
Involvement of Oxidative Stress in the Development of Subcellular Defects and Heart Disease


1
St. Boniface Hospital Albrechtsen Research Centre, Institute of Cardiovascular Sciences, Department of Physiology and Pathophysiology, Max Rady College of Medicine, University of Manitoba, Winnipeg, MB R2H 2A6, Canada
2
Centre of Experimental Medicine, Institute for Heart Research, Slovak Academy of Sciences, Dubravska cesta 9, 84104 Bratislava, Slovakia
3
Department of Pharmacology and Toxicology, Faculty of Pharmacy, Comenius University Bratislava, Odbojarov 10, 83232 Bratislava, Slovakia
*
Author to whom correspondence should be addressed.
Biomedicines 2022, 10(2), 393; https://doi.org/10.3390/biomedicines10020393
Submission received: 12 January 2022
/
Revised: 1 February 2022
/
Accepted: 2 February 2022
/
Published: 7 February 2022
(This article belongs to the Special Issue Oxidative Stress and Inflammation: From Mechanisms to Therapeutic Approaches 3.0)
Abstract
:It is now well known that oxidative stress promotes lipid peroxidation, protein oxidation, activation of proteases, fragmentation of DNA and alteration in gene expression for producing myocardial cell damage, whereas its actions for the induction of fibrosis, necrosis and apoptosis are considered to result in the loss of cardiomyocytes in different types of heart disease. The present article is focused on the discussion concerning the generation and implications of oxidative stress from various sources such as defective mitochondrial electron transport and enzymatic reactions mainly due to the activation of NADPH oxidase, nitric oxide synthase and monoamine oxidase in diseased myocardium. Oxidative stress has been reported to promote excessive entry of Ca2+ due to increased permeability of the sarcolemmal membrane as well as depressions of Na+-K+ ATPase and Na+-Ca2+ exchange systems, which are considered to increase the intracellular of Ca2+. In addition, marked changes in the ryanodine receptors and Ca2+-pump ATPase have been shown to cause Ca2+-release and depress Ca2+ accumulation in the sarcoplasmic reticulum as a consequence of oxidative stress. Such alterations in sarcolemma and sarcoplasmic reticulum are considered to cause Ca2+-handling abnormalities, which are associated with mitochondrial Ca2+-overload and loss of myofibrillar Ca2+-sensitivity due to oxidative stress. Information regarding the direct effects of different oxyradicals and oxidants on subcellular organelles has also been outlined to show the mechanisms by which oxidative stress may induce Ca2+-handling abnormalities. These observations support the view that oxidative stress plays an important role in the genesis of subcellular defects and cardiac dysfunction in heart disease.
1. Introduction
Several clinical and experimental investigations have shown that oxidative stress plays a critical role in the pathogenesis of heart disease [1,2,3,4,5,6,7,8,9,10,11,12,13]. It is also becoming evident that the development of oxidative stress is invariably associated with the occurrence of cardiac dysfunction in different types of cardiovascular diseases such as atherosclerosis, myocardial infarction, hypertension, diabetes and various types of cardiomyopathies [14,15,16,17,18,19,20,21,22,23,24]. These studies have revealed that oxidative stress may induce cardiac remodeling, fibrosis, apoptosis, necrosis, metabolic defects and Ca2+-handling abnormalities in cardiomyocytes as well as endothelial dysfunction. Furthermore, various antioxidants have been observed to exert beneficial effects in improving cardiac function and attenuating heart disease [25,26,27,28,29,30,31]. The present article is focused on updating the existing information regarding the generation and implications of oxidative stress during the development of cardiovascular disorders. It is planned to emphasize the involvement of oxidative stress as a mechanism for inducing cardiac dysfunction as well as abnormalities in subcellular organelles and Ca2+-handling in cardiomyocytes in chronic myocardial infarction. Although oxidative stress is known to modify different proteins in subcellular organelles by affecting cardiac gene expression and various signal transduction mechanisms as well as by activating different proteases, no effort is made to deal with these issues in this article. On the other hand, some evidence is presented in this review to show that oxyradicals and some oxidants exert direct effects on subcellular organelles, and thus may explain the role of oxidative stress in the development of subcellular abnormalities, Ca2+-handling defects and cardiac dysfunction in heart disease.
2. Generation of Oxidative Stress in Heart Disease
It is now well known that the occurrence of oxidative stress is mainly a consequence of excessive formation of reactive oxygen species (ROS) including superoxide radicals and hydroxyl radicals as well as oxidants such as hydrogen peroxide (H2O2) and hypochlorous acid (HOCl) and/or reduction in the activities of endogenous antioxidants such as superoxide dismutase, glutathione peroxidase and catalase [1,32,33,34,35]. The transcription factors such as KLF9 (Kruppel-like factor 9) and Nrf2 (nuclear factor erythroid-2 related factor 2), which control the development of oxidative stress and the expression of antioxidant genes, have been reported to be activated in heart disease, respectively [36,37,38]. It should be pointed out that excessively produced nitric oxide (NO) due to the activation of NO synthase is known to combine with superoxide radicals to form peroxynitrite and produce nitrosative stress [39,40,41]. Furthermore, inflammatory cytokines such as tumor necrosis factor-alpha (TNF-α) have been shown to promote the development of both oxidative stress and nitrosative stress due to the activation of myeloperoxidase (for the generation of superoxide radicals) and NO synthase [42,43,44,45,46]. It is also noteworthy that the production of nitrosative stress is considered to be a major cause for the endothelial dysfunction in heart disease [47,48,49,50]. In fact, both oxidative stress and nitrosative stress have been demonstrated to activate the nuclear enzyme poly (ADP-ribose) polymerase to cause the fragmentation of DNA strands as well as initiate lipid peroxidation, protein oxidation and endothelial dysfunction in diseased myocardium.
