
Androgen-Induced MIG6 Regulates Phosphorylation of Retinoblastoma Protein and AKT to Counteract Non-Genomic AR Signaling in Prostate Cancer Cells

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatments
2.2. RNA-Sequencing and Transcriptome Analysis
2.3. Cellular Senescence Assays
2.4. Antibodies and Western Blotting
2.5. Immunofluorescence Staining
2.6. Quantitative Reverse Transcription PCR (qRT-PCR)
2.7. ChIP-Seq and ChIP-Seq Analysis
2.8. Statistical Analysis
3. Results
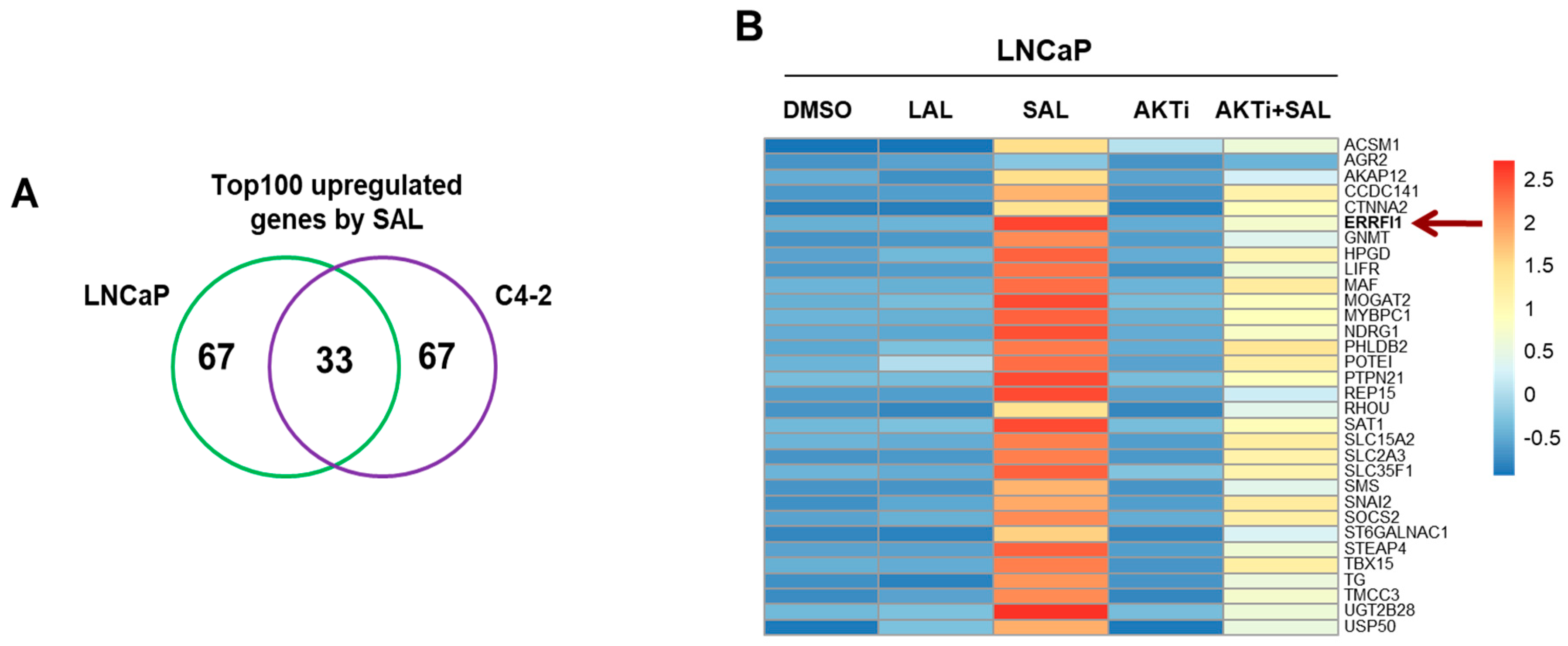
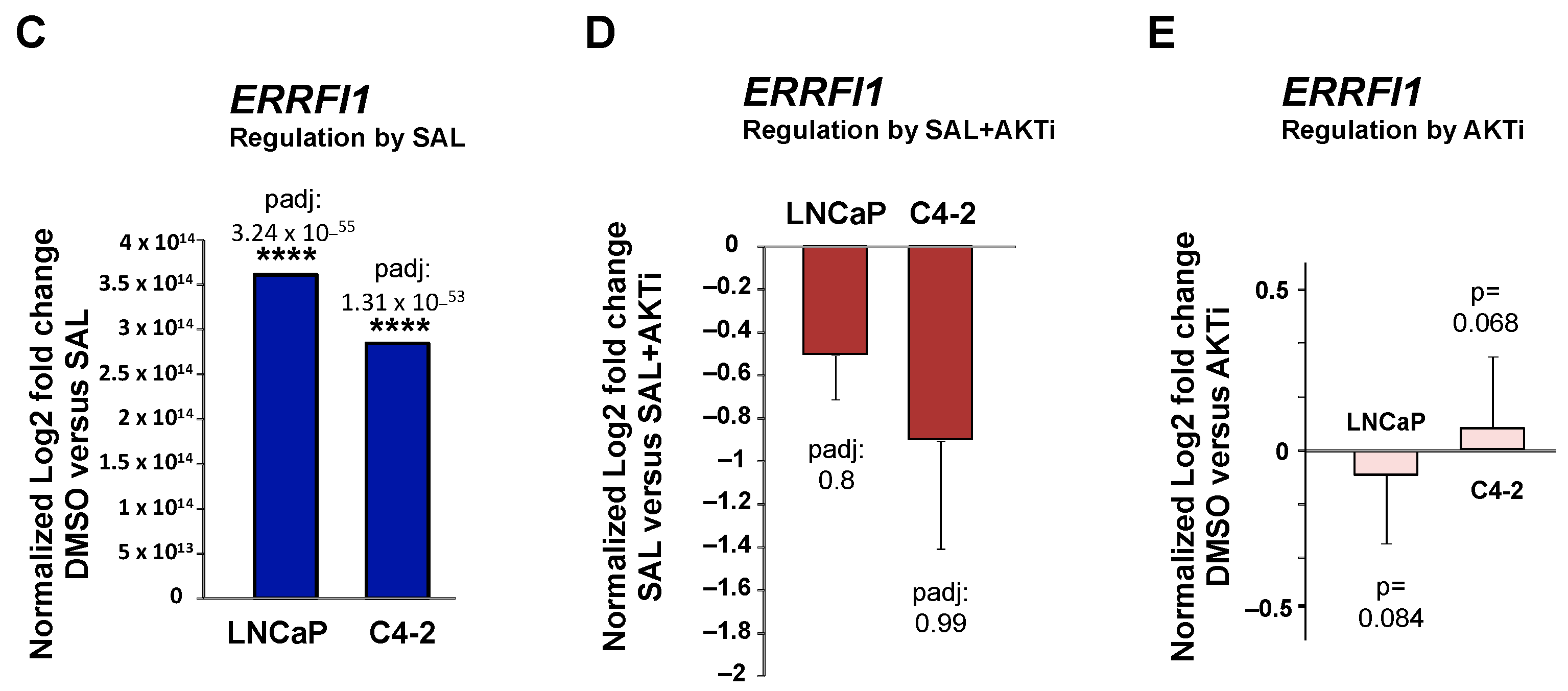
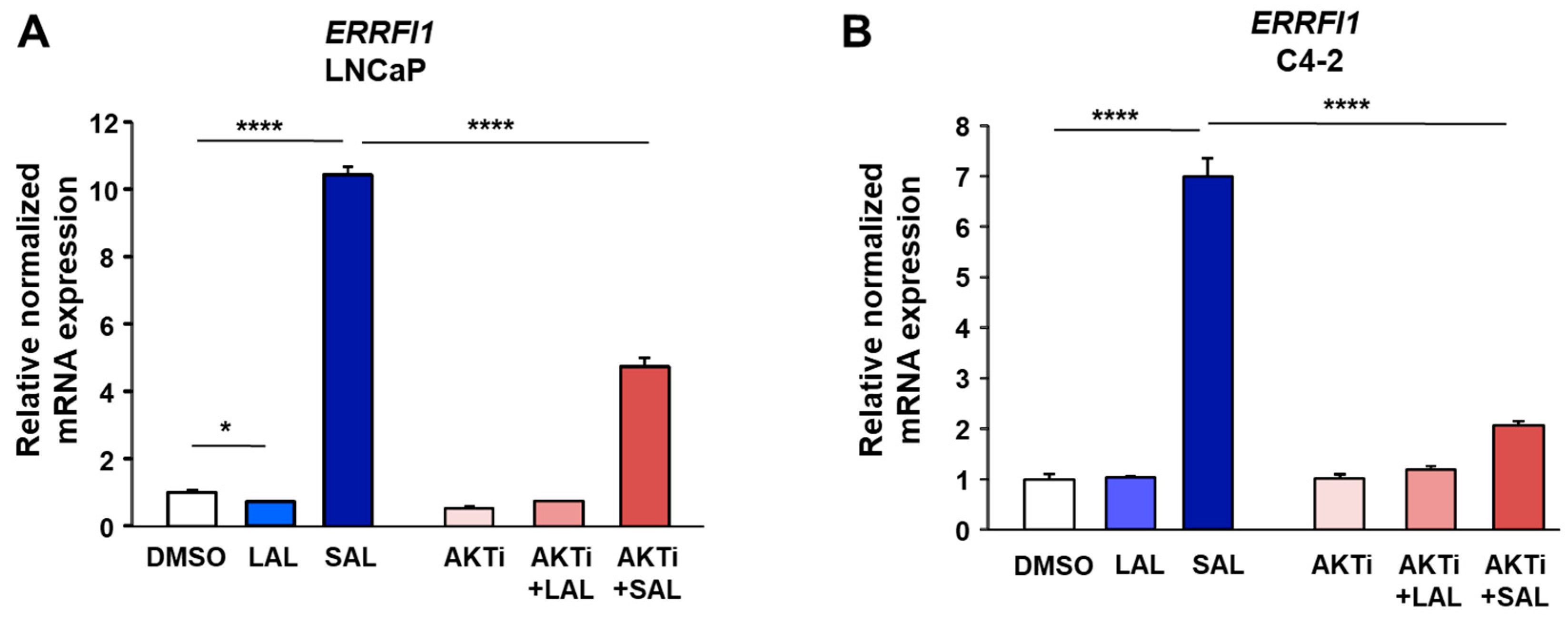
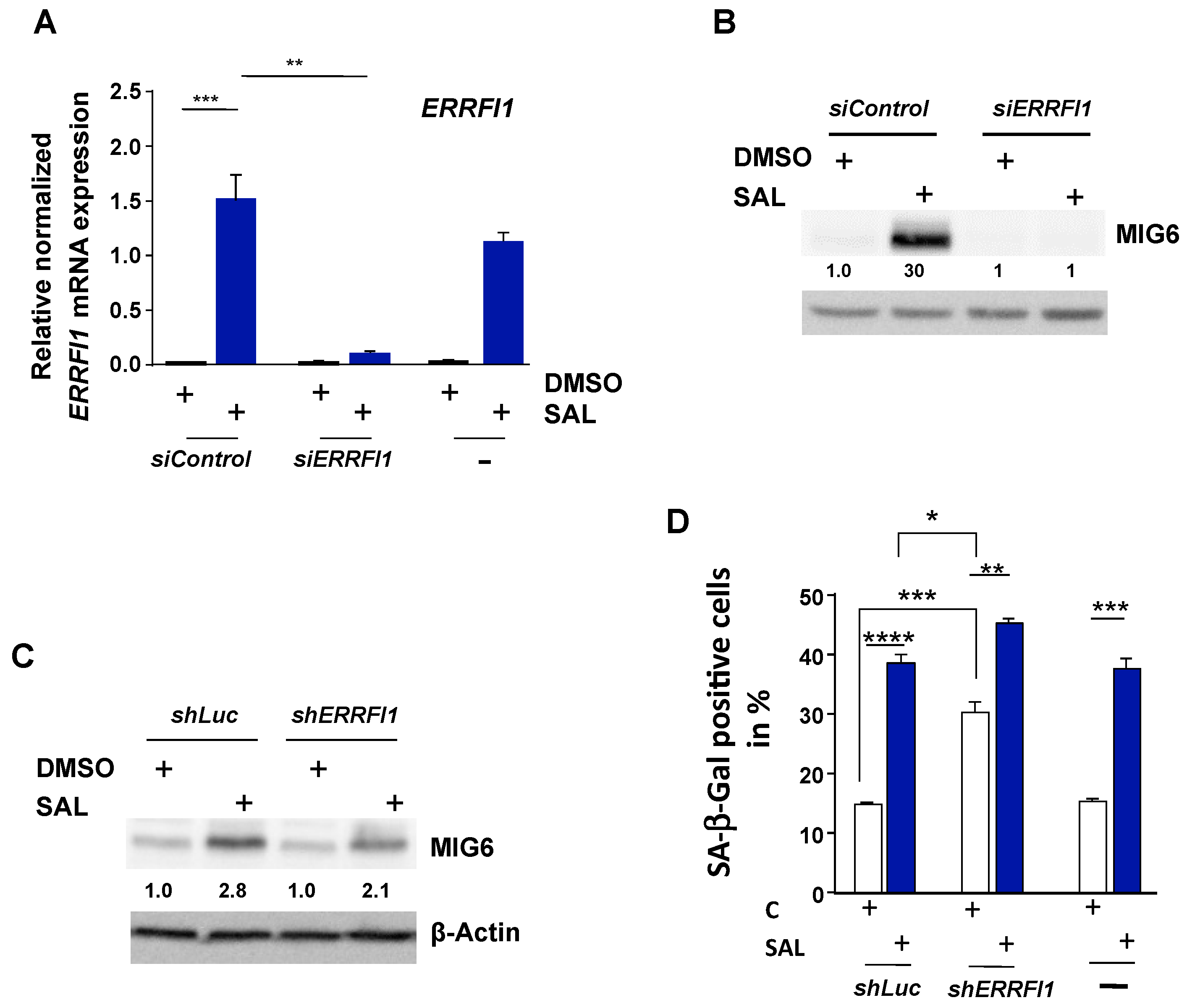
3.1. RNA-seq Identifies ERRFI1 Being Upregulated by SAL in Both LNCaP and C4-2 Cells
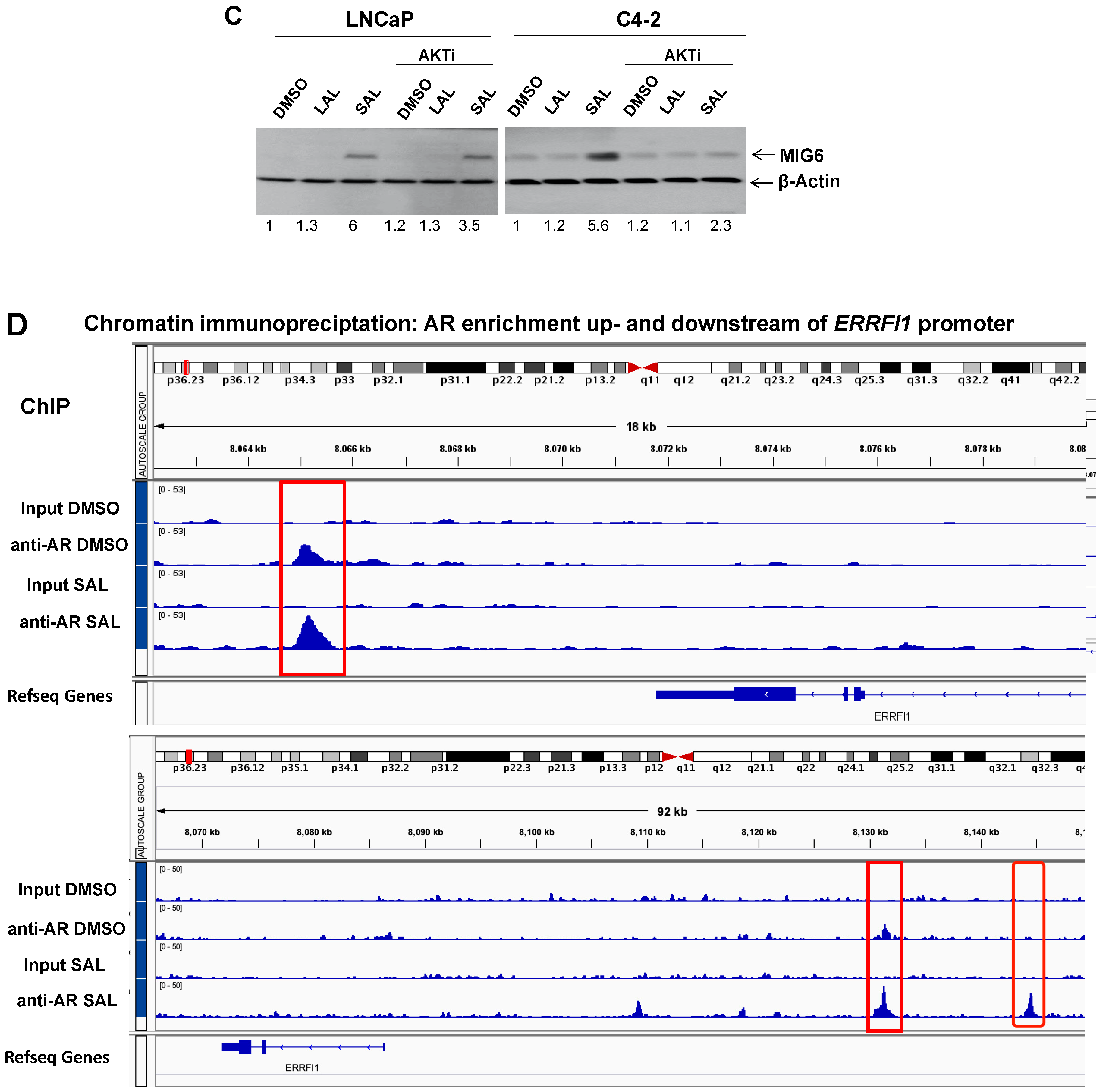
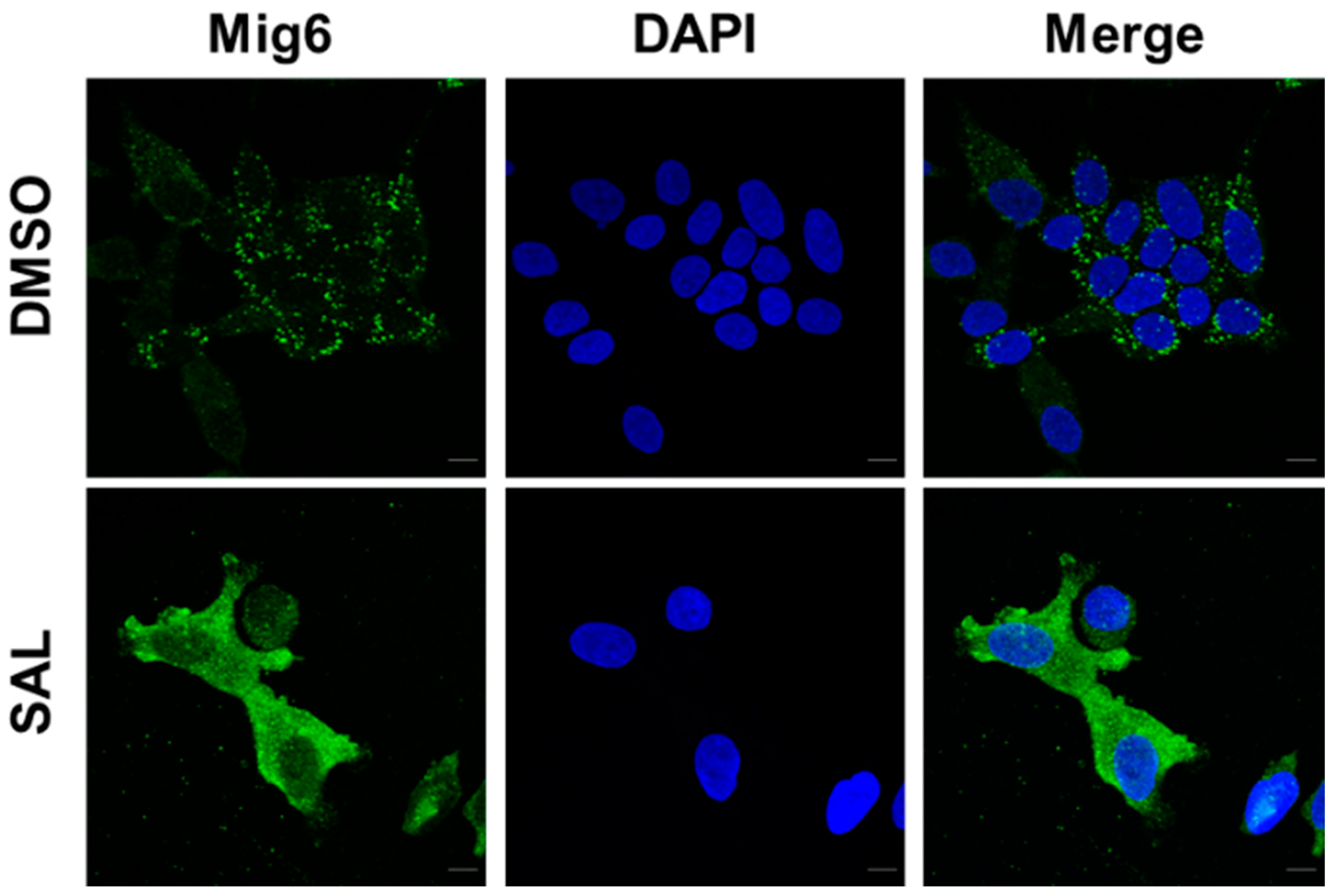
3.2. SAL Treatment Enhances Cytosolic MIG6 Levels
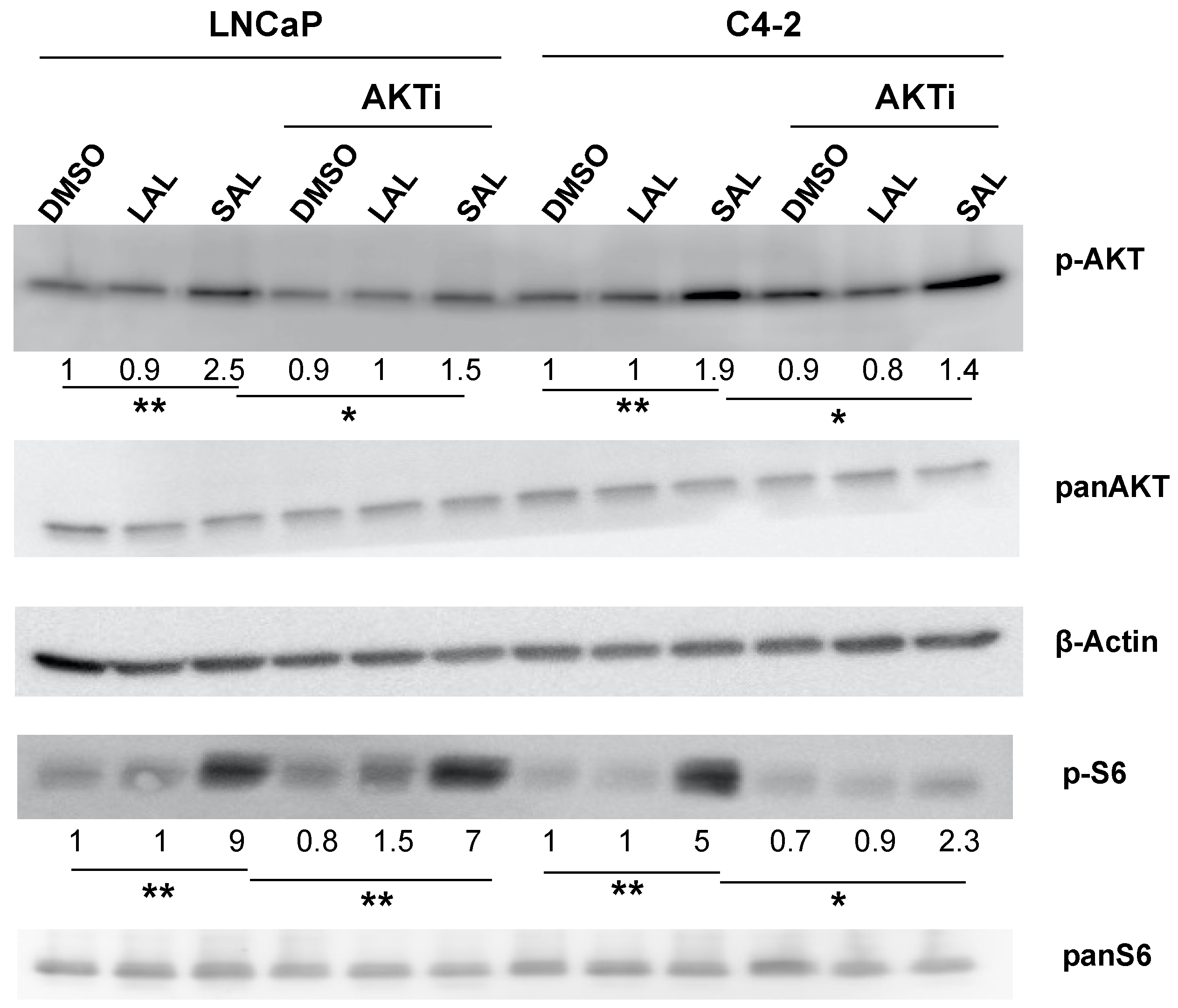
3.3. AKTi Reduces SAL-Enhanced p-AKT and p-S6 Levels
3.4. Knockdown of ERRFI1 Increases SA-β-Gal Positive Cells
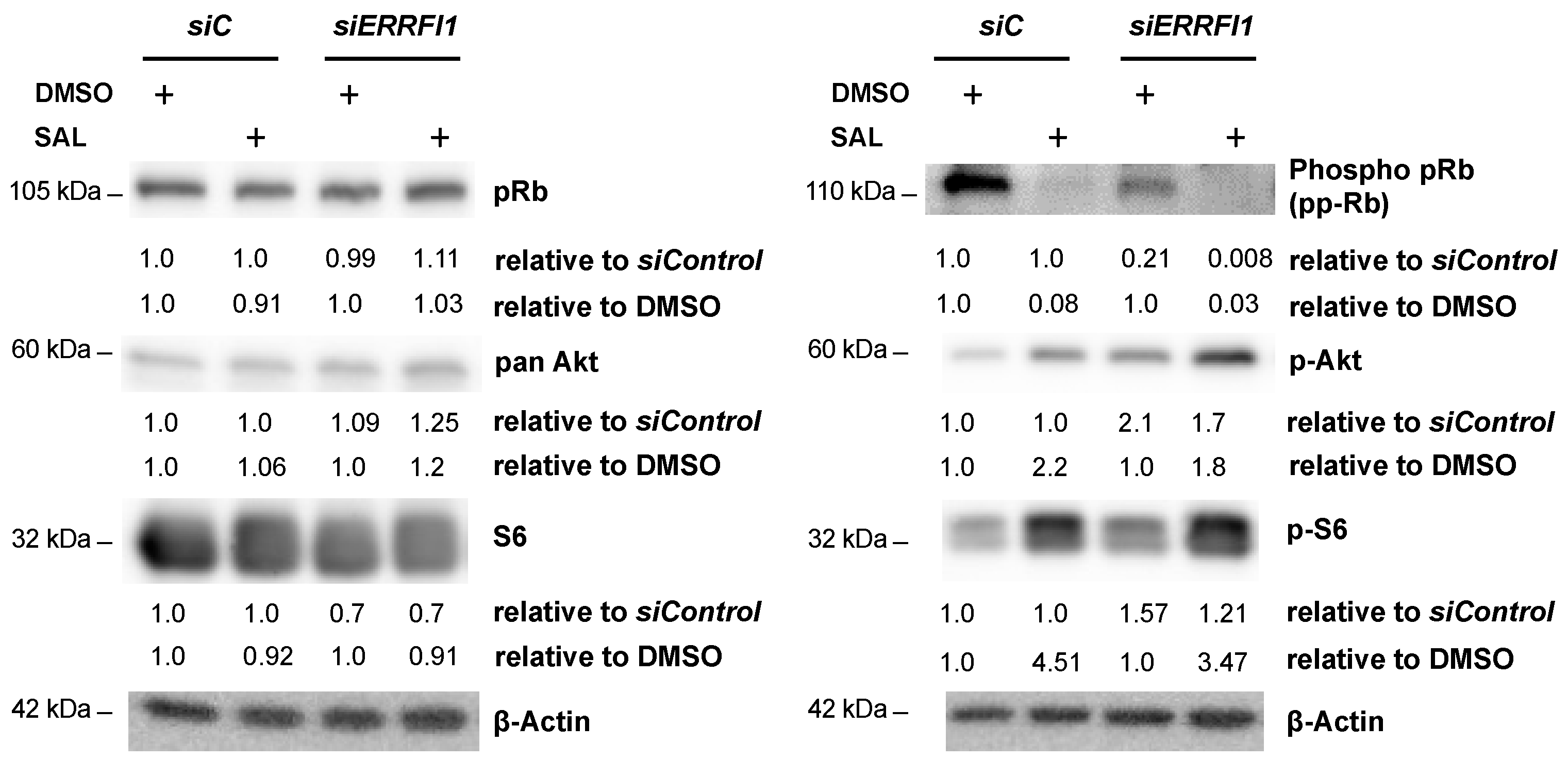
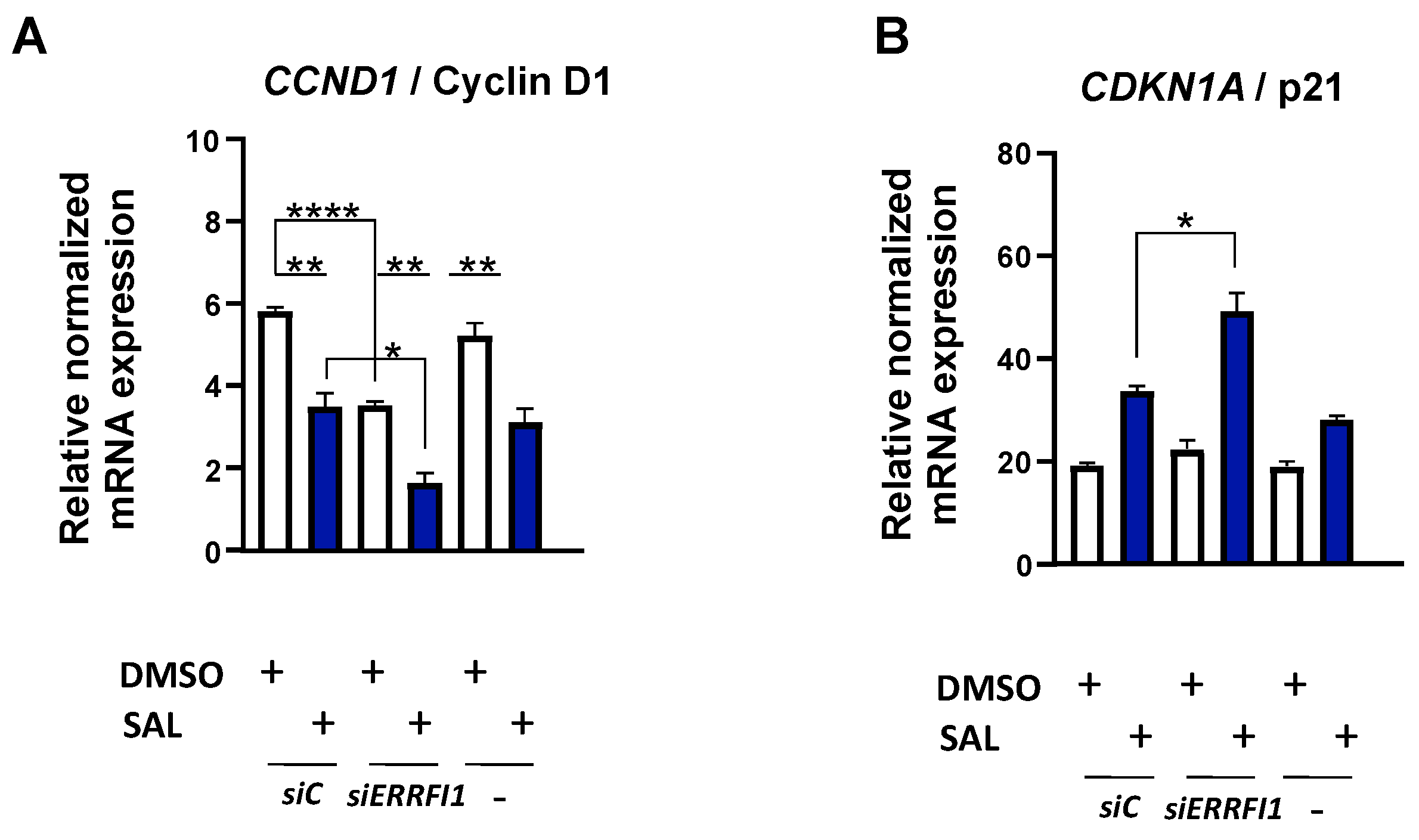
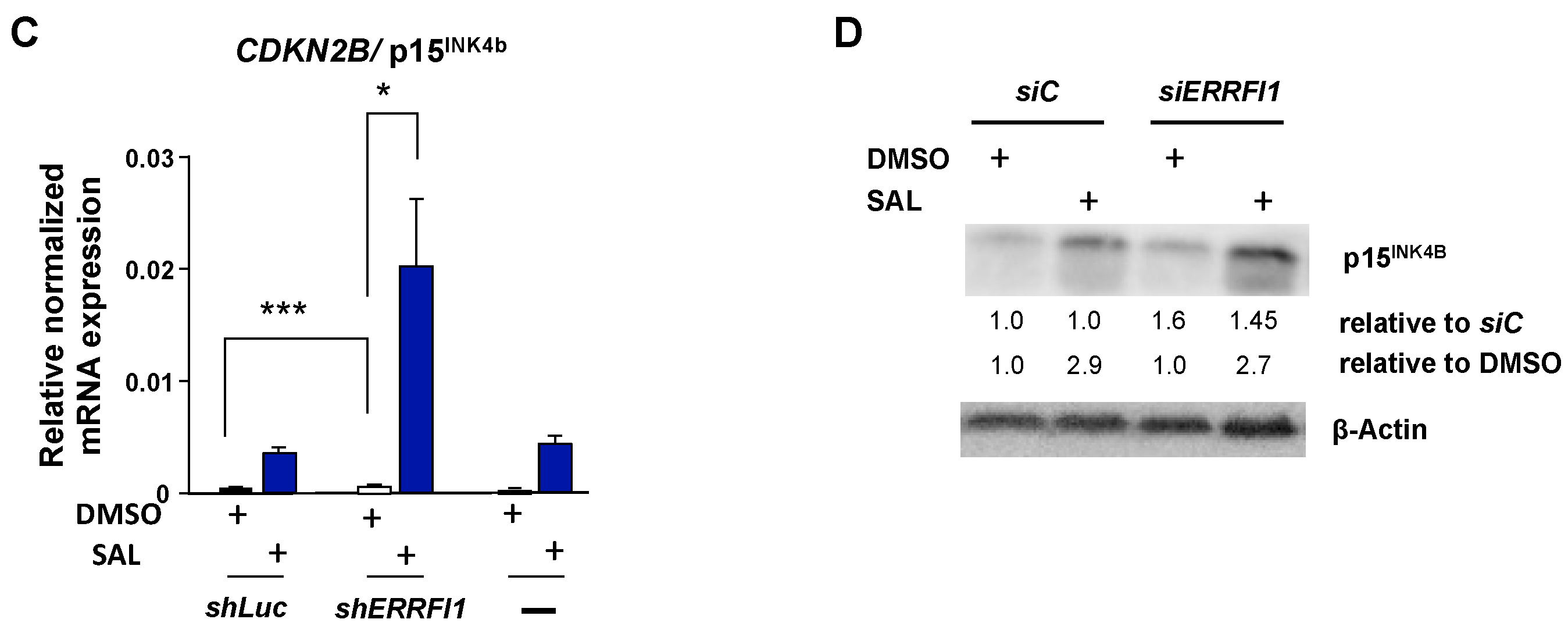
3.5. ERRFI1 Knockdown Represses CCND1 and Induces the Expression of CDKN2B/p15INK4b
