
Pro-Calcifying Role of Enzymatically Modified LDL (eLDL) in Aortic Valve Sclerosis via Induction of IL-6 and IL-33


Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Aortic Valvular Tissue
2.2. Histological and Immunhohistochemical Analysis
2.3. VIC/Myofibroblast Isolation and Culture
2.4. Treatment of VICs/Myofibroblasts
2.5. Lipoprotein Isolation and Modifications
2.6. Calcification Assay
2.7. RNA Isolation and Real-Time Quantitative PCR
2.8. Western Blot Analyses
2.9. MTT Assay
2.10. Statistical Analyses
3. Results
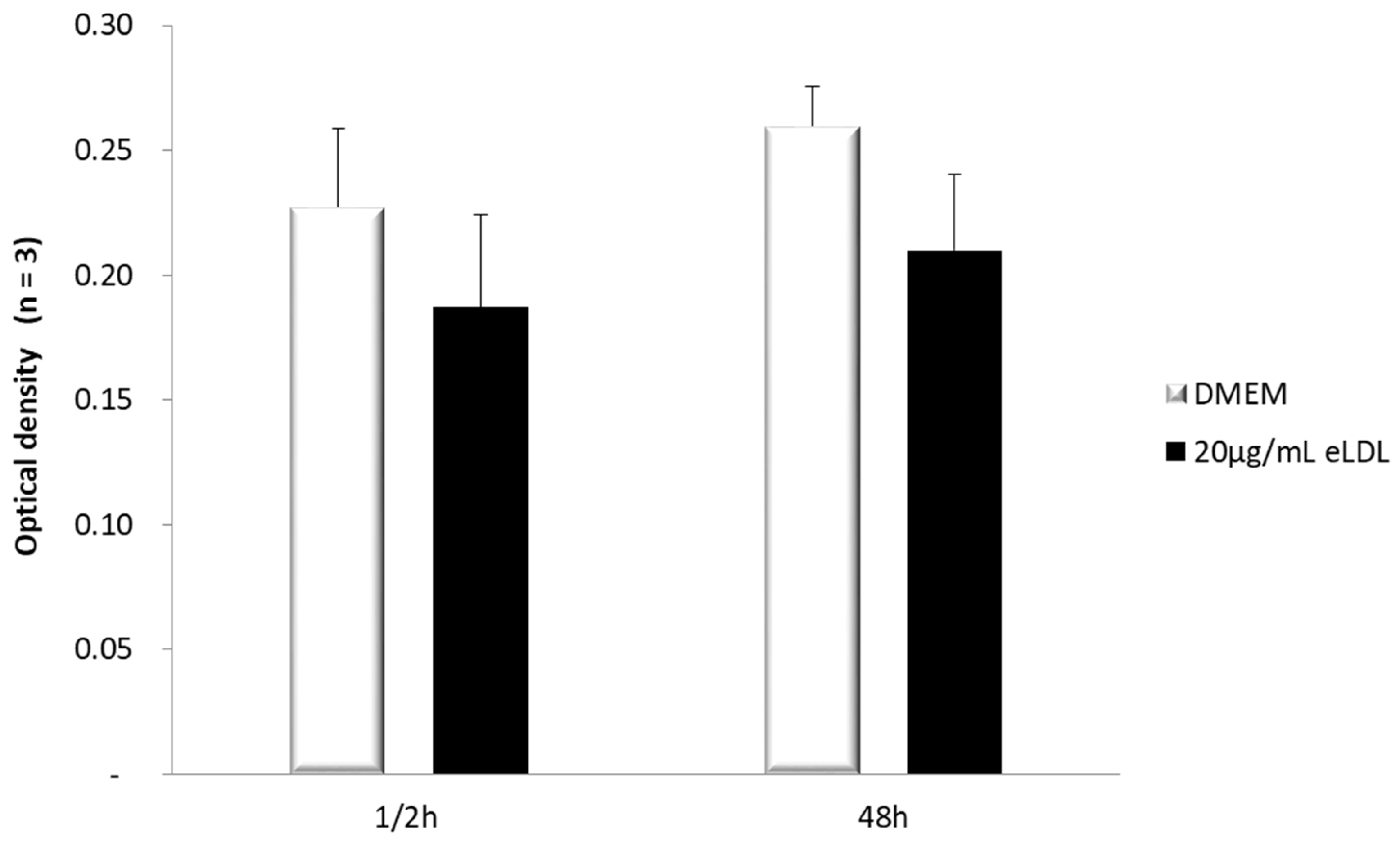
3.1. Estimation of Cell Viability in VICs/Myofibroblasts
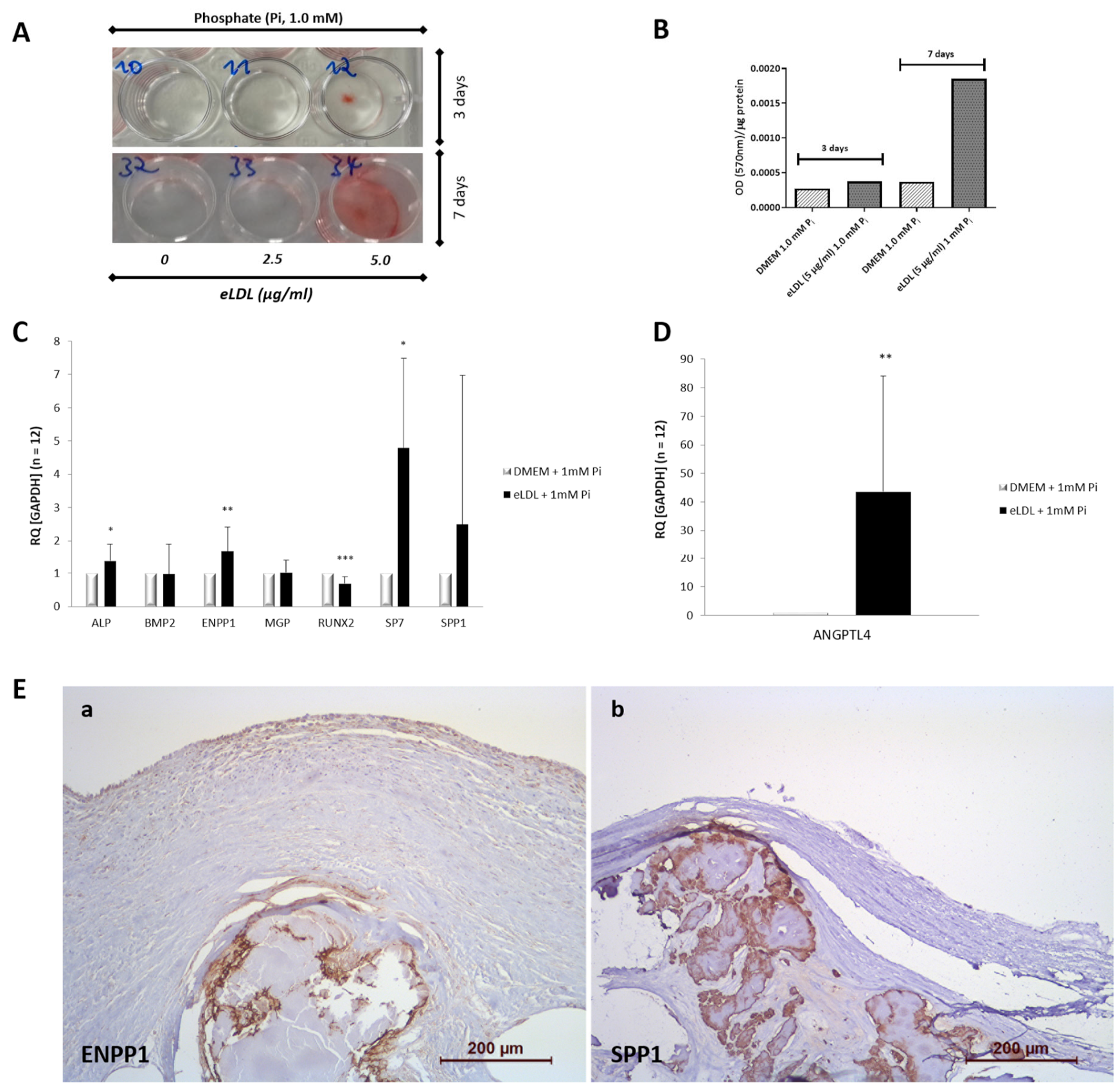
3.2. eLDL Upregulates Phosphate Induced Calcification in Cultured VICs/Myofibroblasts
3.3. eLDL and the Expression of Calcification-Related Genes
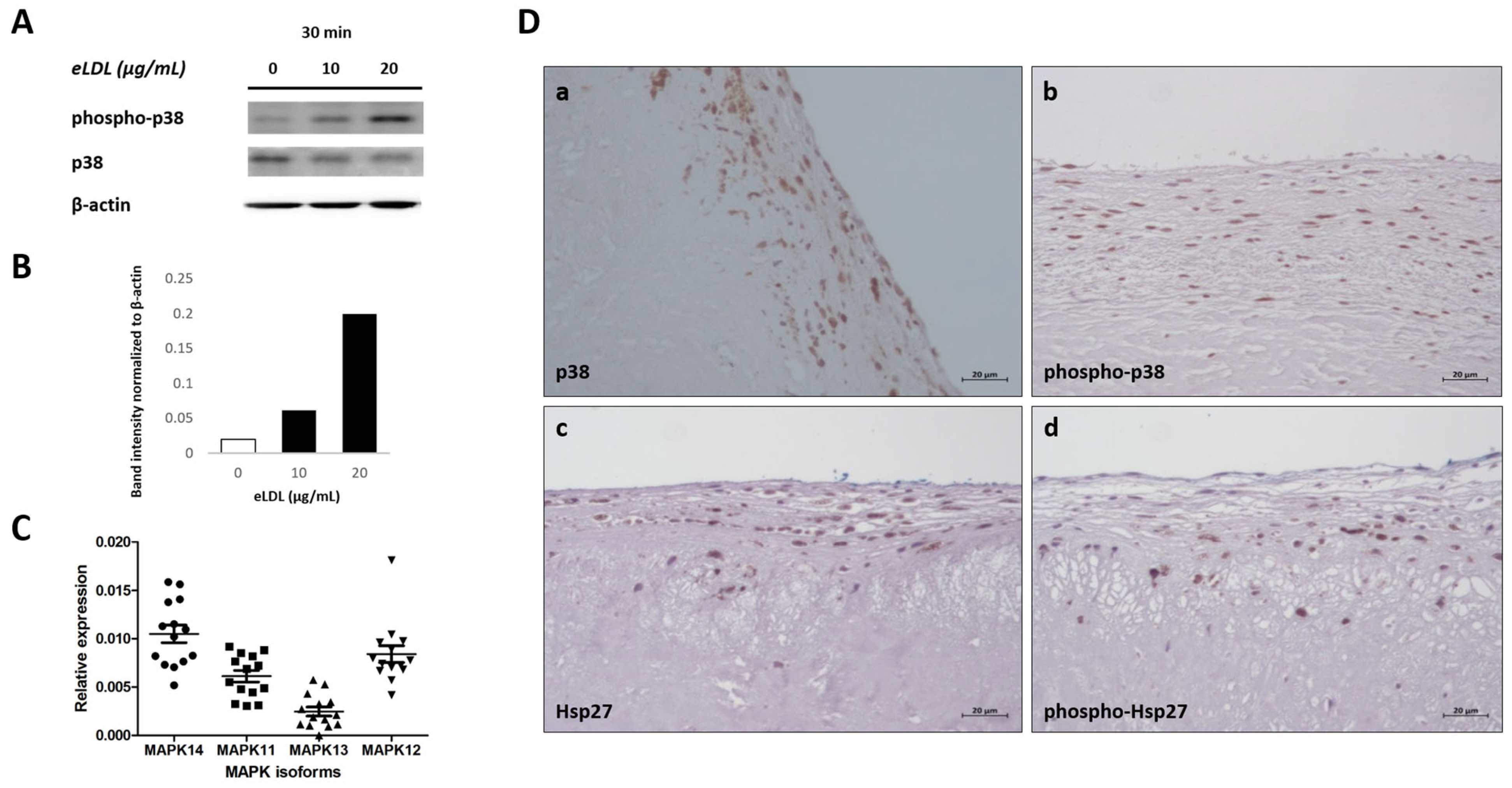
3.4. eLDL and p38 MAPK in Human VICs/Myofibroblasts
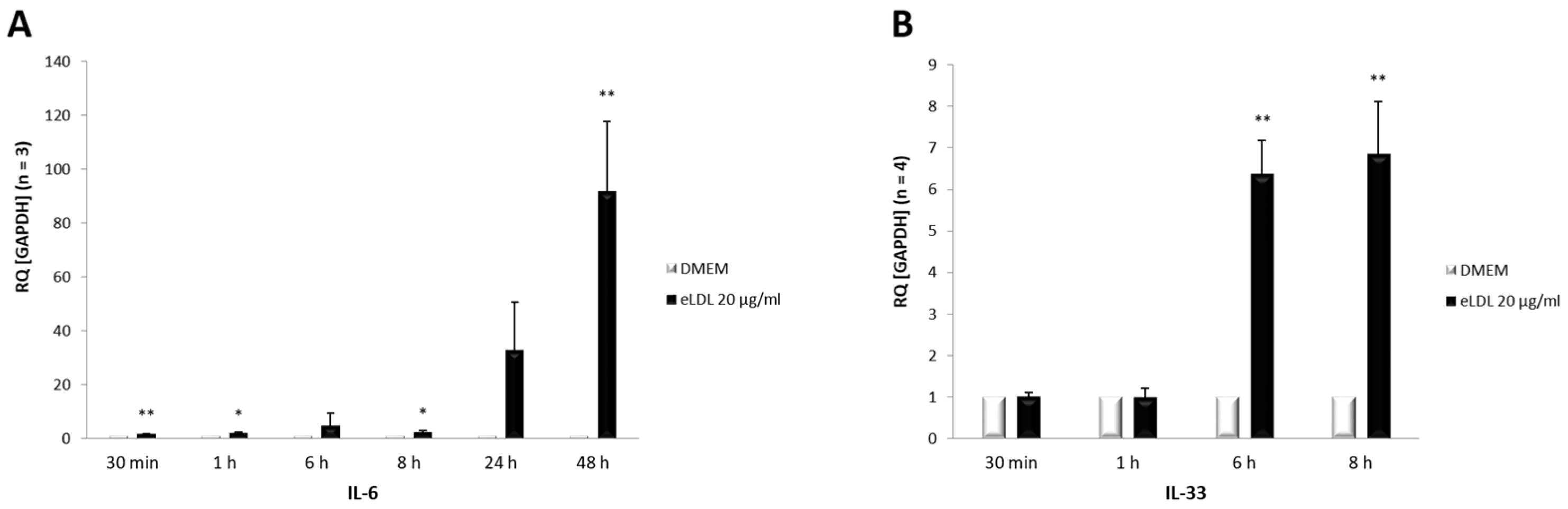
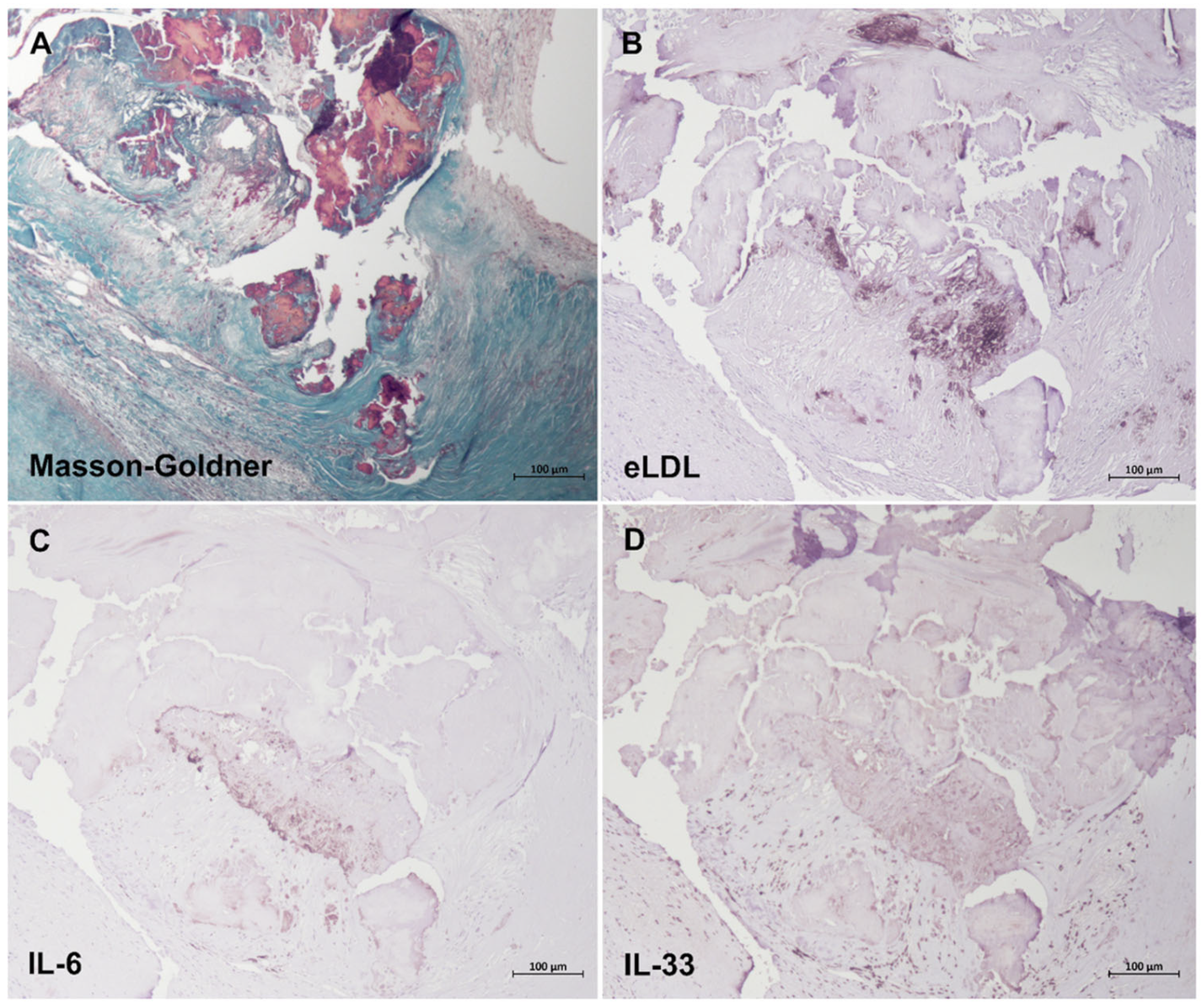
3.5. Induction of Both IL-6 and IL-33 by eLDL
3.6. Colocalization of eLDL with IL-6 and IL-33
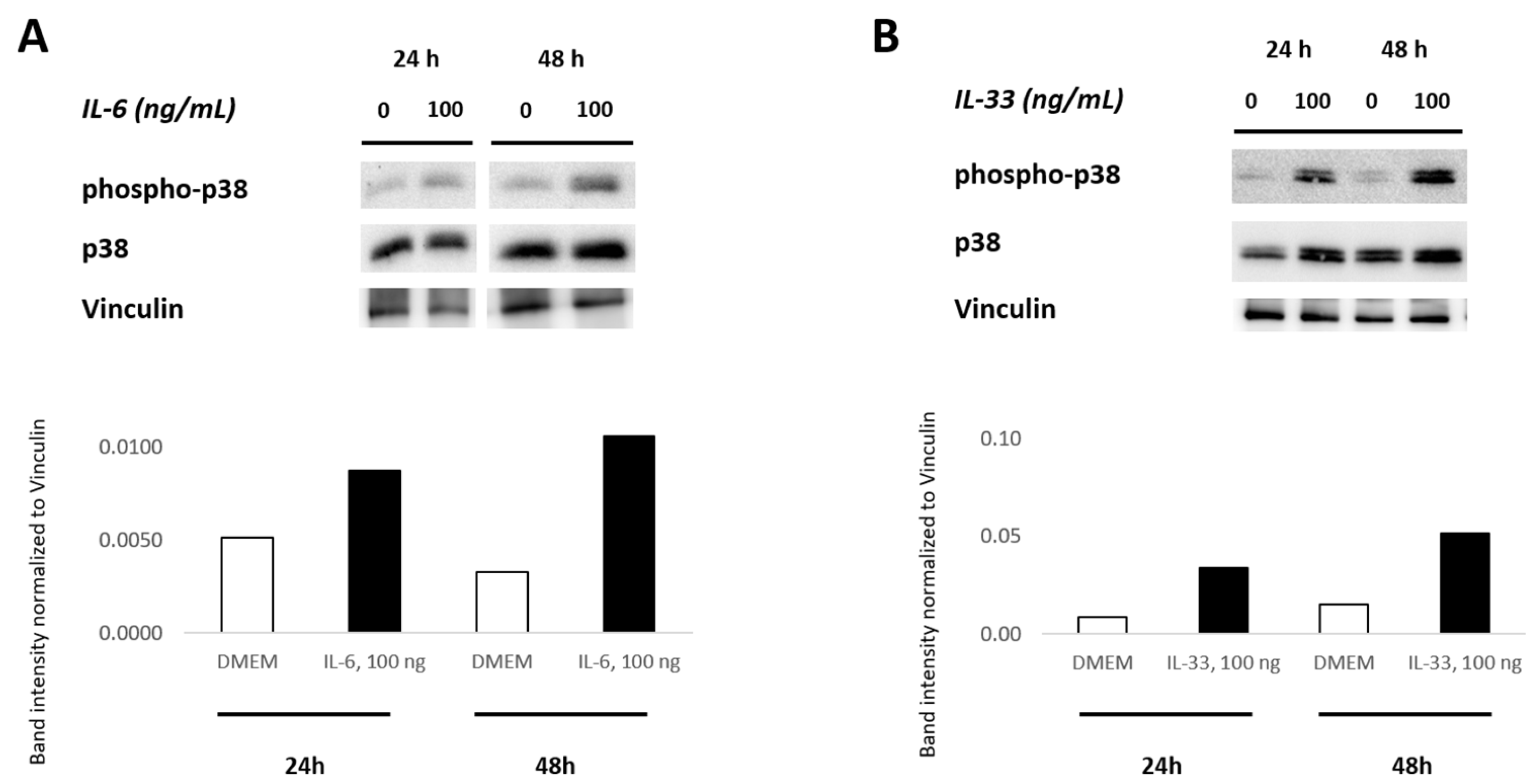
3.7. IL-6 and IL-33 Activate the p38 MAPK Pathway
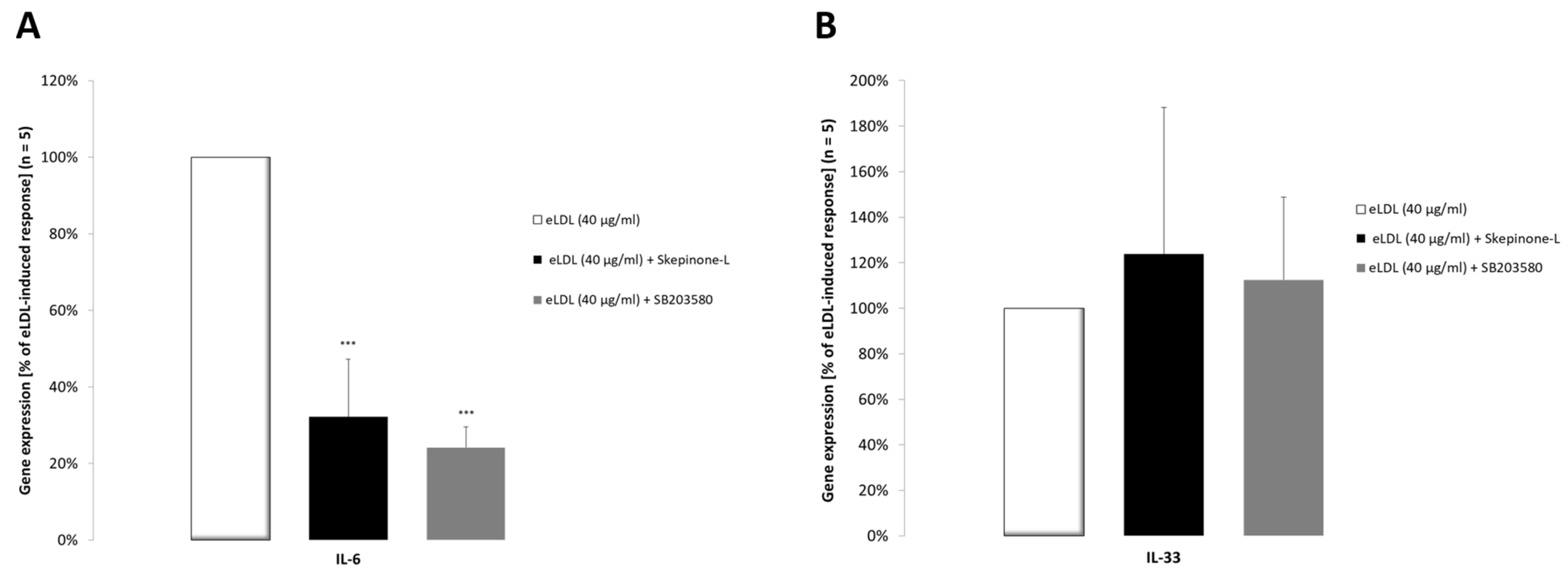
3.8. The p38 MAPK Pathway Is Involved in Increased IL-6 Cytokine Expression
3.9. IL-33 Induces IL-6 Cytokine Expression in Primary VICs/Myofibroblasts
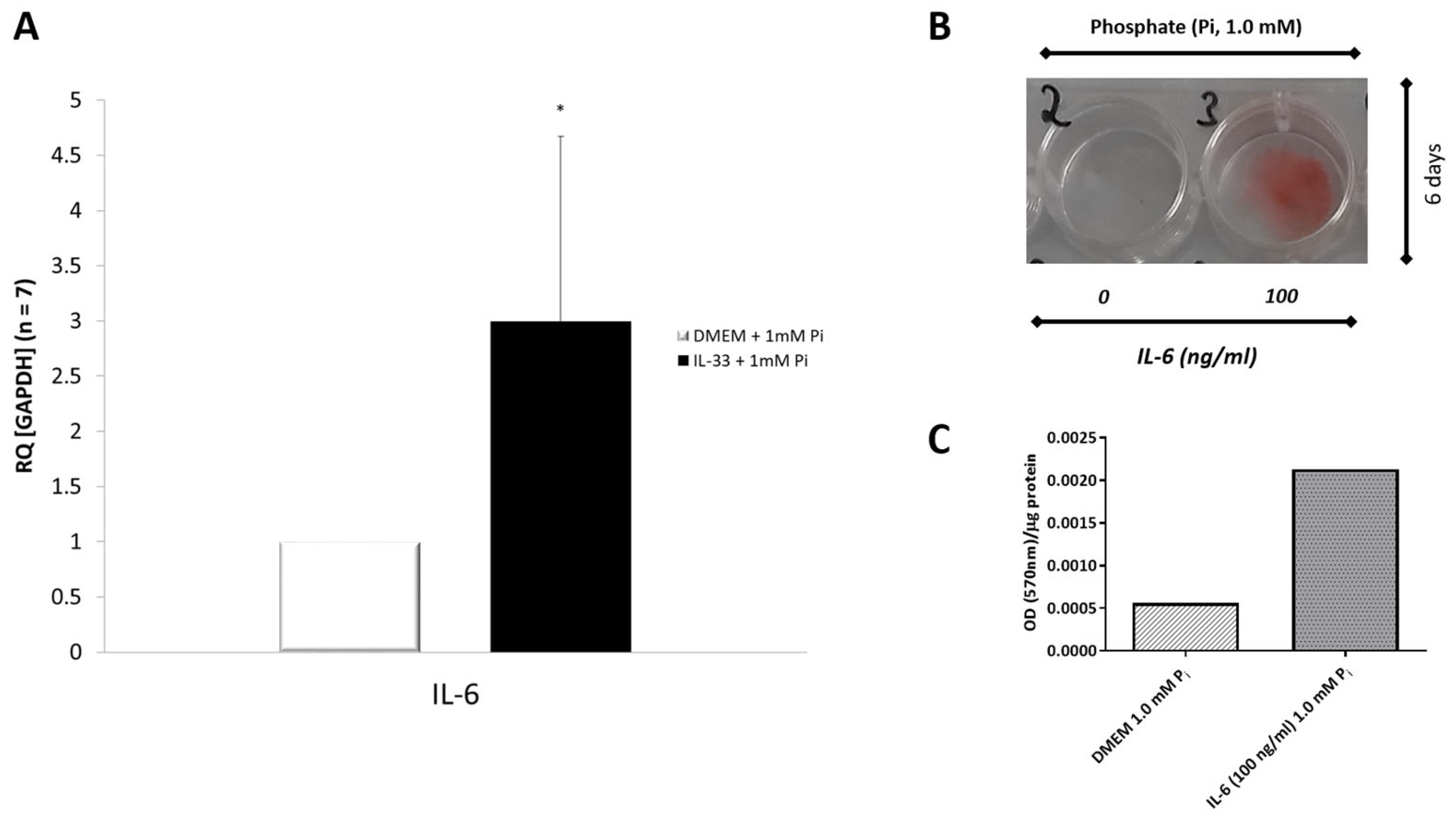
3.10. Effects of IL-6 on Phosphate-Induced Calcification in VICs/Myofibroblasts
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nkomo, V.T.; Gardin, J.M.; Skelton, T.N.; Gottdiener, J.S.; Scott, C.G.; Enriquez-Sarano, M. Burden of valvular heart diseases: A population-based study. Lancet 2006, 368, 1005–1011. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.; Pesce, M. The Complex Interplay of Inflammation, Metabolism, Epigenetics, and Sex in Calcific Disease of the Aortic Valve. Front. Cardiovasc. Med. 2021, 8, 791646. [Google Scholar] [CrossRef] [PubMed]
- Alushi, B.; Curini, L.; Christopher, M.R.; Grubitzch, H.; Landmesser, U.; Amedei, A.; Lauten, A. Calcific Aortic Valve Disease-Natural History and Future Therapeutic Strategies. Front. Pharmacol. 2020, 11, 685. [Google Scholar] [CrossRef] [PubMed]
- Dweck, M.R.; Boon, N.A.; Newby, D.E. Calcific aortic stenosis: A disease of the valve and the myocardium. J. Am. Coll. Cardiol. 2012, 60, 1854–1863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajamannan, N.M.; Evans, F.J.; Aikawa, E.; Grande-Allen, K.J.; Demer, L.L.; Heistad, D.D.; Simmons, C.A.; Masters, K.S.; Mathieu, P.; O’Brien, K.D.; et al. Calcific aortic valve disease: Not simply a degenerative process: A review and agenda for research from the National Heart and Lung and Blood Institute Aortic Stenosis Working Group. Executive summary: Calcific aortic valve disease-2011 update. Circulation 2011, 124, 1783–1791. [Google Scholar] [CrossRef] [Green Version]
- Iung, B.; Baron, G.; Butchart, E.G.; Delahaye, F.; Gohlke-Bärwolf, C.; Levang, O.W.; Tornos, P.; Vanoverschelde, J.-L.; Vermeer, F.; Boersma, E.; et al. A prospective survey of patients with valvular heart disease in Europe: The Euro Heart Survey on Valvular Heart Disease. Eur. Heart J. 2003, 24, 1231–1243. [Google Scholar] [CrossRef] [Green Version]
