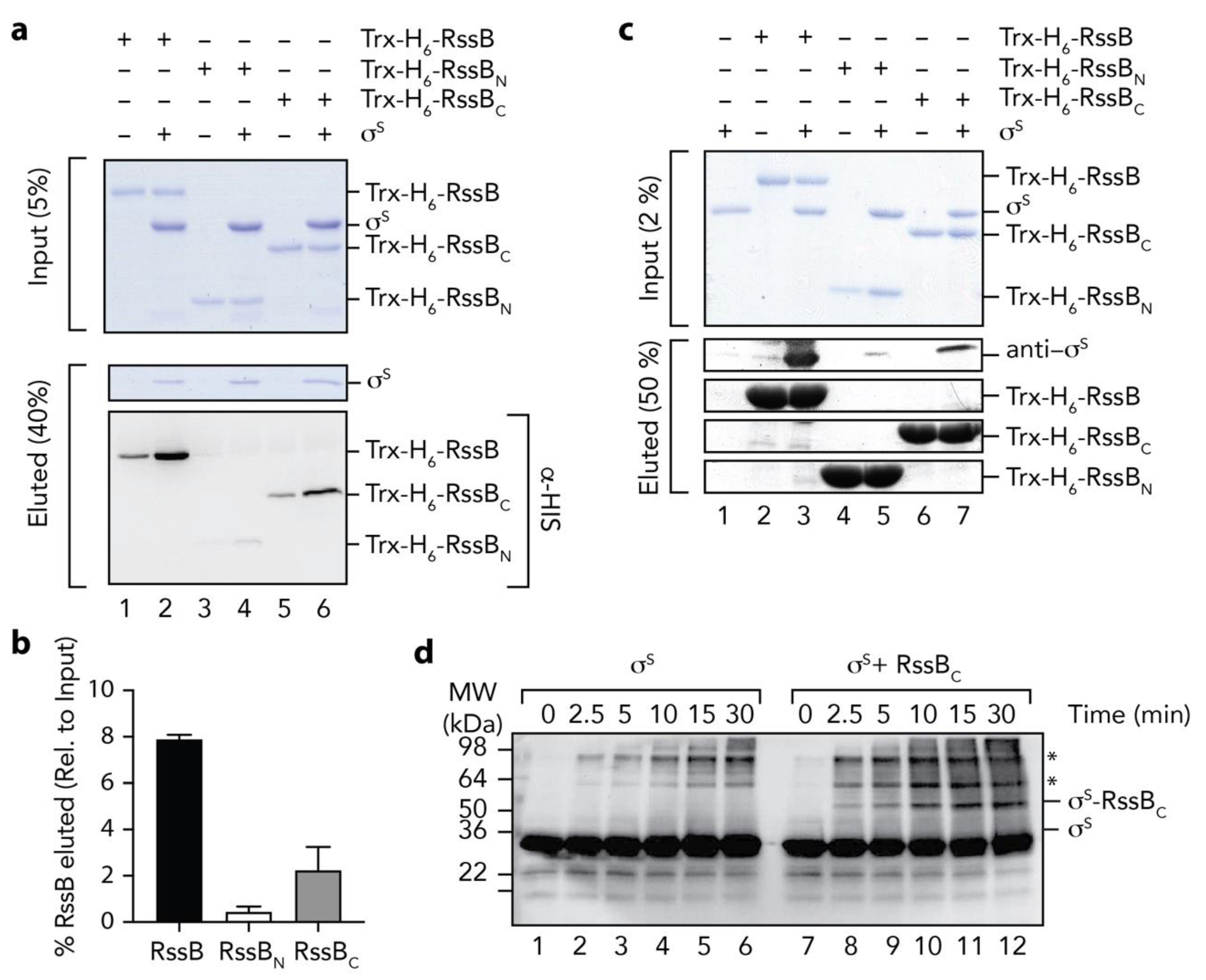
3.5. The C-Terminal Domain of RssB is Required for σs Docking
3.6. Mutations in RssBN Regulate Phosphorylation Dependent σs Binding
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Loewen, P.C.; Hu, B.; Strutinsky, J.; Sparling, R. Regulation in the rpoS regulon of Escherichia coli. Can. J. Microbiol. 1998, 44, 707–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hengge-Aronis, R. Back to log phase: Sigma S as a global regulator in the osmotic control of gene expression in Escherichia coli. Mol. Microbiol. 1996, 21, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Stokes, N.R.; Murray, H.D.; Subramaniam, C.; Gourse, R.L.; Louis, P.; Bartlett, W.; Miller, S.; Booth, I.R. A role for mechanosensitive channels in survival of stationary phase: Regulation of channel expression by RpoS. Proc. Natl. Acad. Sci. USA 2003, 100, 15959–15964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottesman, S. Trouble is coming: Signaling pathways that regulate general stress responses in bacteria. J. Biol. Chem. 2019, 294, 11685–11700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micevski, D.; Dougan, D.A. Proteolytic regulation of stress response pathways in Escherichia coli. Subcell. Biochem. 2013, 66, 105–128. [Google Scholar] [CrossRef]
- Dougan, D.A.; Truscott, K.N.; Zeth, K. The bacterial N-end rule pathway: Expect the unexpected. Mol. Microbiol. 2010, 76, 545–558. [Google Scholar] [CrossRef]
- Gur, E.; Ottofueling, R.; Dougan, D.A. Machines of destruction - AAA+ proteases and the adaptors that control them. Subcell. Biochem. 2013, 66, 3–33. [Google Scholar] [CrossRef]
- Varshavsky, A. N-degron and C-degron pathways of protein degradation. Proc. Natl. Acad. Sci. USA 2019, 116, 358–366. [Google Scholar] [CrossRef] [Green Version]
- Flynn, J.M.; Neher, S.B.; Kim, Y.I.; Sauer, R.T.; Baker, T.A. Proteomic discovery of cellular substrates of the ClpXP protease reveals five classes of ClpX-recognition signals. Mol. Cell 2003, 11, 671–683. [Google Scholar] [CrossRef]
- Gottesman, S.; Roche, E.; Zhou, Y.; Sauer, R.T. The ClpXP and ClpAP proteases degrade proteins with carboxy-terminal peptide tails added by the SsrA-tagging system. Genes Dev. 1998, 12, 1338–1347. [Google Scholar] [CrossRef]
- Erbse, A.; Schmidt, R.; Bornemann, T.; Schneider-Mergener, J.; Mogk, A.; Zahn, R.; Dougan, D.A.; Bukau, B. ClpS is an essential component of the N-end rule pathway in Escherichia coli. Nature 2006, 439, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Dougan, D.A.; Weber-Ban, E.; Bukau, B. Targeted delivery of an ssrA-tagged substrate by the adaptor protein SspB to its cognate AAA+ protein ClpX. Mol. Cell 2003, 12, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Levchenko, I.; Grant, R.A.; Wah, D.A.; Sauer, R.T.; Baker, T.A. Structure