Although ROS are considered to be mainly generated as a by-product of defects in mitochondrial metabolism and uncoupling of electron transport in heart disease, several enzymes such as xanthine oxidase, NADPH oxidase, nitric oxidase synthase, myeloperoxidase and monoamine oxidase are also involved in this process [51,52,53,54,55]. ROS production due to impaired electron transport in mitochondria is balanced by mitochondrial antioxidant enzymes such as superoxide dismutase and glutathione peroxidase but is augmented by different chronic pathological conditions [56,57,58,59]. Mitochondrial uncoupling proteins, which promote the leakage of protons across the inner mitochondrial membrane, are considered to be the promoters of mitochondrial ROS generation [58]. Furthermore, mitochondria were shown to integrate ROS signals from other cellular sources and promote the development of oxidative stress through a process termed as “ROS-induced ROS release” involving mitochondrial ion channels [59]. Increased myocardial fatty acid uptake has been shown to promote palmitoyl carnitine oxidation, increase formation of ROS and induce mitochondrial structural remodeling [60]. In addition, exposure of mitochondria for a prolonged period to palmitate was observed to enhance ROS generation associated with mitochondrial fission [60]. Angiotensin II has also been reported to promote ROS generation and produce mitochondrial DNA deletion as well as autophagy in cardiomyocytes [61]. In fact, chronic increase in ROS in mitochondria has been demonstrated to produce mitochondrial oxidative stress, which is associated with mitochondrial DNA damage [62,63]. Overexpression of mitochondrial transcription factor A (TFAM) and genes for mitochondrial antioxidant, peroxiredoxin-3 (PRX-3), were observed to attenuate ROS-induced mitochondrial oxidative stress, DNA deletion and decrease in oxidative phosphorylation activities [64,65].
The family of NADPH oxidase (NOX) are transmembrane proteins, which are involved in the generation of ROS by transferring electrons from NADPH to molecular oxygen, and serve as signaling molecules for inducing cardiac hypertrophy, apoptosis, fibrosis and heart failure [66,67,68]. ROS are released upon the activation of phagocytic NOS by acute myocardial infarction or reperfusion injury whereas abnormal stimulation of nonphagocytic NOS by angiotensin II, catecholamines and TNF-α has been implicated in cardiac hypertrophy collagen deposition, metalloprotease activation, fibrosis and heart failure [69]. The cascade of events triggered by increased activity of NOS includes lipid peroxidation and activation of mitogen-activated protein kinases (ERK 1/2, JNK and p38) for the occurrence of adverse cardiac remodeling [70]. It should be pointed out that NOX is present in different isoforms in multiple cell types; two isoforms (NOX2 and NOX4) are mainly expressed in cardiomyocytes [71,72,73,74]. The NOX2 isoform is localized in the sarcolemmal membrane and plays an important role in mediating angiotensin II-induced cardiac hypertrophy whereas the NOX4 isoform is localized in mitochondria, sarcoplasmic reticulum as well as nucleus and mediates adverse cardiac remodeling and heart failure due to pressure overload [72]. Inhibition of NOX2 was observed to prevent palmitate-induced abnormalities in mitochondrial respiration, ROS generation and Ca2+-overload [74]. Studies in mouse model of NOX4 knockout have revealed that NOX4 contributes to heart failure due to coronary ligation by increasing the inflammatory cytokine levels via enhancing the soluble epoxide hydrolase (a potent regulator of inflammation) [75], and in fact, NOX4 is a major source of mitochondrial ROS production in heart failure due to pressure-overload [76]. Apocynin, an inhibitor of NOX was found to attenuate oxidative stress, cardiomyocyte apoptosis and heart failure due to myocardial infarction in rabbits [77]. Furthermore, this agent ameliorated cardiac dysfunction, attenuated cardiac fibrosis and restored defects in Ca2+-handling activities of the sarcoplasmic reticulum in rabbits subjected to combined volume and pressure overload [78].