- Goody, P.R.; Hosen, M.R.; Christmann, D.; Niepmann, S.T.; Zietzer, A.; Adam, M.; Bönner, F.; Zimmer, S.; Nickenig, G.; Jansen, F. Aortic Valve Stenosis: From Basic Mechanisms to Novel Therapeutic Targets. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 885–900. [Google Scholar] [CrossRef]
- Yutzey, K.E.; Demer, L.L.; Body, S.C.; Huggins, G.S.; Towler, D.A.; Giachelli, C.M.; Hofmann-Bowman, M.A.; Mortlock, D.P.; Rogers, M.B.; Sadeghi, M.M.; et al. Calcific aortic valve disease: A consensus summary from the Alliance of Investigators on Calcific Aortic Valve Disease. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2387–2393. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, R.A.; Otto, C.M.; Bonow, R.O.; Carabello, B.A.; Erwin, I.I.I.J.P.; Guyton, R.A.; O’Gara, P.T.; Ruiz, C.E.; Skubas, N.J.; Sorajja, P.; et al. 2014 AHA/ACC guideline for the management of patients with valvular heart disease: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J. Am. Coll. Cardiol. 2014, 63, e57–e185. [Google Scholar] [CrossRef] [Green Version]
- Zheng, K.H.; Tzolos, E.; Dweck, M.R. Pathophysiology of Aortic Stenosis and Future Perspectives for Medical Therapy. Cardiol. Clin. 2019, 38, 1–12. [Google Scholar] [CrossRef]
- Pawade, T.A.; Newby, D.E.; Dweck, M.R. Calcification in Aortic Stenosis: The Skeleton Key. J. Am. Coll. Cardiol. 2015, 66, 561–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- New, S.E.P.; Aikawa, E. Molecular imaging insights into early inflammatory stages of arterial and aortic valve calcification. Circ. Res. 2011, 108, 1381–1391. [Google Scholar] [CrossRef] [PubMed]
- Aikawa, E.; Nahrendorf, M.; Figueiredo, J.L.; Swirski, F.K.; Shtatland, T.; Kohler, R.H.; Jaffer, F.A.; Aikawa, M.; Weissleder, R. Osteogenesis associates with inflammation in early-stage atherosclerosis evaluated by molecular imaging in vivo. Circulation 2007, 116, 2841–2850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reustle, A.; Torzewski, M. Role of p38 MAPK in Atherosclerosis and Aortic Valve Sclerosis. Int. J. Mol. Sci. 2018, 19, 3761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lackner, K.; Han, S.-R.; Torzewski, M.; Husmann, M.; Bhakdi, S. Beyond cholesterol: The enigma of atherosclerosis revisited. Thromb. Haemost. 2004, 91, 639–645. [Google Scholar] [CrossRef]
- Torzewski, M. Enzymatically modified LDL, atherosclerosis and beyond: Paving the way to acceptance. Front. Biosci. (Landmark Ed.) 2018, 23, 1257–1271. [Google Scholar] [CrossRef] [Green Version]
- Bhakdi, S.; Dorweiler, B.; Kirchmann, R.; Torzewski, J.; Weise, E.; Tranum-Jensen, J.; Walev, I.; Wieland, E. On the pathogenesis of atherosclerosis: Enzymatic transformation of human low density lipoprotein to an atherogenic moiety. J. Exp. Med. 1995, 182, 1959–1971. [Google Scholar] [CrossRef]
- Torzewski, M.; Suriyaphol, P.; Paprotka, K.; Spath, L.; Ochsenhirt, V.; Schmitt, A.; Han, S.R.; Husmann, M.; Gerl, V.B.; Bhakdi, S.; et al. Enzymatic modification of low-density lipoprotein in the arterial wall: A new role for plasmin and matrix metalloproteinases in atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2130–2136. [Google Scholar] [CrossRef] [Green Version]