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Vickman, R.E.; Franco, O.E.; Moline, D.C.; Vander Griend, D.J.; Thumbikat, P.; Hayward, S.W. The role of the androgen receptor in prostate development and benign prostatic hyperplasia: A review. Asian J. Urol. 2020, 7, 191–202. [Google Scholar] [CrossRef]
- Parker, C.; Castro, E.; Fizazi, K.; Heidenreich, A.; Ost, P.; Procopio, G.; Tombal, B.; Gillessen, S. ESMO Guidelines Committee Prostate cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2020, 31, 1119–1134. [Google Scholar] [PubMed]
- Hessenkemper, W.; Roediger, J.; Bartsch, S.; Houtsmuller, A.B.; van Royen, M.; Petersen, I.; Grimm, M.-O.; Baniahmad, A. A natural androgen receptor antagonist induces cellular senescence in prostate cancer cells. Mol. Endocrinol. 2014, 28, 1831–1840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fousteris, M.A.; Schubert, U.; Roell, D.; Roediger, J.; Bailis, N.; Nikolaropoulos, S.S.; Baniahmad, A.; Giannis, A. 20-Aminosteroids as a novel class of selective and complete androgen receptor antagonists and inhibitors of prostate cancer cell growth. Bioorg. Med. Chem. 2010, 18, 6960–6969. [Google Scholar] [CrossRef]
- Roell, D.; Rösler, T.W.; Hessenkemper, W.; Kraft, F.; Hauschild, M.; Bartsch, S.; Abraham, T.E.; Houtsmuller, A.B.; Matusch, R.; van Royen, M.E.; et al. Halogen-substituted anthranilic acid derivatives provide a novel chemical platform for androgen receptor antagonists. J. Steroid Biochem. Mol. Biol. 2019, 188, 59–70. [Google Scholar] [CrossRef]