Another major source of ROS production is a monoamine oxidase (MAO), which participates in degradation of neurotransmitters such as norepinephrine, epinephrine and dopamine as well as serotonin [79,80]. This enzyme is localized in mitochondria and is involved in the production of H2O during the process of oxidative breakdown of catecholamines and serotonin. It should be noted that the levels of plasma catecholamines and serotonin are elevated in different types of heart disease and these may partly generate ROS during their degradation by MAO. In fact, MAO-A has been shown to play a key role in the development of acute and chronic heart diseases [81]. Exposure of cardiomyocytes to MAO-A has been reported to block autophagic flux with the accumulation of LC311, p62 and ubiquitylated proteins leading to mitochondrial fission and cellular necrosis [82]. Furthermore, the activation of MAO-A was found to result in the accumulation of lysosomal proteins (cathepsin D and Lamp 1), reduction in lysosomal acidification and blockade of the nuclear translocation of transcription factor-EB (TFEB), a regulator of autophagy and lysosome biogenesis [82]. It is also pointed out that MAO was found to be overactivated in ischemic heart disease [83], and its enhanced activity was shown to contribute to adverse cardiac remodeling and heart failure due to pressure overload [84]. Furthermore, the activation of MAO has been observed to depress mitochondrial function and result in heart failure as a consequence of oxidative stress in chronic diabetes [85].
3. Implications of Oxidative Stress in Heart Disease
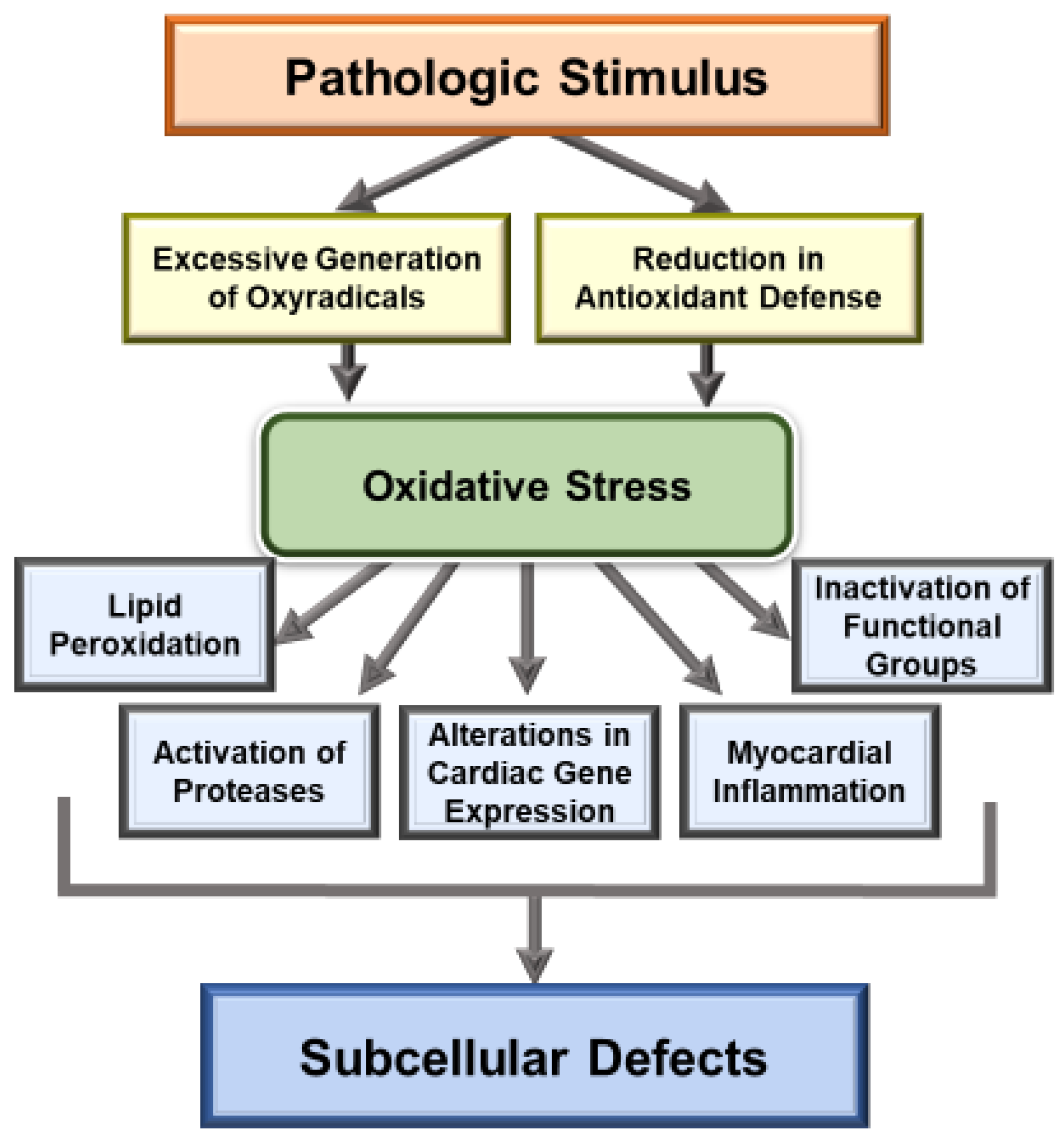