of a delivery protein for an AAA+ protease in complex with a peptide degradation tag. Mol. Cell 2003, 12, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Wah, D.A.; Levchenko, I.; Rieckhof, G.E.; Bolon, D.N.; Baker, T.A.; Sauer, R.T. Flexible linkers leash the substrate binding domain of SspB to a peptide module that stabilizes delivery complexes with the AAA+ ClpXP protease. Mol. Cell 2003, 12, 355–363. [Google Scholar] [CrossRef]
- Wojtyra, U.A.; Thibault, G.; Tuite, A.; Houry, W.A. The N-terminal zinc binding domain of ClpX is a dimerization domain that modulates the chaperone function. J. Biol. Chem. 2003, 278, 48981–48990. [Google Scholar] [CrossRef] [Green Version]
- Muffler, A.; Fischer, D.; Altuvia, S.; Storz, G.; Hengge-Aronis, R. The response regulator RssB controls stability of the sigma(S) subunit of RNA polymerase in Escherichia coli. EMBO J. 1996, 15, 1333–1339. [Google Scholar] [CrossRef]
- Pratt, L.A.; Silhavy, T.J. The response regulator SprE controls the stability of RpoS. Proc. Natl. Acad. Sci. USA 1996, 93, 2488–2492. [Google Scholar] [CrossRef] [Green Version]
- Klauck, E.; Lingnau, M.; Hengge-Aronis, R. Role of the response regulator RssB in sigma recognition and initiation of sigma proteolysis in Escherichia coli. Mol. Microbiol. 2001, 40, 1381–1390. [Google Scholar] [CrossRef]
- Zhou, Y.; Gottesman, S.; Hoskins, J.R.; Maurizi, M.R.; Wickner, S. The RssB response regulator directly targets sigma(S) for degradation by ClpXP. Genes Dev. 2001, 15, 627–637. [Google Scholar] [CrossRef] [Green Version]
- Bougdour, A.; Wickner, S.; Gottesman, S. Modulating RssB activity: IraP, a novel regulator of sigma(S) stability in Escherichia coli. Genes Dev. 2006, 20, 884–897. [Google Scholar] [CrossRef] [Green Version]
- Merrikh, H.; Ferrazzoli, A.E.; Bougdour, A.; Olivier-Mason, A.; Lovett, S.T. A DNA damage response in Escherichia coli involving the alternative sigma factor, RpoS. Proc. Natl. Acad. Sci. USA 2009, 106, 611–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battesti, A.; Hoskins, J.R.; Tong, S.; Milanesio, P.; Mann, J.M.; Kravats, A.; Tsegaye, Y.M.; Bougdour, A.; Wickner, S.; Gottesman, S. Anti-adaptors provide multiple modes for regulation of the RssB adaptor protein. Genes Dev. 2013, 27, 2722–2735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorich, V.; Brugger, C.; Tripathi, A.; Hoskins, J.R.; Tong, S.; Suhanovsky, M.M.; Sastry, A.; Wickner, S.; Gottesman, S.; Deaconescu, A.M. Structural basis for inhibition of a response regulator of sigma(S) stability by a ClpXP antiadaptor. Genes Dev. 2019, 33, 718–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micevski, D.; Zammit, J.E.; Truscott, K.N.; Dougan, D.A. Anti-adaptors use distinct modes of binding to inhibit the RssB-dependent turnover of RpoS (sigma(S)) by ClpXP. Front. Mol. Biosci. 