Calcium Export from Neurons and Multi-Kinase Signaling Cascades Contribute to Ouabain Neuroprotection in Hyperhomocysteinemia

 ,
, 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Material and Animals
2.2. Primary Cortical Culture
2.3. Quantitation of Cell Viability
2.4. Western Blot Analysis
2.5. Calcium Imaging
2.6. Mitochondrial Membrane Potential Imaging
2.7. Analysis and Statistics
3. Results
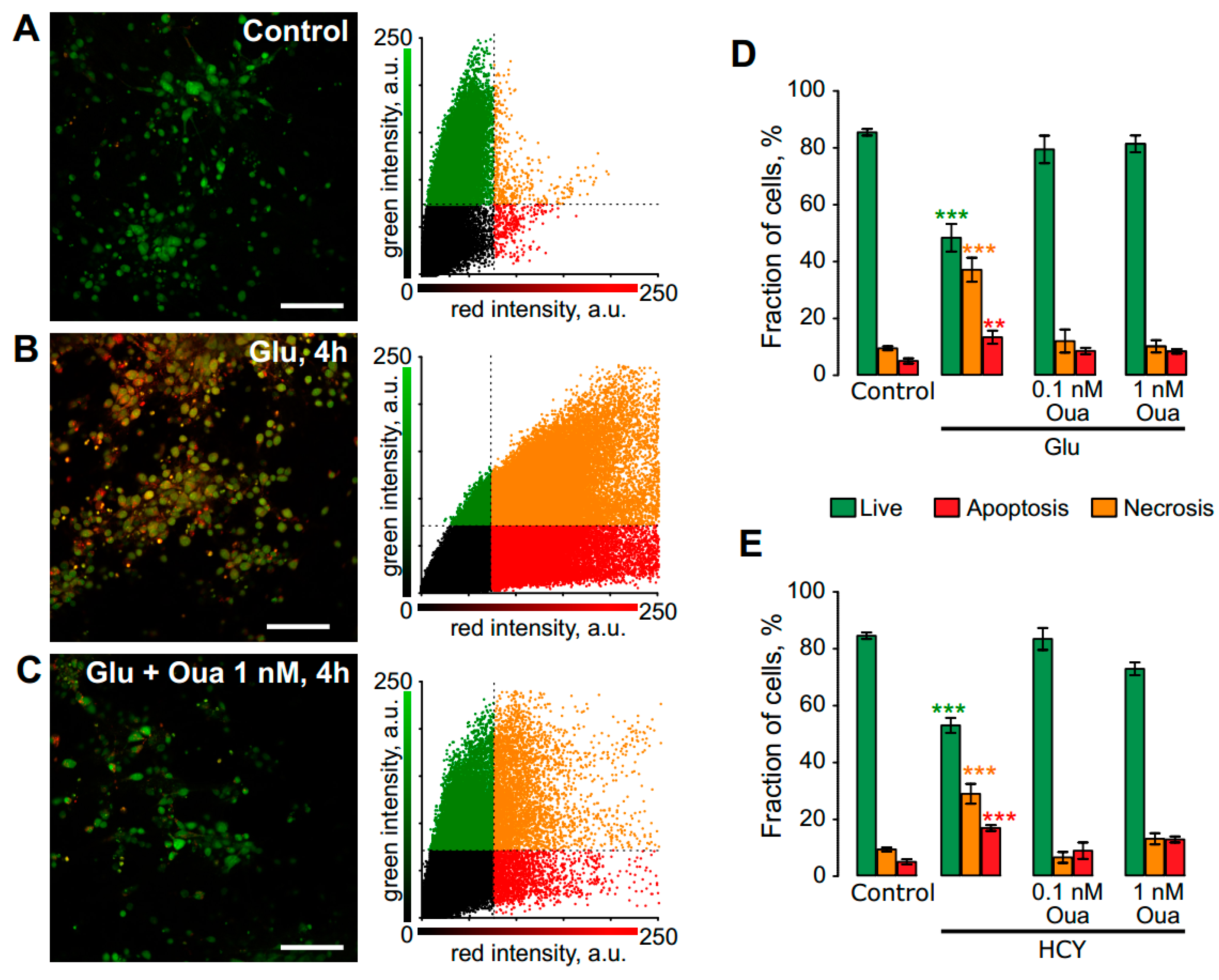
3.1. Ouabain Enhances the Viability of Neurons in Short Excitotoxic Stress
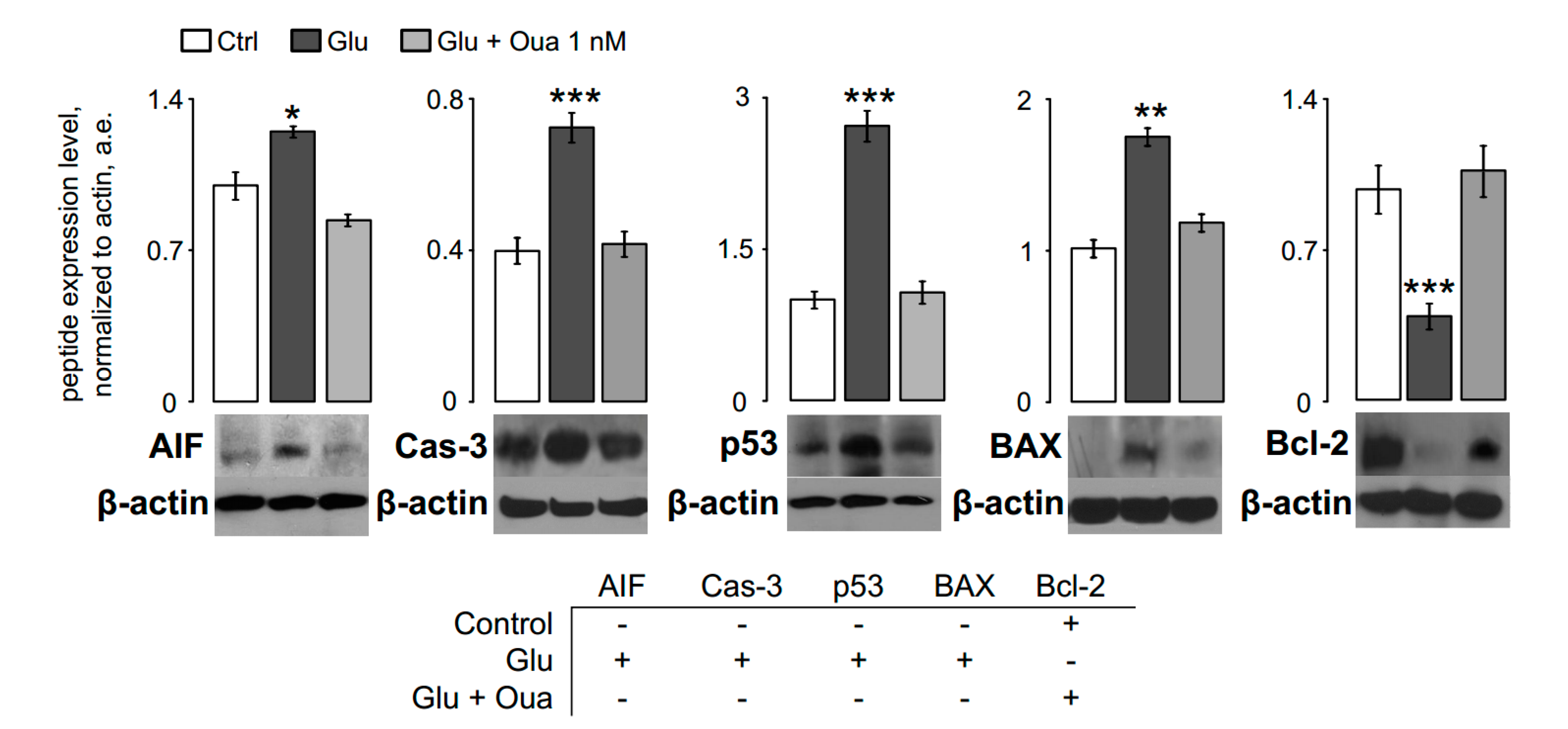
3.2. Ouabain Prevents Pro-Apoptotic Proteins Expression in Short Excitotoxic Insults
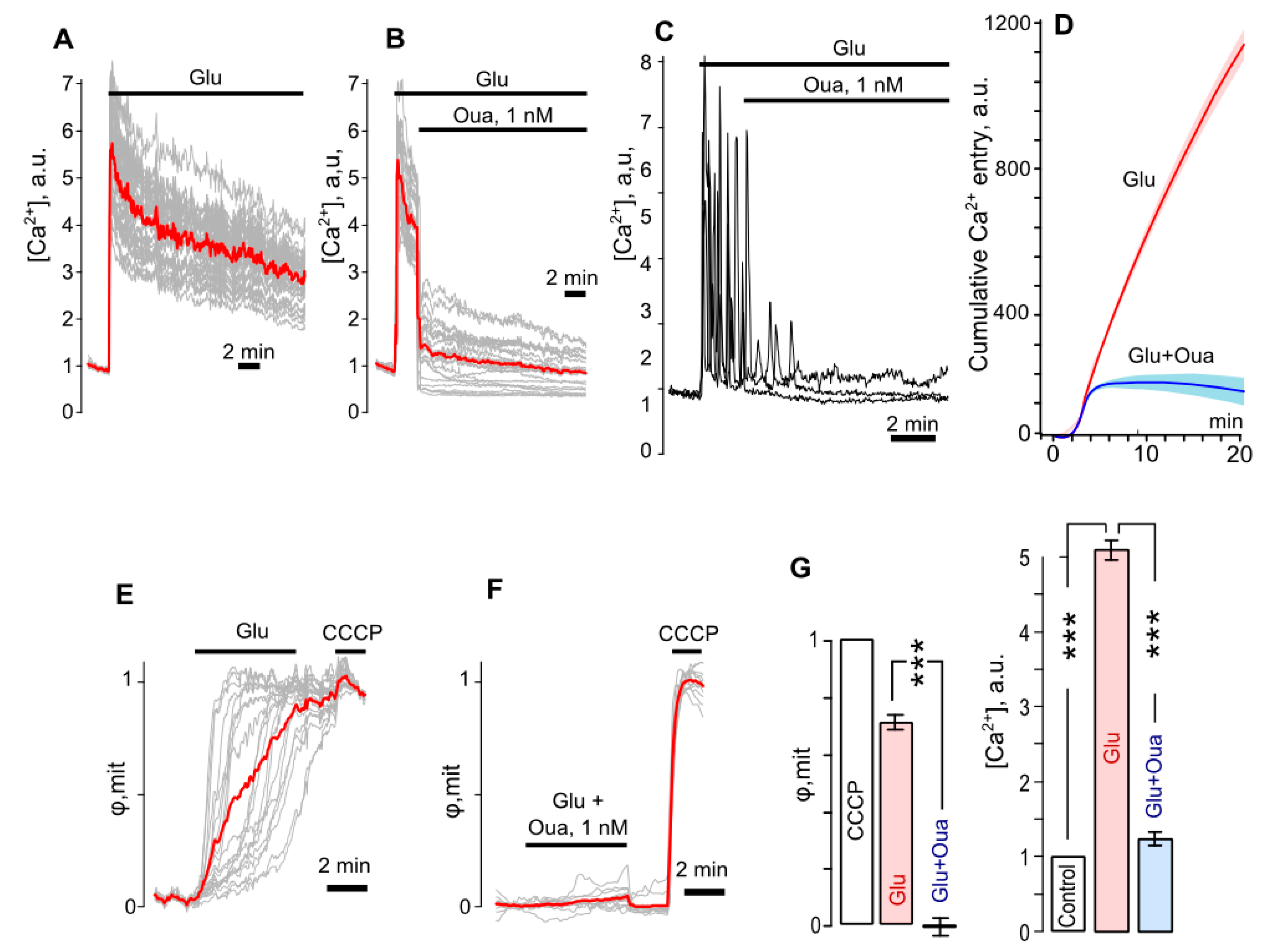
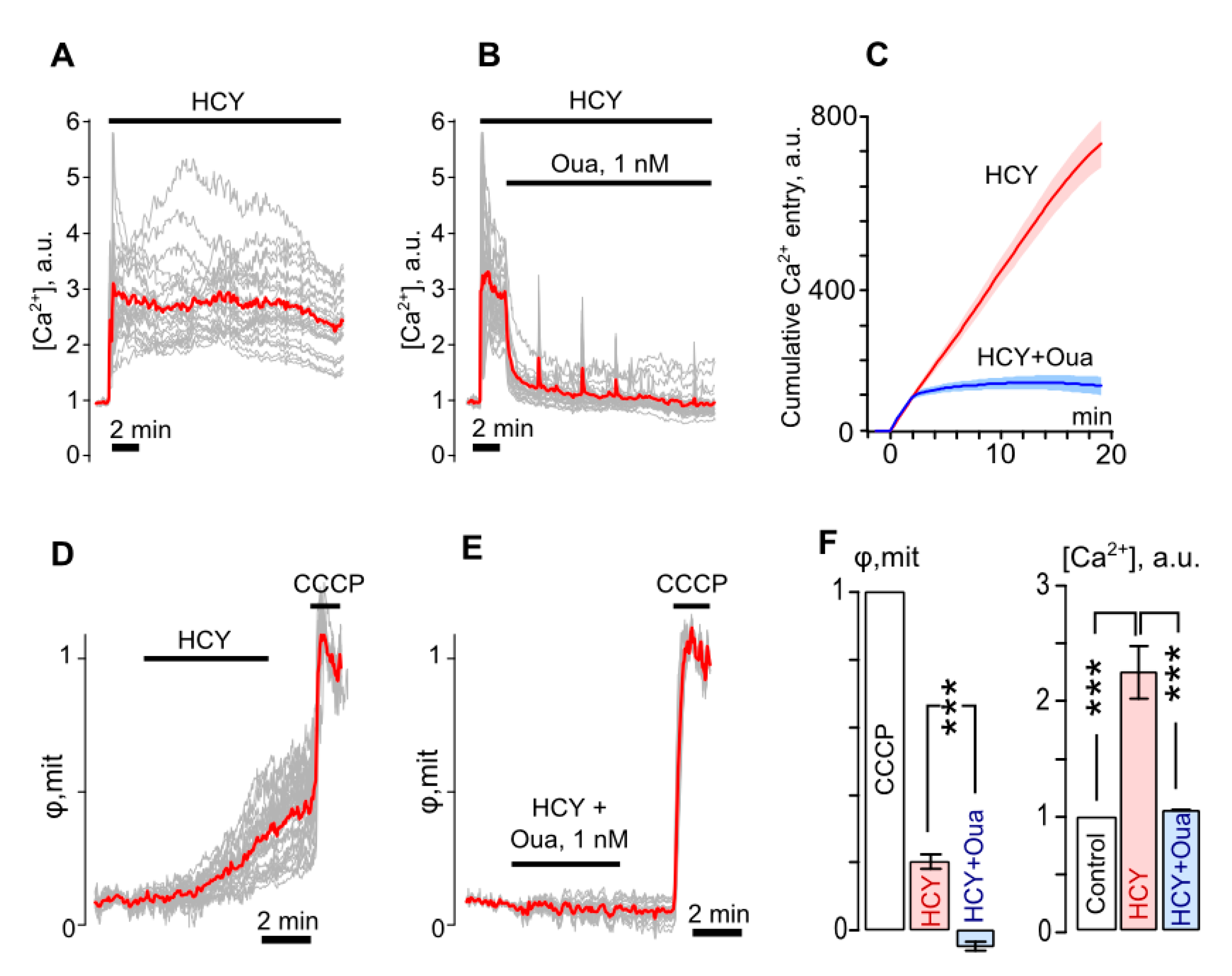
3.3. Ouabain Suppresses Intracellular Ca2+ Overload and Mitochondrial Dysfunction of Neurons
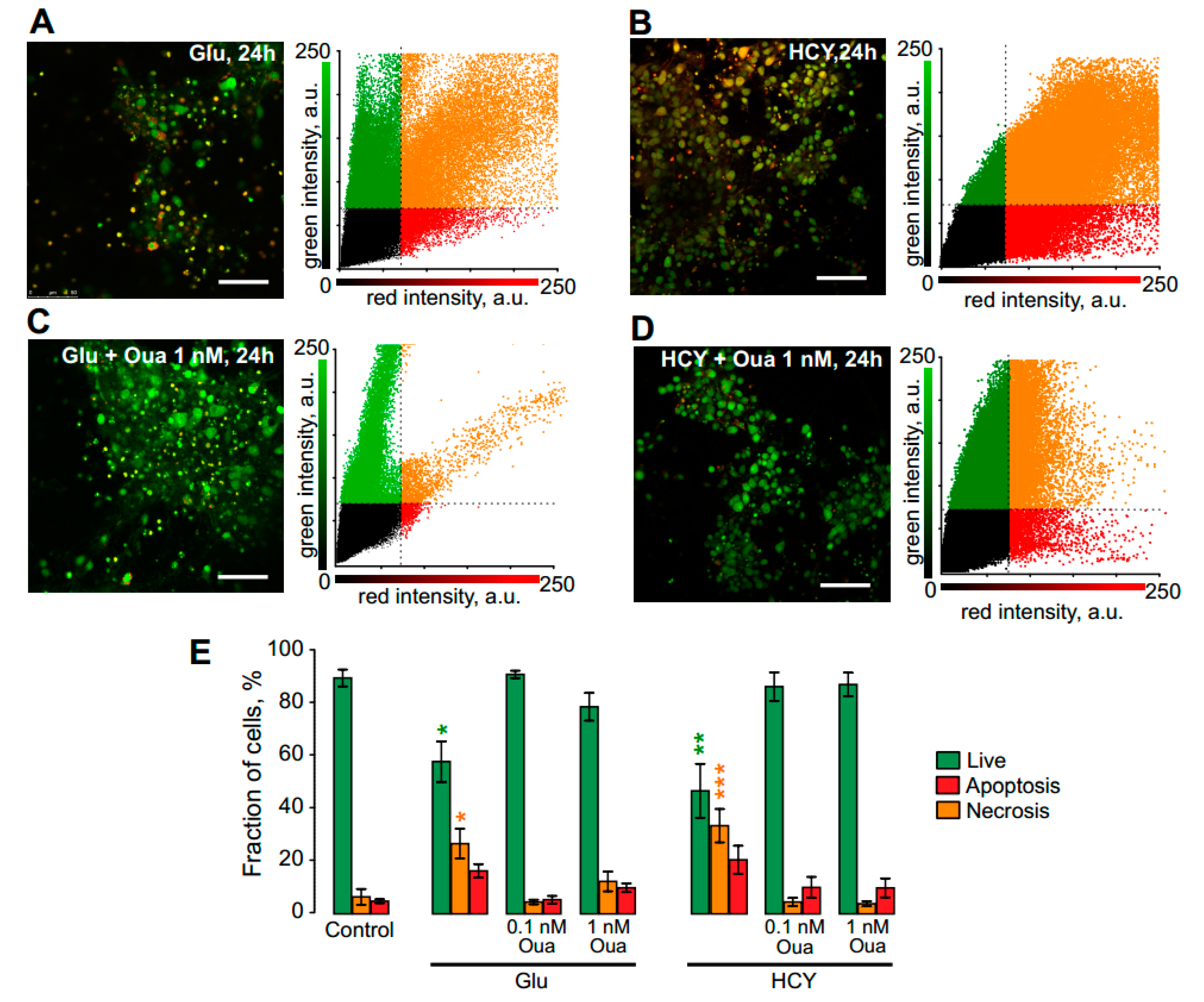
3.4. Ouabain Enhances the Viability of Neurons in Long Excitotoxic Stress Triggering Multi-Kinase Signaling Cascades
4. Discussion
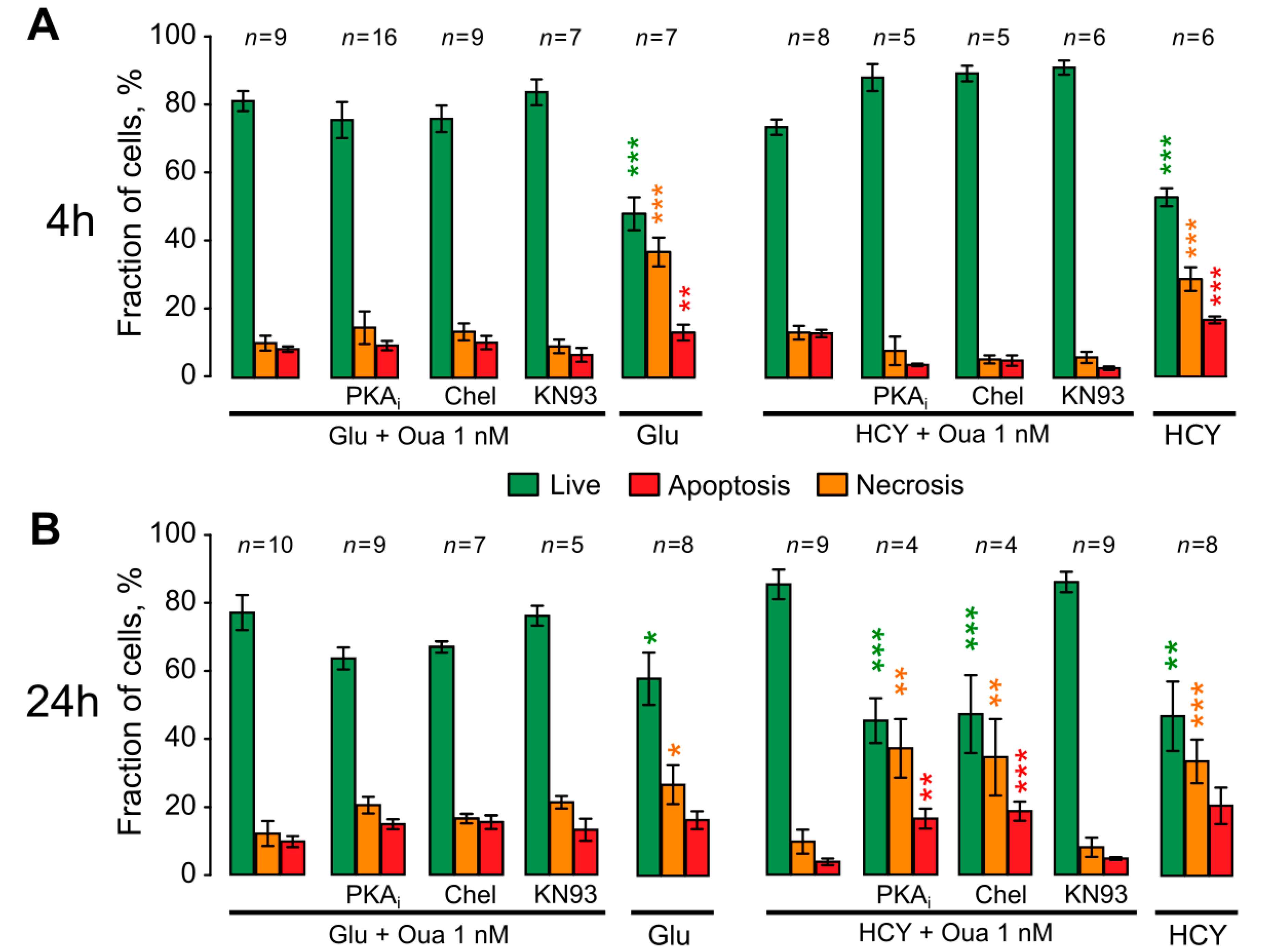
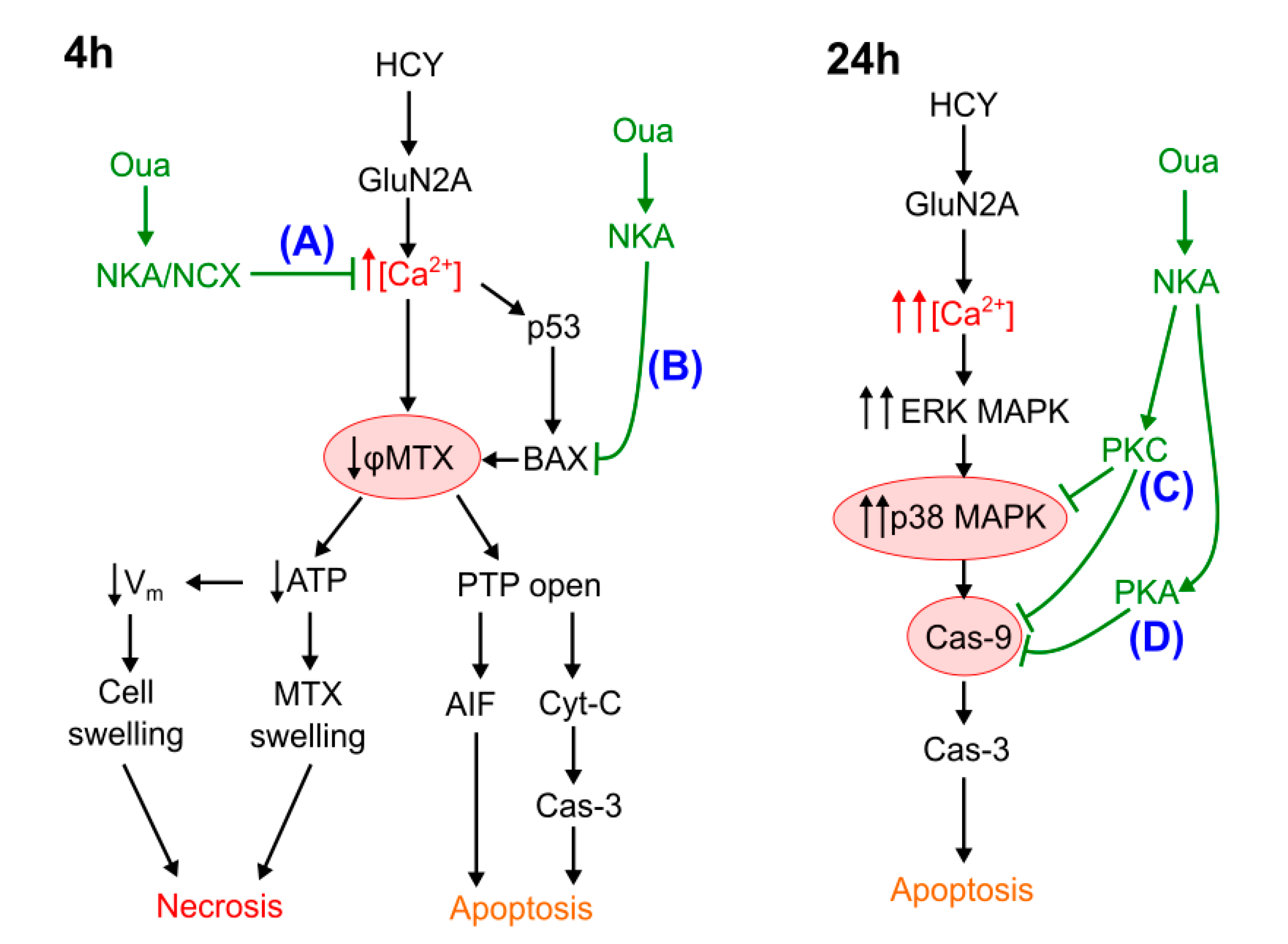
4.1. Necrosis at Short-Term Excitotoxic Stress
4.2. Apoptosis at Short-Term Excitotoxic Stress
4.3. Apoptosis at Long-Term Excitotoxic Stress
4.4. The Role of PKC and PKA in Ouabain Effects against Hyperhomocysteinemia
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Refsum, H.; Ueland, P.M.; Nygard, O.; Vollset, S.E. Homocysteine and cardiovascular disease. Annu. Rev. Med. 1998, 49, 31–62. [Google Scholar] [CrossRef]
- Sacco, R.L.; Roberts, J.K.; Jacobs, B.S. Homocysteine as a risk factor for ischemic stroke: An epidemiological story in evolution. Neuroepidemiology 1998, 17, 167–173. [Google Scholar] [CrossRef]
- Huang, G.; Dragan, M.; Freeman, D.; Wilson, J.X. Activation of catechol-O-methyltransferase in astrocytes stimulates homocysteine synthesis and export to neurons. Glia 2005, 51, 47–55. [Google Scholar] [CrossRef]
- Hu, X.W.; Qin, S.M.; Li, D.; Hu, L.F.; Liu, C.F. Elevated homocysteine levels in levodopa-treated idiopathic Parkinson’s disease: A meta-analysis. Acta Neurol. Scand. 2013, 128, 73e82. [Google Scholar] [CrossRef]
- Miller, J.W. Homocysteine, Alzheimer’s disease and cognitive function. Nutrition 2000, 16, 675–677. [Google Scholar] [CrossRef]
- Surtees, R.; Leung, D.Y.M.; Bowron, A.; Leonard, J. Cerebrospinal fluid and plasma total homocysteine and related metabolites in children with cystathionine b-synthase deficiency: The effect of treatment. Pediatr. Res. 1997, 42, 577–582. [Google Scholar] [CrossRef] [Green Version]