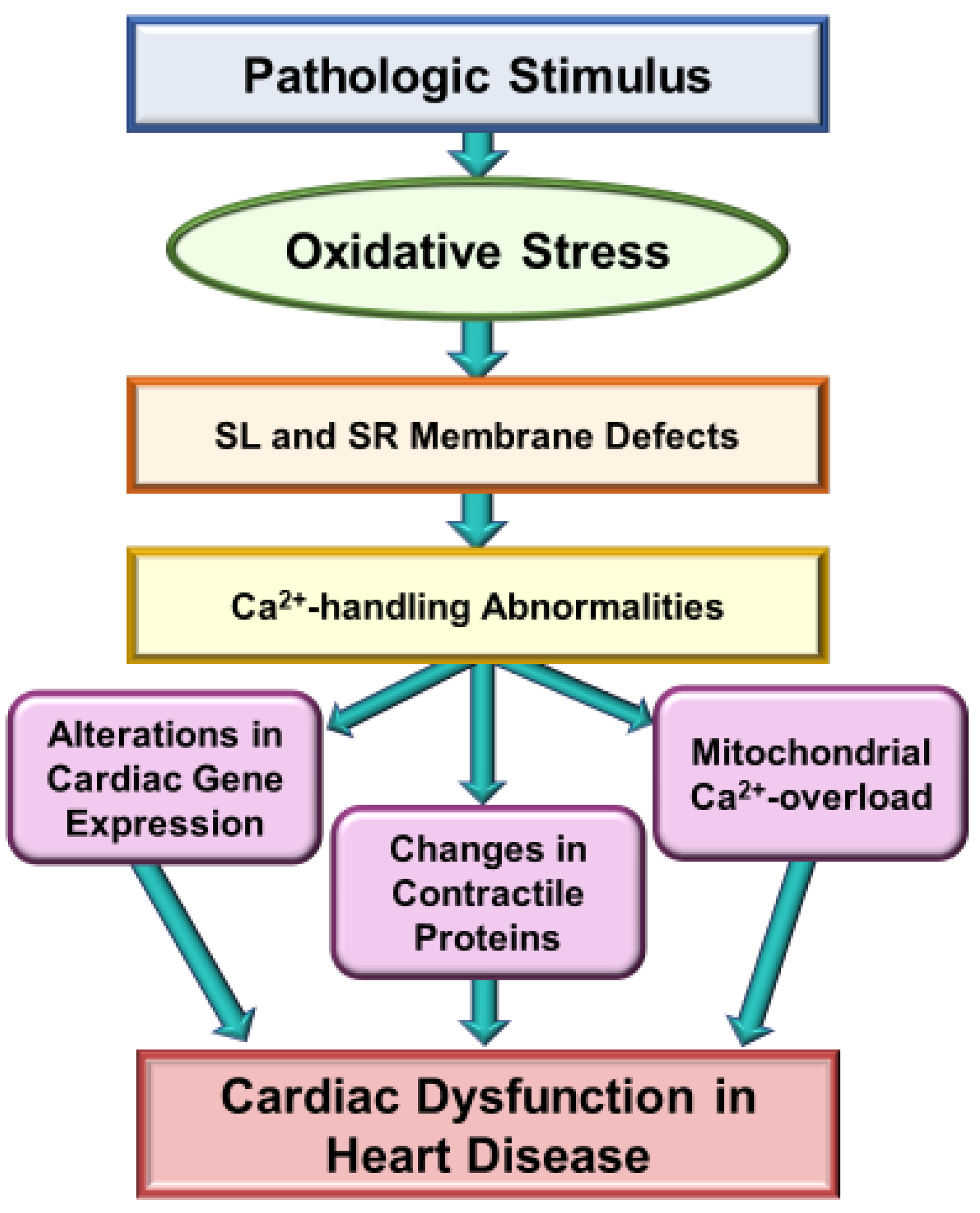
Extensive research over the past several decades has revealed that oxidative stress may be a major mechanism for the genesis of cardiac dysfunction, subcellular defects and Ca2+-handling abnormalities in diverse cardiovascular disorders [1,86,87,88,89,90]. Such a role of both oxidative stress and nitrosative stress is mainly based on observations regarding the association of increased levels of various biomarkers of these stress factors and defects in subcellular organelles such as sarcolemma, sarcoplasmic reticulum, mitochondria and myofibrils [91,92,93,94]. Particularly, there occurs an increase in the level of inflammatory cytokines, depression in the activities of endogenous antioxidant enzymes and down-regulation of antioxidant defense mechanisms, including the Nrf2 pathway, which are known to promote the development of oxidative stress in diseased myocardium [1,35,38,42,43,44]. It is pointed out that the relationship of oxidative stress and inflammatory cytokines is of complex nature as both of these are known to promote the formation of each other in the diseased myocardium. Nonetheless, different antioxidant agents such as several vitamins, resveratrol and pterostilbene, as well as activation of the Nrf2-associated antioxidant pathway, have been shown to improve cardiac function, subcellular defects and Ca2+-handling abnormalities in heart disease [31,95,96,97,98,99]. Some of the major mechanisms for the induction of subcellular defects due to oxidative stress are depicted in Figure 1 and whereas those for the occurrence of Ca2+-handling abnormalities and cardiac dysfunction in heart disease are shown in Figure 2.