- Torzewski, M.; Klouche, M.; Hock, J.; Meßner, M.; Dorweiler, B.; Torzewski, J.; Gabbert, H.E.; Bhakdi, S. Immunohistochemical Demonstration of Enzymatically Modified Human LDL and Its Colocalization With the Terminal Complement Complex in the Early Atherosclerotic Lesion. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 369–378. [Google Scholar] [CrossRef] [Green Version]
- Torzewski, M. The Initial Human Atherosclerotic Lesion and Lipoprotein Modification—A Deep Connection. Int. J. Mol. Sci. 2021, 22, 11488. [Google Scholar] [CrossRef]
- Twardowski, L.; Cheng, F.; Michaelsen, J.; Winter, S.; Hofmann, U.; Schaeffeler, E.; Müller, S.; Sonnenberg, M.; Steuer, K.; Otto, C.M.; et al. Enzymatically Modified Low-Density Lipoprotein Is Present in All Stages of Aortic Valve Sclerosis: Implications for Pathogenesis of the Disease. J. Am. Heart Assoc. 2015, 4, e002156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Liao, H.; Wang, N.; Ma, K.-S.; Verna, L.K.; Shyy, J.Y.-J.; Chien, S.; Stemerman, M.B. LDL-Activated p38 in Endothelial Cells Is Mediated by Ras. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1159–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dersch, K.; Ichijo, H.; Bhakdi, S.; Husmann, M. Fatty acids liberated from low-density lipoprotein trigger endothelial apoptosis via mitogen-activated protein kinases. Cell Death Differ. 2005, 12, 1107–1114. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Twardowski, L.; Fehr, S.; Aner, C.; Schaeffeler, E.; Joos, T.; Knorpp, T.; Dorweiler, B.; Laufer, S.; Schwab, M.; et al. Selective p38α MAP kinase/MAPK14 inhibition in enzymatically modified LDL-stimulated human monocytes: Implications for atherosclerosis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2017, 31, 674–686. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Zhou, H.; Feng, T.; Wu, R.; Sun, X.; Guan, N.; Qu, L.; Gao, Z.; Yan, J.; Xu, N.; et al. β-Glucan attenuates inflammatory responses in oxidized LDL-induced THP-1 cells via the p38 MAPK pathway. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 248–255. [Google Scholar] [CrossRef]
- Hakala, J.K.; Lindstedt, K.A.; Kovanen, P.T.; Pentikäinen, M.O. Low-density lipoprotein modified by macrophage-derived lysosomal hydrolases induces expression and secretion of IL-8 via p38 MAPK and NF-kappaB by human monocyte-derived macrophages. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2504–2509. [Google Scholar] [CrossRef] [Green Version]
- El Husseini, D.; Boulanger, M.-C.; Mahmut, A.; Bouchareb, R.; Laflamme, M.-H.; Fournier, D.; Pibarot, P.; Bossé, Y.; Mathieu, P. P2Y2 receptor represses IL-6 expression by valve interstitial cells through Akt: Implication for calcific aortic valve disease. J. Mol. Cell. Cardiol. 2014, 72, 146–156. [Google Scholar] [CrossRef]
- He, Y.-B.; Guo, J.-H.; Wang, C.; Zhu, D.; Lu, L.-M. IL-33 promotes the progression of nonrheumatic aortic valve stenosis via inducing differential phenotypic transition in valvular interstitial cells. J. Cardiol. 2020, 75, 124–133. [Google Scholar] [CrossRef]
- Miller, A.M.; Xu, D.; Asquith, D.L.; Denby, L.; Li, Y.; Sattar, N.; Baker, A.H.; McInnes, I.B.; Liew, F.Y. IL-33 reduces the development of atherosclerosis. J. Exp. Med. 2008, 205, 339–346. [Google Scholar] [CrossRef]