2015, 2, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, C.N.; Ruiz, N.; Silhavy, T.J. RpoS proteolysis is regulated by a mechanism that does not require the SprE (RssB) response regulator phosphorylation site. J. Bacteriol. 2004, 186, 7403–7410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, L.; Baumann, U.; Reymond, J.L. An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 2004, 32, e115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catanzariti, A.M.; Soboleva, T.A.; Jans, D.A.; Board, P.G.; Baker, R.T. An efficient system for high-level expression and easy purification of authentic recombinant proteins. Protein Sci. 2004, 13, 1331–1339. [Google Scholar] [CrossRef]
- Bouche, S.; Klauck, E.; Fischer, D.; Lucassen, M.; Jung, K.; Hengge-Aronis, R. Regulation of RssB-dependent proteolysis in Escherichia coli: A role for acetyl phosphate in a response regulator-controlled process. Mol. Microbiol. 1998, 27, 787–795. [Google Scholar] [CrossRef]
- Rudiger, S.; Germeroth, L.; Schneider-Mergener, J.; Bukau, B. Substrate specificity of the DnaK chaperone determined by screening cellulose-bound peptide libraries. EMBO J. 1997, 16, 1501–1507. [Google Scholar] [CrossRef] [Green Version]
- Geissler, A.; Chacinska, A.; Truscott, K.N.; Wiedemann, N.; Brandner, K.; Sickmann, A.; Meyer, H.E.; Meisinger, C.; Pfanner, N.; Rehling, P. The mitochondrial presequence translocase: An essential role of Tim50 in directing preproteins to the import channel. Cell 2002, 111, 507–518. [Google Scholar] [CrossRef] [Green Version]
- Harlow, E.; Lane, D. Using Antibodies: A Laboratory Manual, 2nd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1999. [Google Scholar]
- Erbse, A.H.; Wagner, J.N.; Truscott, K.N.; Spall, S.K.; Kirstein, J.; Zeth, K.; Turgay, K.; Mogk, A.; Bukau, B.; Dougan, D.A. Conserved residues in the N-domain of the AAA+ chaperone ClpA regulate substrate recognition and unfolding. FEBS J. 2008, 275, 1400–1410. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. Integration, scaling, space-group assignment and post-refinement. Acta Cryst. D Biol. Cryst. 2010, 66, 133–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Experimental phasing with SHELXC/D/E: Combining chain tracing with density modification. Acta Cryst. D Biol. Cryst. 2010, 66, 479–485. [Google Scholar] [CrossRef] [Green Version]