- Pungsrinont, T.; Sutter, M.F.; Ertingshausen, M.C.C.M.; Lakshmana, G.; Kokal, M.; Khan, A.S.; Baniahmad, A. Senolytic compounds control a distinct fate of androgen receptor agonist- and antagonist-induced cellular senescent LNCaP prostate cancer cells. Cell Biosci. 2020, 10, 59. [Google Scholar] [CrossRef] [Green Version]
- Roediger, J.; Hessenkemper, W.; Bartsch, S.; Manvelyan, M.; Huettner, S.S.; Liehr, T.; Esmaeili, M.; Foller, S.; Petersen, I.; Grimm, M.-O.; et al. Supraphysiological androgen levels induce cellular senescence in human prostate cancer cells through the Src-Akt pathway. Mol. Cancer. 2014, 13, 214. [Google Scholar] [CrossRef] [Green Version]
- Mirzakhani, K.; Kallenbach, J.; Rasa, S.M.M.; Ribaudo, F.; Ungelenk, M.; Ehsani, M.; Gong, W.; Gassler, N.; Leeder, M.; Grimm, M.O.; et al. The androgen receptor-lncRNASAT1-AKT-p15 axis mediates androgen-induced cellular senescence in prostate cancer cells. Oncogene 2021, 41, 1071–1077. [Google Scholar] [CrossRef]
- Teply, B.A.; Qiu, F.; Antonarakis, E.S.; Carducci, M.A.; Denmeade, S.R. Risk of development of visceral metastases subsequent to abiraterone vs placebo: An analysis of mode of radiographic progression in COU-AA-302. Prostate 2019, 79, 929–933. [Google Scholar] [CrossRef]