- McLaren, J.E.; Michael, D.R.; Salter, R.C.; Ashlin, T.G.; Calder, C.J.; Miller, A.M.; Liew, F.Y.; Ramji, D.P.; Li, X.L.; Ménoret, S.; et al. IL-33 Reduces Macrophage Foam Cell Formation. J. Immunol. 2010, 185, 1222–1229. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.-L.; Shen, D.-L.; Zhu, K.; Tang, J.-N.; Wang, X.-F.; Zhang, L.; Zhang, J.-Y. Characterization of interleukin-33 and matrix metalloproteinase-28 in serum and their association with disease severity in patients with coronary heart disease. Coron. Artery Dis. 2014, 25, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Warren, B.A.; Yong, J.L. Calcification of the aortic valve: Its progression and grading. Pathology 1997, 29, 360–368. [Google Scholar] [CrossRef]
- Schlotter, F.; Halu, A.; Goto, S.; Blaser, M.C.; Body, S.C.; Lee, L.H.; Higashi, H.; DeLaughter, D.M.; Hutcheson, J.D.; Vyas, P.; et al. Spatiotemporal Multi-Omics Mapping Generates a Molecular Atlas of the Aortic Valve and Reveals Networks Driving Disease. Circulation 2018, 138, 377–393. [Google Scholar] [CrossRef] [PubMed]
- Chellan, B.; Rojas, E.; Zhang, C.; Bowman, M.A.H. Enzyme-modified non-oxidized LDL (ELDL) induces human coronary artery smooth muscle cell transformation to a migratory and osteoblast-like phenotype. Sci. Rep. 2018, 8, 11954. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Carver, W. Effects of interleukin-33 on cardiac fibroblast gene expression and activity. Cytokine 2012, 58, 368–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Su, H.; Yan, M.; Zhang, L.; Tang, J.; Li, Q.; Gu, X.; Gong, Q. Interleukin-33 Promotes Cell Survival via p38 MAPK-Mediated Interleukin-6 Gene Expression and Release in Pediatric AML. Front. Immunol. 2020, 11, 595053. [Google Scholar] [CrossRef] [PubMed]
- Bhakdi, S.; Torzewski, M.; Paprotka, K.; Schmitt, S.; Barsoom, H.; Suriyaphol, P.; Han, S.R.; Lackner, K.J.; Husmann, M. Possible protective role for C-reactive protein in atherogenesis: Complement activation by modified lipoproteins halts before detrimental terminal sequence. Circulation 2004, 109, 1870–1876. [Google Scholar] [CrossRef] [Green Version]
- Prosdocimo, D.A.; Wyler, S.C.; Romani, A.M.; O’Neill, W.C.; Dubyak, G.R.; Martínez-Moreno, J.M.; Muñoz-Castañeda, J.R.; Herencia, C.; de Oca, A.M.; Estepa, J.C.; et al. Regulation of vascular smooth muscle cell calcification by extracellular pyrophosphate homeostasis: Synergistic modulation by cyclic AMP and hyperphosphatemia. Am. J. Physiol. Physiol. 2010, 298, C702–C713. [Google Scholar] [CrossRef] [Green Version]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Rutsch, F.; Ruf, N.; Vaingankar, S.; Toliat, M.R.; Suk, A.; Höhne, W.; Schauer, G.; Lehmann, M.; Roscioli, T.; Schnabel, D.; et al. Mutations in ENPP1 are associated with ‘idiopathic’ infantile arterial calcification. Nat. Genet. 2003, 34, 379–381. [Google Scholar] [CrossRef]
- Hofmann Bowman, M.A.; McNally, E.M. Genetic pathways of vascular calcification. Trends Cardiovasc. Med. 2012, 22, 93–98. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Liu, Y.; Wang, X.; New, L.; Han, J.; Brunk, U.T. Activation of the p38 MAP kinase pathway is required for foam cell formation from macrophages exposed to oxidized LDL. APMIS 2002, 110, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.D.; Weiss, R.M.; Heistad, D.D. Calcific aortic valve stenosis: Methods, models, and mechanisms. Circ. Res. 2011, 108, 1392–1412. [Google Scholar] [CrossRef] [PubMed]