- Lipton, S.A.; Kim, W.K.; Choi, Y.B.; Kumar, S.; D’Emilia, D.M.; Rayudu, P.V.; Arnelle, D.R.; Stamler, J.S. Neurotoxicity associated with dual actions of homocysteine at the N-methyl-d-aspartate receptor. Proc. Natl. Acad. Sci. USA 1997, 94, 5923–5928. [Google Scholar] [CrossRef] [Green Version]
- Poddar, R.; Paul, S. Novel crosstalk between ERK MAPK and p38 MAPK leads to homocysteine-NMDA receptor-mediated neuronal cell death. J. Neurochem. 2013, 124, 558–570. [Google Scholar] [CrossRef] [Green Version]
- Tan, B.; Venketasubramanian, N.; Vrooman, H.; Cheng, C.Y.; Wong, T.Y.; Ikram, M.K.; Chen, C.; Hilal, S. Homocysteine and Cerebral Atrophy: The Epidemiology of Dementia in Singapore Study. J. Alzheimers Dis. 2018, 62, 877–885. [Google Scholar] [CrossRef]
- Abushik, P.A.; Niittykoski, M.; Giniatullina, R.; Shakirzyanova, A.; Bart, G.; Fayuk, D.; Sibarov, D.A.; Antonov, S.M.; Giniatullin, R. The role of NMDA and mGluR5 receptors in calcium mobilization and neurotoxicity of homocysteine in trigeminal and cortical neurons and glial cells. J. Neurochem. 2014, 129, 264–274. [Google Scholar] [CrossRef]
- Bolton, A.D.; Phillips, M.A.; Constantine-Paton, M. Homocysteine reduces NMDAR desensitization and differentially modulates peak amplitude of NMDAR currents, depending on GluN2 subunit composition. J. Neurophysiol. 2013, 110, 1567–1582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sibarov, D.A.; Abushik, P.A.; Giniatullin, R.; Antonov, S.M. GluN2A subunit-containing NMDA receptors are the preferential neuronal targets of homocysteine. Front. Cell. Neurosci. 2016, 10, 246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sibarov, D.A.; Giniatullin, R.; Antonov, S.M. High sensitivity of cerebellar neurons to homocysteine is determined by expression of GluN2C and GluN2D subunits of NMDA receptors. Biochem. Biophys. Res. Commun. 2018, 506, 648–652. [Google Scholar] [CrossRef]
- Rajagopal, S.; Fitzgerald, A.A.; Deep, S.N.; Paul, S.; Poddar, R. Role of GluN2A NMDA receptor in homocysteine-induced prostaglandin E2 release from neurons. J. Neurochem. 2019, 150, 44–55. [Google Scholar] [CrossRef]
- Jindal, A.; Rajagopal, S.; Winter, L.; Miller, J.W.; Jacobsen, D.W.; Brigman, J.; Allan, A.M.; Paul, S.; Poddar, R. Hyperhomocysteinemia leads to exacerbation of ischemic brain damage: Role of GluN2A NMDA receptors. Neurobiol. Dis. 2019, 127, 287–302. [Google Scholar] [CrossRef]