It is now well known that oxidative stress increases the intracellular Ca2+ in cardiomyocytes by promoting the entry of Ca2+ upon affecting the sarcolemmal membrane as well as by inducing changes in Ca2+-release and Ca2+-uptake activities in the sarcoplasmic reticulum under different pathological conditions [1,91]. Exposure of the heart to high levels of circulating angiotensin II or catecholamines for a prolonged period as well as in chronic myocardial infarction have been shown to induce abnormal Ca2+ -handling associated with mitochondrial Ca2+-overload, depression of mitochondrial function, generation of oxidative stress, activation of proteases, fragmentation of DNA and impaired cardiovascular function [1,91,95]. The loss of myofibrillar Ca2+-sensitivity due to prolonged oxidative stress and Ca2+-handling abnormalities is considered to explain cardiac dysfunction as a consequence of myofilament derangements and myofibrillar degeneration [91,99,100]. The oxidation of myofibrillar proteins such as actin, myosin and troponin has been shown to be accompanied by depressed ATPase activities as a consequence of oxidative stress [91,101]. Prolonged inhibition of xanthine oxidase was demonstrated to prevent myofibrillar oxidation and preserve cardiac function in a transgenic model of cardiomyopathy [102]. Failing hearts due to myocardial infarction exhibited a marked depression in myofibrillar Ca2+-stimulated ATPase activity due to the modification of myosin gene expression as a consequence of both oxidative stress and Ca2+-handling abnormalities in cardiomyocytes [91].
While excessive entry of extracellular Ca2+ through the sarcolemmal membrane has been shown to occur for the development of Ca2+-handling abnormalities in heart failure due to myocardial infarction, the exact contribution of Ca2+-influx and Ca2+-efflux mechanisms has not been fully established [91]. In this regard, the density of voltage dependent Ca2+-channels, which are associated with beat-to-heat Ca2+-influx through sarcolemma, was decreased but the activity of sarcolemmal Ca2+-pump, which is involved in Ca2+-efflux, was unaltered in the failing heart [1,91]. On the other hand, marked depression in sarcolemmal Na+-K+ ATPase in the infarcted heart has indicated that the Ca2+ entry in cardiomyocytes may increase indirectly through the Na+-Ca2+ exchange mechanism. In fact, the sarcolemmal Na+-Ca2+ exchange activity was also decreased, which may impair Ca2+-efflux (via the reverse mode) and contribute in the development of Ca2+-handling abnormalities in failing cardiomyocyte [103]. Furthermore, depression of the sarcolemmal Na+-K+ ATPase activity by cardiac glycosides has been shown to cause abnormal Ca2+-cycling and impair mitochondrial energetics in guinea pig cardiomyocytes [104]. Although oxidative stress can be seen to induce Ca2+-entry through the sarcolemmal membrane by increasing its permeability due to lipid peroxidation, changes in the sarcolemmal phospholipid composition by other mechanisms cannot be overlooked. Particularly, different oxyradical generating systems have been shown to decrease phospholipid N-methylation, which is known to determine the membrane fluidity [105]. Furthermore, oxidative stress has been demonstrated to modify the sarcolemmal activities of both phospholipases C and D, which may directly or indirectly participate in the occurrence of Ca2+-handling abnormalities in cardiomyocytes [91,106].
It needs to be emphasized that mitochondria is not only a major source for the production of cellular ATP but is also a major contributor for the production of oxyradicals in cardiomyocytes, and these processes are regulated by the intracellular concentration of Ca2+ [107,108]. Ca2+-handling abnormalities associated with oxidative stress are known to induce mitochondrial Ca2+-overload and impair mitochondrial function for the production of energy [109,110,111]. On the other hand, an increase in the mitochondrial ATPase inhibitory factor-1 due to oxidative stress has been shown to disrupt mitochondrial Ca2+-handling whereas a loss of mitochondrial Ca2+ uniporter has been reported to trigger arrhythmias possibly by affecting the Ca2+-handling function of the sarcoplasmic reticulum [112,113]. It is pointed out that the sarcoplasmic reticulum, by virtue of its ability to release and accumulate Ca2+ on a beat-to-heat basis, is known to play a major role in Ca2+-handling in cardiomyocytes, and has been indicated to serve as a critical target for oxidative stress. Both oxidative stress and nitrosative stress have been demonstrated to promote Ca2+-leak by modifying ryanodine receptors in the sarcoplasmic reticulum in various types of failing hearts [114,115,116]. Alterations in the sarcoplasmic reticulum Ca2+-pump ATPase and ryanodine receptors in heart failure are considered to occur due to oxidative stress as these were corrected by an antioxidant, edaravone, as well as by carvedilol (due to its antioxidant properties) [117,118].