- Nam, H.K.; Emmanouil, E.; Hatch, N.E. Deletion of the Pyrophosphate Generating Enzyme ENPP1 Rescues Craniofacial Abnormalities in the TNAP-/- Mouse Model of Hypophosphatasia and Reveals FGF23 as a Marker of Phenotype Severity. Front. Dent. Med. 2022, 3, 846962. [Google Scholar] [CrossRef] [PubMed]
- Orriss, I.R.; Arnett, T.R.; Russell, R.G.G. Pyrophosphate: A key inhibitor of mineralisation. Curr. Opin. Pharmacol. 2016, 28, 57–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hessle, L.; Johnson, K.A.; Anderson, H.C.; Narisawa, S.; Sali, A.; Goding, J.W.; Terkeltaub, R.; Millán, J.L. Tissue-nonspecific alkaline phosphatase and plasma cell membrane glycoprotein-1 are central antagonistic regulators of bone mineralization. Proc. Natl. Acad. Sci. USA 2002, 99, 9445–9449. [Google Scholar] [CrossRef]
- Eaton, R.H.; Moss, D.W. Organic pyrophosphates as substrates for human alkaline phosphatases. Biochem. J. 1967, 105, 1307–1312. [Google Scholar] [CrossRef] [Green Version]
- Rachow, J.W.; Ryan, L.M. Inorganic pyrophosphate metabolism in arthritis. Rheum. Dis. Clin. N. Am. 1988, 14, 289–302. [Google Scholar] [CrossRef]
- Nakashima, K.; Zhou, X.; Kunkel, G.; Zhang, Z.; Deng, J.M.; Behringer, R.R.; de Crombrugghe, B. The Novel Zinc Finger-Containing Transcription Factor Osterix Is Required for Osteoblast Differentiation and Bone Formation. Cell 2002, 108, 17–29. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Liao, H.; Cao, Z. Role of Osterix and MicroRNAs in Bone Formation and Tooth Development. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2016, 22, 2934–2942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, W.; Zhang, D.; Wang, D.; Xu, R.; Tang, L.; Zhao, M.; Xu, R. MicroRNA-638 inhibits human aortic valve interstitial cell calcification by targeting Sp7. J. Cell. Mol. Med. 2019, 23, 5292–5302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.; Ao, L.; Song, Y.; Babu, A.; Yang, X.; Wang, M.; Weyant, M.J.; Dinarello, C.A.; Cleveland, J.C., Jr.; Fullerton, D.A. Expression of functional Toll-like receptors 2 and 4 in human aortic valve interstitial cells: Potential roles in aortic valve inflammation and stenosis. Am. J. Physiol. Cell Physiol. 2008, 294, C29–C35. [Google Scholar] [CrossRef]
- Aikawa, E.; Nahrendorf, M.; Sosnovik, D.; Lok, V.M.; Jaffer, F.A.; Aikawa, M.; Weissleder, R. Multimodality molecular imaging identifies proteolytic and osteogenic activities in early aortic valve disease. Circulation 2007, 115, 377–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirrig, E.E.; Hinton, R.B.; Yutzey, K.E. Differential expression of cartilage and bone-related proteins in pediatric and adult diseased aortic valves. J. Mol. Cell. Cardiol. 2011, 50, 561–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajamannan, N.M.; Subramaniam, M.; Rickard, D.; Stock, S.R.; Donovan, J.; Springett, M.; Orszulak, T.; Fullerton, D.A.; Tajik, A.J.; Bonow, R.O.; et al. Human aortic valve calcification is associated with an osteoblast phenotype. Circulation 2003, 107, 2181–2184. [Google Scholar] [CrossRef]
- Rutsch, F.; Vaingankar, S.; Johnson, K.; Goldfine, I.; Maddux, B.; Schauerte, P.; Kalhoff, H.; Sano, K.; Boisvert, W.A.; Superti-Furga, A.; et al. PC-1 Nucleoside Triphosphate Pyrophosphohydrolase Deficiency in Idiopathic Infantile Arterial Calcification. Am. J. Pathol. 2001, 158, 543–554. [Google Scholar] [CrossRef] [Green Version]