- Deep, S.N.; Mitra, S.; Rajagopal, S.; Paul, S.; Poddar, R. GluN2A-NMDA receptor-mediated sustained Ca2+ influx leads to homocysteine-induced neuronal cell death. J. Biol. Chem. 2019, 294, 11154–11165. [Google Scholar] [CrossRef]
- Kim, M.J.; Dunah, A.W.; Wang, Y.T.; Sheng, M. Differential roles of NR2A- and NR2B-containing NMDA receptors in Ras-ERK signaling and AMPA receptor trafficking. Neuron 2005, 46, 745–760. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wong, T.P.; Aarts, M.; Rooyakkers, A.; Liu, L.; Lai, T.W.; Wu, D.C.; Lu, J.; Tymianski, M.; Craig, A.M.; et al. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J. Neurosci. 2007, 27, 2846–2857. [Google Scholar] [CrossRef] [Green Version]
- Hardingham, G.E.; Fukunaga, Y.; Bading, H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci. 2002, 5, 405–414. [Google Scholar] [CrossRef]
- Yakovlev, A.V.; Kurmashova, E.; Zakharov, A.; Sitdikova, G.F. Network-driven activity and neuronal excitability in hippocampus of neonatal rats with prenatal hyperhomocysteinemia. Bionanoscience 2018, 8, 304–309. [Google Scholar] [CrossRef]
- Montecinos-Oliva, C.; Arrázola, M.S.; Jara, C.; Tapia-Rojas, C.; Inestrosa, N.C. Hormetic-like effects of l-homocysteine on synaptic structure, function, and Aβ aggregation. Pharmaceuticals 2020, 13, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sibarov, D.A.; Bolshakov, A.E.; Abushik, P.A.; Krivoi, I.I.; Antonov, S.M. Na+, K+-ATPase functionally interacts with the plasma membrane Na+, Ca2+ exchanger to prevent Ca2+ overload and neuronal apoptosis in excitotoxic stress. J. Pharmacol. Exp. Ther. 2012, 343, 596–607. [Google Scholar] [CrossRef] [PubMed]
- Abushik, P.A.; Sibarov, D.A.; Eaton, M.J.; Skatchkov, S.N.; Antonov, S.M. Kainate-induced calcium overload of cortical neurons in vitro: Dependence on expression of AMPAR GluA2-subunit and down-regulation by subnanomolar ouabain. Cell Calcium 2013, 54, 95–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanova, M.A.; Sitnikova, L.S.; Stepanenko, Y.D.; Karelina, T.V.; Sibarov, D.A.; Abushik, P.A.; Antonov, S.M. Forskolin as a neuroprotector and modulator of glutamate-evoked Ca2+ entry into cerebellar neurons. Biochem. Suppl. Ser. A Membr. Cell Biol. 2018, 12, 390–393. [Google Scholar] [CrossRef]
- Pierre, S.V.; Xie, Z. The Na,K-ATPase receptor complex: Its organization and membership. Cell Biochem. Biophys. 2006, 46, 303–316. [Google Scholar] [CrossRef]