4. Evidence for the Direct Action of Oxidative Stress on Subcellular Organelles
Although the development of oxidative stress has been shown to be associated with subcellular defects for the occurrence of Ca2+-handling abnormalities and subsequent cardiac dysfunction, it is not clear whether these effects of oxidative stress on subcellular organelles are of any direct or indirect nature. In order to gain some information in this regard, sarcolemmal membranes, sarcoplasmic reticulum, mitochondria and myofibrils were isolated from control hearts; these subcellular organelles were incubated with different oxyradical generating systems and oxidants for 30 min and their activities were determined. The results in Table 1 indicate that the incubation of sarcolemma with superoxide radical generating mixture, H2O2 and hydroxyl radical generating mixture depressed Na+-K+ ATPase and Na+-Ca2+ exchange activities. These changes were associated with increased malondialdehyde (MDA) content and reduced sulfhydryl groups (SH-groups). The effects of superoxide radicals were attenuated by superoxide dismutase (SOD), the effects of H2O2 were attenuated by catalase and those of hydroxyl radicals were reduced by mannitol [119,120]. The sarcolemmal ATP-dependent Ca2+ uptake activity, Ca2+-stimulated ATPase activity and Mg2+-ATPase activity were also depressed by superoxide radicals, H2O2 and hydroxyl radicals and these effects were attenuated by their scavengers, SOD, catalase and mannitol, respectively (Table 2) [120]. Furthermore, the density, unlike the affinity of Ca2+-binding and both low and high affinities of ATP-binding, were depressed whereas the activity of Ca2+-ecto ATPase (which serves as a Ca2+-gating mechanism) was increased by superoxide radicals, H2O2 and hydroxyl radicals (Table 3) [121,122]. Superoxide radicals and H2O2 were also observed to depress the sarcoplasmic reticulum Ca2+-release, Ca2+-uptake and Ca2+-pump ATPase activities whereas the myofibrillar Ca2+-stimulated ATPase activity and SH-group content were reduced and Mg2+-ATPase activity was increased (Table 4) [123,124,125]. Furthermore, both superoxide radicals and H2O2 depressed mitochondrial state 3 respiration, RCI value and ADP-to-O ratio, indicating impaired mitochondrial function (Table 4) [126]. The effects of incubating sarcolemma and myofibrils with a potent oxidant, HOCl, are shown in Table 5 [125,127]. The depressions in sarcolemmal Na+-K+ ATPase as well as SH-group content and increase in MDA content by HOCl were attenuated by the presence of its scavenger, methionine [127]. Furthermore, HOCl was observed to increase myofibrillar Mg2+-ATPase and decrease Ca2+-stimulated ATPase activities, these effects of HOCl were attenuated by methionine [125]. It is pointed out that most of the changes in subcellular activities induced by different oxyradical generating systems and oxidants under in vitro conditions are similar to those seen in failing hearts due to chronic myocardial infarction. Furthermore, the present in vitro observations indicate that the activities of different subcellular organelles are affected directly by oxyradicals and oxidants, thus, support the view that oxidative stress can induce subcellular organelles and Ca2+-handling abnormalities in cardiomyocytes for the occurrence of cardiac dysfunction in heart disease.
5. Concluding Remarks
An in-depth analysis of the literature regarding the pathophysiology and pharmacotherapy of different types of cardiovascular diseases has revealed that oxidative stress is one of the most critical factors, which is involved in the pathogenesis of cardiac dysfunction. It is also evident that the occurrence of oxidative stress represents a disbalance between the excessive formation of oxyradicals and the activities of antioxidant defense mechanisms. Furthermore, it has been demonstrated that the generation of oxyradicals in diseased myocardium is a consequence of defects in the mitochondrial electron transport system as well as the by-product of reactions involving different enzymes including NADPH oxidase, xanthine oxidase, nitric oxide synthase and monoamine oxidase. Although the contribution of each source to produce oxidative stress is not clear, it seems that the involvement of each source for the generation of oxyradicals may differ from one disease to the other and may be specific for the stage and type of heart disease. In addition, oxidative stress has been shown to produce a wide variety of changes in various metabolic pathways and signal transduction systems for the induction of fibrosis, necrosis and apoptosis as well as the depression of cardiac gene expression and activation of different proteolytic enzymes in heart disease. However, alterations in subcellular organelles such as sarcolemma, sarcoplasmic reticulum, mitochondria and myofibrils may be more related to the development of Ca2+-handling abnormalities and cardiac dysfunction due to oxidative stress in heart disease. Particularly, there occurs an excessive entry of Ca2+ due to increased membrane permeability (as a consequence of changes in lipid composition and activation of the Ca2+-gating mechanism) as well as depression in the sarcolemmal Na+-K+ ATPase and Na+-Ca2+ exchange systems due to oxidative stress. Furthermore, oxidative stress promotes the release of Ca2+ (due to defect in ryanodine receptors) and depresses Ca2+-uptake (due to changes in Ca2+-pump ATPase) in the sarcoplasmic reticulum. Such Ca2+-handling alterations in both sarcolemma and sarcoplasmic reticulum can be seen to induce mitochondrial Ca2+-overload and depress the process of energy production. Prolonged Ca2+-handling abnormalities in diseased myocardium will also cause derangements of myofilaments and loss of myofibrillar Ca2+-sensitivity. Taken together, all these defects in subcellular organelles can be seen to result in cardiac dysfunction due to the development of oxidative stress in heart disease. It should be emphasized that it is not our intention to exclude the participation of several other pathogenic factors in the pathogenesis of cardiac dysfunction but the information in this article provides evidence in support of the concept that oxidative stress may induce cardiac dysfunction due to subcellular defects and Ca2+-handling abnormalities in heart disease.