- Ruf, N.; Uhlenberg, B.; Terkeltaub, R.; Nürnberg, P.; Rutsch, F. The mutational spectrum of ENPP1 as arising after the analysis of 23 unrelated patients with generalized arterial calcification of infancy (GACI). Hum. Mutat. 2005, 25, 98. [Google Scholar] [CrossRef]
- Schurgers, L.J.; Cranenburg, E.C.M.; Vermeer, C. Matrix Gla-protein: The calcification inhibitor in need of vitamin K. Thromb. Haemost. 2008, 100, 593–603. [Google Scholar]
- Chiyoya, M.; Seya, K.; Yu, Z.; Daitoku, K.; Motomura, S.; Imaizumi, T.; Fukuda, I.; Furukawa, K.-I. Matrix Gla protein negatively regulates calcification of human aortic valve interstitial cells isolated from calcified aortic valves. J. Pharmacol. Sci. 2018, 136, 257–265. [Google Scholar] [CrossRef]
- Proudfoot, D.; Shanahan, C.M. Molecular mechanisms mediating vascular calcification: Role of matrix Gla protein. Nephrology 2006, 11, 455–461. [Google Scholar] [CrossRef]
- Georgiadi, A.; Lichtenstein, L.; Degenhardt, T.; Boekschoten, M.V.; van Bilsen, M.; Desvergne, B.; Müller, M.; Kersten, S. Induction of cardiac Angptl4 by dietary fatty acids is mediated by peroxisome proliferator-activated receptor beta/delta and protects against fatty acid-induced oxidative stress. Circ. Res. 2010, 106, 1712–1721. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S.; Lichtenstein, L.; Steenbergen, E.; Mudde, K.; Hendriks, H.F.J.; Hesselink, M.K.; Schrauwen, P.; Müller, M. Caloric Restriction and Exercise Increase Plasma ANGPTL4 Levels in Humans via Elevated Free Fatty Acids. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Staiger, H.; Haas, C.; Machann, J.; Werner, R.; Weisser, M.; Schick, F.; Machicao, F.; Stefan, N.; Fritsche, A.; Häring, H.U. Muscle-derived angiopoietin-like protein 4 is induced by fatty acids via peroxisome proliferator-activated receptor (PPAR)-delta and is of metabolic relevance in humans. Diabetes 2009, 58, 579–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, K.; Shimizugawa, T.; Ono, M.; Furukawa, H. Angiopoietin-like protein 4 is a potent hyperlipidemia-inducing factor in mice and inhibitor of lipoprotein lipase. J. Lipid Res. 2002, 43, 1770–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kersten, S. Role and mechanism of the action of angiopoietin-like protein ANGPTL4 in plasma lipid metabolism. J. Lipid Res. 2021, 62, 100150. [Google Scholar] [CrossRef] [PubMed]
- Aryal, B.; Rotllan, N.; Araldi, E.; Ramírez, C.M.; He, S.; Chousterman, B.G.; Fenn, A.M.; Wanschel, A.; Madrigal-Matute, J.; Warrier, N.; et al. ANGPTL4 deficiency in haematopoietic cells promotes monocyte expansion and atherosclerosis progression. Nat. Commun. 2016, 7, 12313. [Google Scholar] [CrossRef] [Green Version]
- Shafi, S.; Codrington, R.; Gidden, L.M.; Ferns, G.A.A. Increased expression of phosphorylated forms of heat-shock protein-27 and p38MAPK in macrophage-rich regions of fibro-fatty atherosclerotic lesions in the rabbit. Int. J. Exp. Pathol. 2016, 97, 56–65. [Google Scholar] [CrossRef]
- Kumar, S.; Boehm, J.; Lee, J.C. p38 MAP kinases: Key signalling molecules as therapeutic targets for inflammatory diseases. Nat. Rev. Drug Discov. 2003, 2, 717–726. [Google Scholar] [CrossRef]