- Aperia, A. New roles for an old enzyme: Na,K-ATPase emerges as an interesting drug target. J. Intern. Med. 2007, 261, 44–52. [Google Scholar] [CrossRef]
- Lingrel, J.B. The physiological significance of the cardiotonic steroid/ ouabain-binding site of the Na, K-ATPase. Annu. Rev. Physiol. 2010, 72, 395–412. [Google Scholar] [CrossRef] [Green Version]
- Reinhard, L.; Tidow, H.; Clausen, M.J.; Nissen, P. Na,K-ATPase as a docking station: Protein-protein complexes of the Na,K-ATPase. Cell. Mol. Life Sci. 2013, 70, 205–222. [Google Scholar] [CrossRef]
- Aperia, A.; Akkuratov, E.E.; Fontana, J.M.; Brismar, H. Na+-K+-ATPase, a new class of plasma membrane receptors. Am. J. Physiol. Cell Physiol. 2016, 310, C491–C495. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Wymore, R.S.; Wang, Y.; Gaudette, G.R.; Krukenkamp, I.B.; Cohen, I.S.; Mathias, R.T. Isoform-specific stimulation of cardiac Na/K pumps by nanomolar concentrations of glycosides. J. Gen. Physiol. 2002, 119, 297–312. [Google Scholar] [CrossRef]
- Bolshakov, A.E.; Sibarov, D.A.; Abushik, P.A.; Krivoi, I.I.; Antonov, S.M. Dose-dependence of antiapoptotic and toxic action of ouabain in neurons of primary cultures of rat cortex. Biochem. Suppl. Ser. A Membr. Cell Biol. 2012, 6, 294–299. [Google Scholar] [CrossRef]
- Haas, M.; Askari, A.; Xie, Z. Involvement of Src and epidermal growth factor receptor in the signal-transducing function of Na+/K+-ATPase. J. Biol. Chem. 2000, 275, 27832–27837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Malmersjö, S.; Li, J.; Ando, H.; Aizman, O.; Uhlén, P.; Mikoshiba, K.; Aperia, A. Distinct role of the N-terminal tail of the Na,K-ATPase catalytic subunit as a signal transducer. J. Biol. Chem. 2006, 281, 21954–21962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldmann, T.; Shahar, M.; Baba, A.; Matsuda, T.; Lichtstein, D.; Rosen, H. The Na+/Ca2+-exchanger: An essential component in the mechanism governing cardiac steroid-induced slow Ca2+ oscillations. Cell Calcium 2011, 50, 424–432. [Google Scholar] [CrossRef]
- Liu, L.; Zhao, X.; Pierre, S.V.; Askari, A. Association of PI3K-Akt signaling pathway with digitalis-induced hypertrophy of cardiac myocytes. Am. J. Physiol. Cell Physiol. 2007, 293, C1489–C1497. [Google Scholar] [CrossRef]