- Vonrhein, C.; Blanc, E.; Roversi, P.; Bricogne, G. Automated structure solution with autoSHARP. Methods Mol. Biol. 2007, 364, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Cowtan, K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Cryst. D Biol. Cryst. 2006, 62, 1002–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langer, G.; Cohen, S.X.; Lamzin, V.S.; Perrakis, A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 2008, 3, 1171–1179. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Cryst. D Biol. Cryst. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zwart, P.H.; Afonine, P.V.; Grosse-Kunstleve, R.W.; Hung, L.W.; Ioerger, T.R.; McCoy, A.J.; McKee, E.; Moriarty, N.W.; Read, R.J.; Sacchettini, J.C.; et al. Automated structure solution with the PHENIX suite. Methods Mol. Biol. 2008, 426, 419–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Gunn, N.J.; Gorman, M.A.; Dobson, R.C.; Parker, M.W.; Mulhern, T.D. Purification, crystallization, small-angle X-ray scattering and preliminary X-ray diffraction analysis of the SH2 domain of the Csk-homologous kinase. Acta Cryst. Sect. F Struct. Biol. Cryst. Commun. 2011, 67, 336–339. [Google Scholar] [CrossRef] [Green Version]
- Petoukhov, M.V.; Franke, D.; Shkumatov, A.V.; Tria, G.; Kikhney, A.G.; Gajda, M.; Gorba, C.; Mertens, H.D.; Konarev, P.V.; Svergun, D.I. New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Cryst. 2012, 45, 342–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volkov, V.V.; Svergun, D.I. Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Cryst. 2003, 36, 860–864. [Google Scholar] [CrossRef] [Green Version]
- Petoukhov, M.V.; Svergun, D.I. Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys. J. 2005, 89, 1237–1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thibault, G.; Tsitrin, Y.; Davidson, T.; Gribun, A.; Houry, W.A. Large nucleotide-dependent movement of the N-terminal domain of the ClpX chaperone. EMBO J. 2006, 25, 3367–3376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, E.Y.; Lee, B.G.; Hong, S.B.; Kim, H.W.; Jeon, H.; Song, H.K. Structural basis of SspB-tail recognition by the zinc binding domain of ClpX. J. Mol. Biol. 2007, 367, 514–526. [Google Scholar] [CrossRef] [PubMed]
- Teh, A.H.; Makino, M.; Hoshino, T.; Baba, S.; Shimizu, N.; Yamamoto, M.; Kumasaka, T. Structure of the RsbX phosphatase involved in the general stress response of Bacillus subtilis. Acta Cryst. D Biol. Cryst. 2015, 71, 1392–1399. [Google Scholar] [CrossRef]
- Studemann, A.; Noirclerc-Savoye, M.; Klauck, E.; Becker, G.; Schneider, D.; Hengge, R. Sequential recognition of two distinct sites in sigma(S) by the proteolytic targeting factor RssB and ClpX. Embo J. 2003, 22, 4111–4120. [Google Scholar] [CrossRef] [Green Version]
- Gao, R.; Bouillet, S.; Stock, A.M. Structural Basis of Response Regulator Function. Annu. Rev. Microbiol. 2019, 73, 175–197. [Google Scholar] [CrossRef] [PubMed]
- Dougan, D.A.; Reid, B.G.; Horwich, A.L.; Bukau, B. ClpS, a substrate modulator of the ClpAP machine. Mol. Cell 2002, 9, 673–683. [Google Scholar] [CrossRef]