- Eder, K.; Guan, H.; Sung, H.Y.; Francis, S.E.; Crossman, D.C.; Kiss-Toth, E. LDL uptake by monocytes in response to inflammation is MAPK dependent but independent of tribbles protein expression. Immunol. Lett. 2008, 116, 178–183. [Google Scholar] [CrossRef]
- Mei, S.; Gu, H.; Ward, A.; Yang, X.; Guo, H.; He, K.; Liu, Z.; Cao, W. p38 Mitogen-activated Protein Kinase (MAPK) Promotes Cholesterol Ester Accumulation in Macrophages through Inhibition of Macroautophagy. J. Biol. Chem. 2012, 287, 11761–11768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, B.; Wu, X.; Sun, S.; Wu, Q.; Mei, C.; Xu, Q.; Wu, J.; He, P. MAPK–PPARα/γ signal transduction pathways are involved in Chlamydia pneumoniae-induced macrophage-derived foam cell formation. Microb. Pathog. 2014, 69–70, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Masters, K.S. Role of the MAPK/ERK pathway in valvular interstitial cell calcification. Am. J. Physiol. Circ. Physiol. 2009, 296, H1748–H1757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, F.; Li, F.; Li, R.; Liu, Z.; Shi, J.; Zhang, C.; Dong, N. Inhibition of PP2A enhances the osteogenic differentiation of human aortic valvular interstitial cells via ERK and p38 MAPK pathways. Life Sci. 2020, 257, 118086. [Google Scholar] [CrossRef]
- Zernecke, A.; Weber, C. Chemokines in atherosclerosis: Proceedings resumed. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 742–750. [Google Scholar] [CrossRef]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef] [Green Version]
- Liew, F.Y.; Girard, J.-P.; Turnquist, H.R. Interleukin-33 in health and disease. Nat. Rev. Immunol. 2016, 16, 676–689. [Google Scholar] [CrossRef]
- Tsioufis, P.; Theofilis, P.; Tsioufis, K.; Tousoulis, D. The Impact of Cytokines in Coronary Atherosclerotic Plaque: Current Therapeutic Approaches. Int. J. Mol. Sci. 2022, 23, 15937. [Google Scholar] [CrossRef]
- Buckley, M.L.; Williams, J.O.; Chan, Y.-H.; Laubertová, L.; Gallagher, H.; Moss, J.W.E.; Ramji, D.P. The interleukin-33-mediated inhibition of expression of two key genes implicated in atherosclerosis in human macrophages requires MAP kinase, phosphoinositide 3-kinase and nuclear factor-κB signaling pathways. Sci. Rep. 2019, 9, 11317. [Google Scholar] [CrossRef] [Green Version]
- Petrova, T.; Pesic, J.; Pardali, K.; Gaestel, M.; Arthur, J.S.C. p38 MAPK signalling regulates cytokine production in IL-33 stimulated Type 2 Innate Lymphoid cells. Sci. Rep. 2020, 10, 3479. [Google Scholar] [CrossRef] [Green Version]
- Ochayon, D.E.; Ali, A.; Alarcon, P.C.; Krishnamurthy, D.; Kottyan, L.C.; Borchers, M.T.; Waggoner, S.N. IL-33 promotes type 1 cytokine expression via p38 MAPK in human NK cells. J. Leukoc. Biol. 2020, 107, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Junco-Vicente, A.; Solache-Berrocal, G.; del Río-García, Á.; Rolle-Sóñora, V.; Areces, S.; Morís, C.; Martín, M.; Rodríguez, I. IL6 gene polymorphism association with calcific aortic valve stenosis and influence on serum levels of interleukin-6. Front. Cardiovasc. Med. 2022, 9, 989539. [Google Scholar] [CrossRef] [PubMed]
- Humphries, S.; Luong, L.; Ogg, M.; Hawe, E.; Miller, G. The interleukin-6 −174 G/C promoter polymorphism is associated with risk of coronary heart disease and systolic blood pressure in healthy men. Eur. Heart J. 2001, 22, 2243–2252. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Value |