- Sibarov, D.A.; Abushik, P.A.; Poguzhelskaya, E.E.; Bolshakov, K.V.; Antonov, S.M. Inhibition of plasma membrane Na/Ca-exchanger by KB-R7943 or lithium reveals its role in Ca-dependent N-methyl-d-aspartate receptor inactivation. J. Pharmacol. Exp. Ther. 2015, 355, 484–495. [Google Scholar] [CrossRef] [Green Version]
- Sibarov, D.A.; Poguzhelskaya, E.E.; Antonov, S.M. Downregulation of calcium-dependent NMDA receptor desensitization by sodium-calcium exchangers: A role of membrane cholesterol. BMC Neurosci. 2018, 19, 73. [Google Scholar] [CrossRef]
- Dvela-Levitt, M.; Ami, H.C.B.; Rosen, H.; Shohami, E.; Lichtstein, D. Ouabain improves functional recovery following traumatic brain injury. J. Neurotrauma 2014, 31, 1942–1947. [Google Scholar] [CrossRef] [Green Version]
- Greco, R.; Tassorelli, C.; Sandrini, G.; Di Bella, P.; Buscone, S.; Nappi, G. Role of calcitonin gene-related peptide and substance P in different models of pain. Cephalalgia 2008, 28, 114–126. [Google Scholar]
- Abushik, P.A.; Bart, G.; Korhonen, P.; Leinonen, H.; Giniatullina, R.; Sibarov, D.A.; Levonen, A.L.; Malm, T.; Antonov, S.M.; Giniatullin, R. Pro-nociceptive migraine mediator CGRP provides neuroprotection of sensory, cortical and cerebellar neurons via multi-kinase signaling. Cephalalgia 2017, 37, 1373–1383. [Google Scholar] [CrossRef]
- Mironova, E.V.; Evstratova, A.A.; Antonov, S.M. A fluorescence vital assay for the recognition and quantification of excitotoxic cell death by necrosis and apoptosis using confocal microscopy on neurons in culture. J. Neurosci. Methods 2007, 163, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Han, E.B.; Stevens, C.F. Development regulates a switch between post- and presynaptic strengthening in response to activity deprivation. Proc. Natl. Acad. Sci. USA 2009, 106, 10817–10822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duchen, M.R. Mitochondria, calcium-dependent neuronal death and neurodegenerative disease. Pflügers Arch. 2012, 464, 111–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, J.M.; Cory, S. The Bcl-2 protein family: Arbiters of cell survival. Science 1998, 281, 1322–1326. [Google Scholar] [CrossRef]
- Khodorov, B. Glutamate-induced deregulation of calcium homeostasis and mitochondrial dysfunction in mammalian central neurones. Prog. Biophys. Mol. Biol. 2004, 86, 279–351. [Google Scholar] [CrossRef]
- Sakaguchi, T.; Kuno, M.; Kawasaki, K. Disparity of cell swelling and rapid neuronal death by excitotoxic insults in rat hippocampal slice cultures. Neurosci. Lett. 1999, 274, 135–138. [Google Scholar] [CrossRef]