Funding
This research was supported by the Slovak Research and Development Agency and the Ministry of Education, Science, Research and Sport of the Slovak Republic, and was funded by grants APVV-15-0607, APVV-20-0242, VEGA 1/0016/20.
Acknowledgments
The infrastructural support for this project was provided by the St. Boniface Hospital Albrechtsen Research Centre. We wish to thank Andrea Opsima for typing this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dhalla, N.S.; Temsah, R.M.; Netticadan, T. Role of oxidative stress in cardiovascular diseases. J. Hypertens 2000, 18, 655–673. [Google Scholar] [CrossRef] [PubMed]
- Takano, H.; Zou, Y.; Hasegawa, H.; Akazawa, H.; Nagai, T.; Komuro, I. Oxidative stress-induced signal transduction pathways in cardiac myocytes: Involvement of ROS in heart diseases. Antioxid. Redox. Signal 2003, 5, 789–794. [Google Scholar] [CrossRef] [PubMed]
- Molavi, B.; Mehta, J.L. Oxidative stress in cardiovascular disease: Molecular basis of its deleterious effects, its detection, and therapeutic considerations. Curr. Opin. Cardiol. 2004, 19, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Kusano, K.F.; Matsubara, H.; Nakamura, Y.; Miura, A.; Nishii, N.; Banba, K.; Nagase, S.; Miyaji, K.; Morita, H.; et al. Relationship between oxidative stress and systolic dysfunction in patients with hypertrophic cardiomyopathy. J. Card. Fail. 2005, 11, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Vijaya Lakshmi, S.V.; Padmaja, G.; Kuppusamy, P.; Kutala, V.K. Oxidative stress in cardiovascular disease. Indian J. Biochem. Biophys. 2009, 46, 421–440. [Google Scholar]
- Hecker, P.A.; Galvao, T.F.; O’Shea, K.M.; Brown, B.H.; Henderson Jr, R.; Riggle, H.; Gupte, S.A.; Stanley, W. High-sugar intake does not exacerbate metabolic abnormalities or cardiac dysfunction in genetic cardiomyopathy. Nutrition 2012, 28, 520–526. [Google Scholar] [CrossRef] [Green Version]
- Munzel, T.; Gori, T.; Keaney Jr, J.F.; Maack, C.; Daiber, A. Pathophysiological role of oxidative stress in systolic and diastolic heart failure and its therapeutic implications. Eur. Heart J. 2015, 36, 2555–2564. [Google Scholar] [CrossRef] [Green Version]
- Munzel, T.; Camici, G.G.; Maack, C.; Bonetti, N.R.; Fuster, V.; Kovacic, J.C. Impact of oxidative stress on the heart and vasculature: Part 2 of a 3-part series. J. Am. Coll. Cardiol. 2017, 70, 212–229. [Google Scholar] [CrossRef]
- Zhou, B.; Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Investig. 2018, 128, 3716–3726. [Google Scholar] [CrossRef] [Green Version]
- Steinhorn, B.; Sorrentino, A.; Badole, S.; Bogdanova, Y.; Belousov, V.; Michel, T. Chemogenetic generation of hydrogen peroxide in the heart induces severe cardiac dysfunction. Nat. Commun. 2018, 9, 4044. [Google Scholar] [CrossRef]
- Romuk, E.; Wojciechowska, C.; Jachec, W.; Nowak, J.; Niedziela, J.; Malinowska-Borowska, J.; Glogowska-Gruszka, A.; Birkner, W.; Rozentryt, P. Comparison of oxidative stress parameters in heart failure patients depending on ischaemic or nonischaemic aetiology. Oxid. Med. Cell. Longev. 2019, 2019, 7156038. [Google Scholar] [CrossRef] [PubMed]
- Fabiani, I.; Aimo, A.; Grigoratos, C.; Castiglione, V.; Gentile, F.; Saccaro, L.F.; Arzilli, C.; Cardinale, D.; Passino, C.; Emdin, M. Oxidative stress and inflammation: Determinants of anthracycline cardiotoxicity and possible therapeutic targets. Heart Fail. Rev. 2021, 26, 881–890. [Google Scholar] [CrossRef]
- Du, Y.; Demillard, L.J.; Ren, J. Catecholamine-induced cardiotoxicity: A critical element in the pathophysiology of stroke-induced heart injury. Life Sci. 2021, 287, 120106. [Google Scholar] [CrossRef]
- Hafstad, A.D.; Nabeebaccus, A.A.; Shah, A.M. Novel aspects of ROS signaling in heart failure. Basic Res. Cardiol. 2013, 108, 359. [Google Scholar] [CrossRef] [PubMed]