- Denmeade, S.R. Bipolar androgen therapy in the treatment of prostate cancer. Clin. Adv. Hematol. Oncol. 2018, 16, 408–411. [Google Scholar] [PubMed]
- Denmeade, S.R.; Isaacs, J.T. Bipolar androgen therapy: The rationale for rapid cycling of supraphysiologic androgen/ablation in men with castration resistant prostate cancer. Prostate 2010, 70, 1600–1607. [Google Scholar] [CrossRef] [Green Version]
- Niu, Y.; Altuwaijr, S.; Lai, K.P.; Wu, C.T.; Ricke, W.A.; Messing, E.M.; Yao, J.; Yeh, S.; Chang, C. Androgen receptor is a tumor suppressor and proliferator in prostate cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 12182–12187. [Google Scholar] [CrossRef] [Green Version]
- Protopopov, A.I.; Li, J.; Winberg, G.; Gizatullin, R.Z.; Kashuba, V.I.; Klein, G.; Zabarovsky, E.R. Human cell lines engineered for tetracycline-regulated expression of tumor suppressor candidate genes from a frequently affected chromosomal region, 3p21. J. Gene Med. 2002, 4, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esmaeili, M.; Jennek, S.; Ludwig, S.; Klitzsch, A.; Kraft, F.; Melle, C.; Baniahmad, A. The tumor suppressor ING1b is a novel corepressor for the androgen receptor and induces cellular senescence in prostate cancer cells. J. Mol. Cell Biol. 2016, 8, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Esmaeili, M.; Pungsrinont, T.; Schaefer, A.; Baniahmad, A. A novel crosstalk between the tumor suppressors ING1 and ING2 regulates androgen receptor signaling. J. Mol. Med. 2016, 94, 1167–1179. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino, L.; Pertica, C.; Fiorini, M.; Talora, C.; Crescenzi, M.; Castellani, L.; Alemà, S.; Benedetti, P.; Segatto, O. Inhibition of ErbB-2 mitogenic and transforming activity by RALT, a mitogen-induced signal transducer which binds to the ErbB-2 kinase domain. Mol. Cell. Biol. 2000, 20, 7735–7750. [Google Scholar] [CrossRef] [Green Version]
- Hackel, P.O.; Gishizky, M.; Ullrich, A. Mig-6 is a negative regulator of the epidermal growth factor receptor signal. Biol. Chem. 2001, 382, 1649–166216. [Google Scholar] [CrossRef]
- Fiorini, M.; Ballarò, C.; Sala, G.; Falcone, G.; Alemà, S.; Segatto, O. Expression of RALT, a feedback inhibitor of ErbB receptors, is subjected to an integrated transcriptional and post-translational control. Oncogene 2002, 21, 6530–6539. [Google Scholar] [CrossRef] [Green Version]
- Reschke, M.; Ferby, I.; Stepniak, E.; Seitzer, N.; Horst, D.; Wagner, E.F.; Ullrich, A. Mitogen-inducible gene-6 is a negative regulator of epidermal growth factor receptor signaling in hepatocytes and human hepatocellular carcinoma. Hepatology 2010, 51, 1383–1390. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Zheng, H.; Scott, K.; Wiedemeyer, R.; Yan, H.; Lim, C.; Huang, J.; Dhakal, S.; Ivanova, E.; Xiao, Y.; et al. Mig-6 controls EGFR trafficking and suppresses gliomagenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 6912–6917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.I.; Du, J.; Shen, W.T.; Whang, E.E.; Donner, D.B.; Griff, N.; He, F.; Moore, F.D., Jr.; Clark, O.H.; Ruan, D.T. Mitogen-inducible gene-6 is a multifunctional adaptor protein with tumor suppressor-like activity in papillary thyroid cancer. J. Clin. Endocrinol. Metab. 2011, 96, E554–E565. [Google Scholar] [CrossRef] [PubMed]
- Cairns, J.; Fridley, B.L.; Jenkins, G.D.; Zhuang, Y.; Yu, J.; Wang, L. Differential roles of ERRFI1 in EGFR and AKT pathway regulation affect cancer proliferation. EMBO Rep. 2018, 19, e44767. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.Y.; Kang, H.B.; Broaddus, R.R.; Risinger, J.I.; Choi, K.C.; Kim, T.H. MIG-6 suppresses endometrial epithelial cell proliferation by inhibiting phospho-AKT. BMC Cancer 2018, 18, 605. [Google Scholar] [CrossRef]
- Vu, H.L.; Rosenbaum, S.; Capparelli, C.; Purwin, T.J.; Davies, M.A.; Berger, A.C.; Aplin, A.E. MIG6 Is MEK Regulated and Affects EGF-Induced Migration in Mutant NRAS Melanoma. J. Investig. Dermatol. 2016, 136, 453–463. [Google Scholar] [CrossRef] [Green Version]
- Kokal, M.; Mirzakhani, K.; Pungsrinont, T.; Baniahmad, A. Mechanisms of Androgen Receptor Agonist- and Antagonist-Mediated Cellular Senescence in Prostate Cancer. Cancers 2020, 12, 1833. [Google Scholar] [CrossRef]