- Isokawa, M. N-methyl-d-aspartic acid-induced and Ca-dependent neuronal swelling and its retardation by brain-derived neurotrophic factor in the epileptic hippocampus. Neuroscience 2005, 131, 801–812. [Google Scholar] [CrossRef]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef]
- Choo, A.M.; Geddes-Klein, D.M.; Hockenberry, A.; Scarsella, D.; Mesfin, M.N.; Singh, P.; Patel, T.P.; Meaney, D.F. NR2A and NR2B subunits differentially mediate MAP kinase signaling and mitochondrial morphology following excitotoxic insult. Neurochem. Int. 2012, 60, 506–516. [Google Scholar] [CrossRef] [Green Version]
- Blaustein, M.P.; Juhaszova, M.; Golovina, V.A.; Church, P.J.; Stanley, E.F. Na/Ca exchanger and PMCA localization in neurons and astrocytes: Functional implications. Ann. N. Y. Acad. Sci. 2002, 976, 356–366. [Google Scholar] [CrossRef]
- Ghavami, S.; Hashemi, M.; Ande, S.R.; Yeganeh, B.; Xiao, W.; Eshraghi, M.; Bus, C.J.; Kadkhoda, K.; Wiechec, E.; Halayko, A.J.; et al. Apoptosis and cancer: Mutations within caspase genes. J. Med. Genet. 2009, 46, 497–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chipuk, J.E.; Kuwana, T.; Bouchier-Hayes, L.; Droin, N.M.; Newmeyer, D.D.; Schuler, M.; Green, D.R. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004, 303, 1010–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Orsi, B.; Kilbride, S.M.; Chen, G.; Perez Alvarez, S.; Bonner, H.P.; Pfeiffer, S.; Plesnila, N.; Engel, T.; Henshall, D.C.; Düssmann, H.; et al. Bax regulates neuronal Ca2+ homeostasis. J. Neurosci. 2015, 35, 1706–1722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Zheng, M.; Li, Z.; Li, R.; Jia, L.; Xiong, X.; Southall, N.; Wang, S.; Xia, M.; Austin, C.P.; et al. Cardiac glycosides inhibit p53 synthesis by a mechanism relieved by Src or MAPK inhibition. Cancer Res. 2009, 69, 6556–6564. [Google Scholar] [CrossRef] [Green Version]
- Burlaka, I.; Liu, X.L.; Rebetz, J.; Arvidsson, I.; Yang, L.; Brismar, H.; Karpman, D.; Aperia, A. Ouabain protects against Shiga toxin-triggered apoptosis by reversing the imbalance between Bax and Bcl-xL. J. Am. Soc. Nephrol. 2013, 24, 1413–1423. [Google Scholar] [CrossRef] [Green Version]
- Lauf, P.K.; Alqahtani, T.; Flues, K.; Meller, J.; Adragna, N.C. Interaction between Na-K-ATPase and Bcl-2 proteins BclXL and Bak. Am. J. Physiol. Cell Physiol. 2015, 308, C51–C60. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.M.; Tang, Q.; Wang, J.Q. Protein kinase C-regulated cAMP response element-binding protein phosphorylation in cultured rat striatal neurons. Brain Res. Bull. 2007, 72, 302–308. [Google Scholar] [CrossRef] [Green Version]