- Kirstein, J.; Moliere, N.; Dougan, D.A.; Turgay, K. Adapting the machine: Adaptor proteins for Hsp100/Clp and AAA+ proteases. Nat. Rev. Microbiol. 2009, 7, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Ninnis, R.L.; Spall, S.K.; Talbo, G.H.; Truscott, K.N.; Dougan, D.A. Modification of PATase by L/F-transferase generates a ClpS-dependent N-end rule substrate in Escherichia coli. EMBO J. 2009, 28, 1732–1744. [Google Scholar] [CrossRef] [PubMed]
- Schlothauer, T.; Mogk, A.; Dougan, D.A.; Bukau, B.; Turgay, K. MecA, an adaptor protein necessary for ClpC chaperone activity. Proc. Natl. Acad. Sci. USA 2003, 100, 2306–2311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuhrmann, J.; Schmidt, A.; Spiess, S.; Lehner, A.; Turgay, K.; Mechtler, K.; Charpentier, E.; Clausen, T. McsB is a protein arginine kinase that phosphorylates and inhibits the heat-shock regulator CtsR. Science 2009, 324, 1323–1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeth, K.; Ravelli, R.B.; Paal, K.; Cusack, S.; Bukau, B.; Dougan, D.A. Structural analysis of the adaptor protein ClpS in complex with the N-terminal domain of ClpA. Nat. Struct. Biol. 2002, 9, 906–911. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.H.; Roman-Hernandez, G.; Grant, R.A.; Sauer, R.T.; Baker, T.A. The molecular basis of N-end rule recognition. Mol. Cell 2008, 32, 406–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuenemann, V.J.; Kralik, S.M.; Albrecht, R.; Spall, S.K.; Truscott, K.N.; Dougan, D.A.; Zeth, K. Structural basis of N-end rule substrate recognition in Escherichia coli by the ClpAP adaptor protein ClpS. EMBO Rep. 2009, 10, 508–514. [Google Scholar] [CrossRef] [Green Version]
- Rivera-Rivera, I.; Roman-Hernandez, G.; Sauer, R.T.; Baker, T.A. Remodeling of a delivery complex allows ClpS-mediated degradation of N-degron substrates. Proc. Natl. Acad. Sci. USA 2014, 111, E3853–E3859. [Google Scholar] [CrossRef] [Green Version]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Micevski, D.; Zeth, K.; Mulhern, T.D.; Schuenemann, V.J.; Zammit, J.E.; Truscott, K.N.; Dougan, D.A. Insight into the RssB-Mediated Recognition and Delivery of σs to the AAA+ Protease, ClpXP. Biomolecules 2020, 10, 615. https://doi.org/10.3390/biom10040615
Micevski D, Zeth K, Mulhern TD, Schuenemann VJ, Zammit JE, Truscott KN, Dougan DA. Insight into the RssB-Mediated Recognition and Delivery of σs to the AAA+ Protease, ClpXP. Biomolecules. 2020; 10(4):615. https://doi.org/10.3390/biom10040615
Chicago/Turabian StyleMicevski, Dimce, Kornelius Zeth, Terrence D. Mulhern, Verena J. Schuenemann, Jessica E. Zammit, Kaye N. Truscott, and David A. Dougan. 2020. "Insight into the RssB-Mediated Recognition and Delivery of σs to the AAA+ Protease, ClpXP" Biomolecules 10, no. 4: 615. https://doi.org/10.3390/biom10040615