|---|---|
| Age, y | 66 (6) |
| BMI, kg/m2 | 25.15 (5.38) |
| Cholesterol, mg/dL | 186 (65.25) |
| Creatinine, mg/dL | 0.9 (0.3) |
| CRP, mg/dL | 0.2 (0.35) |
| HbA1c, %Hb | 5.8 (0.4) |
| LDL, mg/dL | 79.0 (65.0) |
| N-terminal of the prohormone brain natriuretic peptide, pg/mL | 496.0 (890.5) |
| Gender | |
| Male | 14 (60.87%) |
| Female | 9 (39.13%) |
| Risk Factor | |
| Hypertension | 12 (52.17%) |
| Smoking | 6 (26.09%) |
| Hyperlipidemia | 4 (17.39%) |
| Hypercholesterolemia | 5 (21.74%) |
| Diabetes mellitus | 5 (21.74%) |
| Antigen | Name | Application | Dilution | Source | Company |
|---|---|---|---|---|---|
| p38 MAPK | p38 MAPK (D13E1) mAb | IHC-P WB | 1:1000 1:1000 | Rabbit | Cell Signaling Technology, Danvers, MA, USA |
| p-p38 MAPK | Phospho-p38 MAPK (Thr180/Tyr182) (D3F9) mAb | IHC-P WB | 1:100 1:1000 | Rabbit | Cell Signaling Technology |
| Hsp27 | HSP27 (G31) mAb | IHC-P | 1:200 | Mouse | Cell Signaling Technology |
| p-Hsp27 | Phosopho-HSP27 (Ser82) (D1H2F6) mAb | IHC-P | 1:200 | Rabbit | Cell Signaling Technology |
| IL-6 | IL-6 Ab polyclonal | IHC-P | 1:100 | Rabbit | Affinity Biosciences, Cincinnati, OH, USA |
| IL-33 | IL-33 monoclonal antibody (12B3C4) | IHC-P | 1:100 | Mouse | Invitrogen by Thermo Fisher Scientific, Waltham, MA, USA |
| eLDL | LDL-8 (AIL-3) mAb | IHC-P | 1:2000 | Mouse | Torzewski et al. [19] |
| Vinculin | Vinculin monoclonal antibody (2B5A7) | WB | 1:1000 | Mouse | Proteintech, Planegg-Martinsried, Germany |
| β-actin | Monoclonal anti-β-actin clone AC-15 | WB | 1:1000 | Mouse | Sigma-Aldrich, St. Louis, MO, USA |
| ENPP1 | Ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1) antibody | IHC-P | 1:25 | Rabbit | Antikörper-online (abbexa), Aachen, Germany |
| SPP1 | Osteopontin (SPP1) antibody | IHC-P | 1:500 | Mouse | Antikörper-online (abbexa) |
| Protein | Gene | Assay ID |
|---|---|---|
| p38α MAPK | MAPK14 | Hs01051152_m1 |
| p38β MAPK | MAPK11 | Hs00177101_m1 |
| p38δ MAPK | MAPK13 | Hs00234085_m1 |
| p38γ MAPK | MAPK12 | Hs00268060_m1 |
| GAPDH | GAPDH | Hs02786624_g1 |
| RUNX2 | RUNX2 | HS01047973_m1 |
| BMP-2 | BMP-2 | Hs00154192_m1 |
| SP7 | SP7 | Hs00541729_m1 |
| ENPP1 | ENPP1 | Hs01054040_m1 |
| MGP | MGP | Hs00969490_m1 |
| SPP1 | SPP1 | Hs00959010_m1 |
| ALPL | ALPL | Hs01029144_m1 |
| ANGPTL4 | ANGPTL4 | Hs01101127_m1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Witz, A.; Effertz, D.; Goebel, N.; Schwab, M.; Franke, U.F.W.; Torzewski, M. Pro-Calcifying Role of Enzymatically Modified LDL (eLDL) in Aortic Valve Sclerosis via Induction of IL-6 and IL-33. Biomolecules 2023, 13, 1091. https://doi.org/10.3390/biom13071091
Witz A, Effertz D, Goebel N, Schwab M, Franke UFW, Torzewski M. Pro-Calcifying Role of Enzymatically Modified LDL (eLDL) in Aortic Valve Sclerosis via Induction of IL-6 and IL-33. Biomolecules. 2023; 13(7):1091. https://doi.org/10.3390/biom13071091
Chicago/Turabian StyleWitz, Annemarie, Denise Effertz, Nora Goebel, Matthias Schwab, Ulrich F. W. Franke, and Michael Torzewski. 2023. "Pro-Calcifying Role of Enzymatically Modified LDL (eLDL) in Aortic Valve Sclerosis via Induction of IL-6 and IL-33" Biomolecules 13, no. 7: 1091. https://doi.org/10.3390/biom13071091