- Ivanova, M.A.; Sitnikova, L.S.; Karelina, T.V.; Sibarov, D.A.; Abushik, P.A.; Antonov, S.M. The Neuroprotective effect of forskolin and its influence of mitochondrial dysfunction in neurons in primary rat cerebellum cultures. Neurosci. Behav. Physiol. 2019, 49, 1096–1099. [Google Scholar] [CrossRef]
- Poddar, R.; Paul, S. Homocysteine-NMDA receptor-mediated activation of extracellular signal-regulated kinase leads to neuronal cell death. J. Neurochem. 2009, 110, 1095–1106. [Google Scholar] [CrossRef] [Green Version]
- Poddar, R.; Chen, A.; Winter, L.; Rajagopal, S.; Paul, S. Role of AMPA receptors in homocysteine-NMDA receptor-induced crosstalk between ERK and p38 MAPK. J. Neurochem. 2017, 142, 560–573. [Google Scholar] [CrossRef] [Green Version]
- Allan, L.A.; Clarke, P.R. Apoptosis and autophagy: Regulation of caspase-9 by phosphorylation. FEBS J. 2009, 276, 6063–6073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legos, J.J.; McLaughlin, B.; Skaper, S.D.; Strijbos, P.J.; Parsons, A.A.; Aizenman, E.; Herin, G.A.; Barone, F.C.; Erhardt, J.A. The selective p38 inhibitor SB-239063 protects primary neurons from mild to moderate excitotoxic injury. Eur. J. Pharmacol. 2002, 447, 37–42. [Google Scholar] [CrossRef]
- Wang, X.; Cui, L.; Joseph, J.; Jiang, B.; Pimental, D.; Handy, D.E.; Liao, R.; Loscalzo, J. Homocysteine induces cardiomyocyte dysfunction and apoptosis through p38 MAPK-mediated increase in oxidant stress. J. Mol. Cell. Cardiol. 2012, 52, 753–760. [Google Scholar] [CrossRef] [Green Version]
- Woolfrey, K.M.; Dell’Acqua, M.L. Coordination of protein phosphorylation and dephosphorylation in synaptic plasticity. J. Biol. Chem. 2015, 290, 28604–28612. [Google Scholar] [CrossRef] [Green Version]
- Brady, S.C.; Allan, L.A.; Clarke, P.R. Regulation of caspase 9 through phosphorylation by protein kinase C zeta in response to hyperosmotic stress. Mol. Cell. Biol. 2005, 25, 10543–10555. [Google Scholar] [CrossRef] [Green Version]
- Serrano, B.P.; Hardy, J.A. Phosphorylation by protein kinase A disassembles the caspase-9 core. Cell Death Differ. 2018, 25, 1025–1039. [Google Scholar] [CrossRef]
- Maher, P. How protein kinase C activation protects nerve cells from oxidative stress-induced cell death. J. Neurosci. 2001, 21, 2929–2938. [Google Scholar] [CrossRef]