- Xie, B.; Zhao, L.; Chen, H.; Jin, B.; Mao, Z.; Yao, Z. The mitogen-inducible gene-6 is involved in regulation of cellular senescence in normal diploid fibroblasts. Biol. Cell 2013, 105, 488–499. [Google Scholar] [CrossRef]
- Milewska, M.; Kolch, W. Mig-6 participates in the regulation of cell senescence and retinoblastoma protein phosphorylation. Cell. Signal. 2014, 26, 1870–1877. [Google Scholar] [CrossRef]
- Basil, P.; Robertson, M.J.; Bingman, W.E., 3rd; Dash, A.K.; Krause, W.C.; Shafi, A.A.; Piyarathna, B.; Coarfa, C.; Weigel, N.L. Cistrome and transcriptome analysis identifies unique androgen receptor (AR) and AR-V7 splice variant chromatin binding and transcriptional activities. Sci. Rep. 2022, 12, 5351. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Liu, M.; Han, D.; Toure, A.A.; Li, M.; Besschetnova, A.; Wang, Z.; Patalano, S.; Macoska, J.A.; Lam, H.M.; et al. Exploiting the tumor-suppressive activity of the androgen receptor by CDK4/6 inhibition in castration-resistant prostate cancer. Mol. Ther. 2022, 30, 1628–1644. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CCND1 (Cyclin D1) | fwd rev | TCAACCTAAGTTCGGTTCCGATG GTCAGCCTCCACACTCTTGC |
| ERRFI1 (MIG6) | fwd rev | GAAGACCTACTGGAGCAGTCG GACTTTTGAGATGGACCATTTTCTG |
| FKBP5 | fwd rev | GAGGAAACGCCGATGATTGGAGAC CATGCCTTGATGACTTGGCCTTTG |
| GAPDH | fwd rev | AGTCCCTGCCACACTCAG TACTTTATTGATGGTACATGACAAGG |
| CDKN1A (p21) | fwd rev | TCGACTTTGTCACCGAGACACCAC T CAGGTCCACATGGTCTTCCTCTG |
| CDKN2B (p15INK4B) | fwd rev | GATGCGTTCACTCCAATGTC CTTTGTCCTCAGTCTTCAGGTT |
| TBP | fwd rev | GGCGTGTGAAGATAACCCAAGG CGCTGGAACTCGTCTCACT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schomann, T.; Mirzakhani, K.; Kallenbach, J.; Lu, J.; Rasa, S.M.M.; Neri, F.; Baniahmad, A. Androgen-Induced MIG6 Regulates Phosphorylation of Retinoblastoma Protein and AKT to Counteract Non-Genomic AR Signaling in Prostate Cancer Cells. Biomolecules 2022, 12, 1048. https://doi.org/10.3390/biom12081048
Schomann T, Mirzakhani K, Kallenbach J, Lu J, Rasa SMM, Neri F, Baniahmad A. Androgen-Induced MIG6 Regulates Phosphorylation of Retinoblastoma Protein and AKT to Counteract Non-Genomic AR Signaling in Prostate Cancer Cells. Biomolecules. 2022; 12(8):1048. https://doi.org/10.3390/biom12081048
Chicago/Turabian StyleSchomann, Tim, Kimia Mirzakhani, Julia Kallenbach, Jing Lu, Seyed Mohammad Mahdi Rasa, Francesco Neri, and Aria Baniahmad. 2022. "Androgen-Induced MIG6 Regulates Phosphorylation of Retinoblastoma Protein and AKT to Counteract Non-Genomic AR Signaling in Prostate Cancer Cells" Biomolecules 12, no. 8: 1048. https://doi.org/10.3390/biom12081048