- Corrêa Gde, R.; Cunha, K.C.; dos Santos, A.A.; de Araujo, E.G. The trophic effect of ouabain on retinal ganglion cell is mediated by EGF receptor and PKC delta activation. Neurochem. Res. 2010, 35, 1343–1352. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ouabain | Live, % | Apoptotic, % | Necrotic, % | ||
|---|---|---|---|---|---|
| Control | n = 8 | 85 ± 1.2 | 9 ± 0.7 | 5 ± 0.9 | |
| +0.1 nM | n = 5 | 85 ± 2.3 | 9 ± 0.5 | 6 ± 1.9 | |
| +1 nM | n = 7 | 84 ± 2.6 | 10 ± 1.1 | 2 ± 1.7 | |
| Glu | n = 7 | 48 ± 4.9 *** | 37 ± 4.2 *** | 14 ± 2.3 ** | |
| +0.1 nM | n = 8 | 79 ± 4.9 | 12 ± 4.1 | 9 ± 1.1 | |
| +1 nM | n = 9 | 81 ± 2.9 | 10 ± 2.2 | 8 ± 0.8 | |
| HCY | n = 6 | 54 ± 2.7 *** | 29 ± 3.5 *** | 17 ± 1 *** | |
| +0.1 nM | n = 7 | 84 ± 3.9 | 7 ± 1.9 | 9 ± 2.9 | |
| +1 nM | n = 8 | 74 ± 2.3 | 13 ± 2 | 13 ± 1.1 |
| Ouabain | Live, % | Apoptotic, % | Necrotic, % | ||
|---|---|---|---|---|---|
| Control | n = 4 | 89 ± 3.2 | 6 ± 3.0 | 5 ± 0.7 | |
| +1 nM | n = 10 | 78 ± 5.2 | 12 ± 3.7 | 10 ± 1.6 | |
| Glu | n = 6 | 57 ± 7.6 * | 27 ± 5.6 * | 16 ± 2.6 | |
| +0.1 nM | n = 8 | 90 ± 1.4 | 5 ± 0.9 | 5 ± 1.5 | |
| +1 nM | n = 10 | 78 ± 5.0 | 11 ± 3.1 | 11 ± 4.0 | |
| HCY | n = 5 | 46 ± 10.1 ** | 33 ± 6.3 *** | 21 ± 5.3 | |
| +0.1 nM | n = 9 | 86 ± 5.4 | 4 ± 1.6 | 10 ± 1.6 | |
| +1 nM | n = 9 | 86 ± 4.4 | 4 ± 0.9 | 10 ± 3.5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ivanova, M.A.; Kokorina, A.D.; Timofeeva, P.D.; Karelina, T.V.; Abushik, P.A.; Stepanenko, J.D.; Sibarov, D.A.; Antonov, S.M. Calcium Export from Neurons and Multi-Kinase Signaling Cascades Contribute to Ouabain Neuroprotection in Hyperhomocysteinemia. Biomolecules 2020, 10, 1104. https://doi.org/10.3390/biom10081104
Ivanova MA, Kokorina AD, Timofeeva PD, Karelina TV, Abushik PA, Stepanenko JD, Sibarov DA, Antonov SM. Calcium Export from Neurons and Multi-Kinase Signaling Cascades Contribute to Ouabain Neuroprotection in Hyperhomocysteinemia. Biomolecules. 2020; 10(8):1104. https://doi.org/10.3390/biom10081104
Chicago/Turabian StyleIvanova, Maria A., Arina D. Kokorina, Polina D. Timofeeva, Tatiana V. Karelina, Polina A. Abushik, Julia D. Stepanenko, Dmitry A. Sibarov, and Sergei M. Antonov. 2020. "Calcium Export from Neurons and Multi-Kinase Signaling Cascades Contribute to Ouabain Neuroprotection in Hyperhomocysteinemia" Biomolecules 10, no. 8: 1104. https://doi.org/10.3390/biom10081104