- Dekleva, M.; Celic, V.; Pencic, B.; Ivanovic, A.M.; Caparevic, Z. Left ventricular diastolic dysfunction is related to oxidative stress and exercise capacity in hypertensive patients with preserved systolic function. Cardiology 2007, 108, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, J.; Lu, L.; Chen, S.S.; Quinn, M.T.; Weber, K.T. Aldosterone-induced inflammation in the rat heart: Role of oxidative stress. Am. J. Pathol. 2002, 161, 1773–1781. [Google Scholar] [CrossRef]
- Wold, L.E.; Ceylan-Isik, A.F.; Ren, J. Oxidative stress and stress signaling: Menace of diabetic cardiomyopathy. Acta Pharmacol. Sin. 2005, 26, 908–917. [Google Scholar] [CrossRef]
- Kayama, Y.; Raaz, U.; Jagger, A.; Matti, A.; Schellinger, I.N.; Sakamoto, M.; Suzuki, H.; Toyama, K.; Spin, J.M.; Tsao, P.S. Diabetic cardiovascular disease induced by oxidative stress. Int. J. Mol. Sci. 2015, 16, 25234–25263. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, Y.; Dong, S.; Zhang, A.; Lu, Y.; Li, Y.; Lv, S.; Zhang, J. Role of oxidative stress in cardiotoxicity of antineoplastic drugs. Life Sci. 2019, 232, 116526. [Google Scholar] [CrossRef]
- Sterba, M.; Popelova, O.; Vavrova, A.; Jirkovsky, E.; Kovarikova, P.; Gersl, V.; Simunek, T. Oxidative stress, redox signaling, and metal chelation in anthracycline cardiotoxicity and pharmacological cardioprotection. Antioxid. Redox. Signal 2013, 18, 899–929. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Enjamoori, R.; Jaiswal, A.; Ray, R.; Seth, S.; Maulik, S.K. Catecholamine-induced myocardial fibrosis and oxidative stress is attenuated by Terminalia arjuna (roxb.). J. Pharm. Pharmacol. 2009, 61, 1529–1536. [Google Scholar] [CrossRef]
- Costa, V.M.; Carvalho, F.; Bastos, M.L.; Carvalho, R.A.; Remiao, F. Contribution of catecholamine reactive intermediates and oxidative stress to the pathologic features of heart diseases. Curr. Med. Chem. 2011, 18, 2272–2314. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, T.; Iwase, M.; Kanazawa, H.; Ichihara, S.; Ichihara, G.; Nagata, K.; Obata, K.; Kitaichi, K.; Yokoi, T.; Watanabe, M.; et al. Serial alterations of beta-adrenergic signaling in dilated cardiomyopathic hamsters: Possible role of myocardial oxidative stress. Circ. J. 2004, 68, 1051–1060. [Google Scholar] [CrossRef] [Green Version]
- Escobales, N.; Crespo, M.J. Angiotensin II-dependent vascular alterations in young cardiomyopathic hamsters: Role for oxidative stress. Vascul. Pharmacol. 2006, 44, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Bruckdorfer, K.R. Antioxidants and CVD. Proc. Nutr. Soc. 2008, 67, 214–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, A.K.; Mehra, N.K.; Swarnakar, N.K. Role of antioxidant for the treatment of cardiovascular diseases: Challenges and opportunities. Curr. Pharm. Des. 2015, 21, 4441–4455. [Google Scholar] [CrossRef]
- Mattera, R.; Benvenuto, M.; Giganti, M.G.; Tresoldi, I.; Pluchinotta, F.R.; Bergante, S.; Tettamanti, G.; Masueilli, L.; Manzari, V.; Modesti, A.; et al. Effects of polyphenols on oxidative stress-mediated injury in cardiomyocytes. Nutrients 2017, 9, 523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riba, A.; Deres, L.; Sumegi, B.; Toth, K.; Szabados, E.; Halmosi, R. Cardioprotective effect of resveratrol in a postinfarction heart failure model. Oxid. Med. Cell. Longev. 2017, 2017, 6819281. [Google Scholar] [CrossRef]
- Reyes, D.R.A.; Gomes, M.J.; Rosa, C.M.; Pagan, L.U.; Damatto, F.C.; Damatto, R.L.; Depra, I.; Campos, D.H.; Fernandez, A.A.H.; Martinez, P.F.; et al. N-acetylcysteine influence on oxidative stress and cardiac remodeling in rats during transition from compensated left ventricular hypertrophy to heart failure. Cell Physiol. Biochem. 2017, 44, 2310–2321. [Google Scholar] [CrossRef]
- Bunsawat, K.; Ratchford, S.M.; Alpenglow, J.K.; Park, S.H.; Jarett, C.L.; Stehlik, J.; Drakos, S.G.; Richardson, R.S.; Wray, D.W. Chronic antioxidant administrations restores macrovascular function in patients with heart failure with reduced ejection fraction. Exp. Physiol. 2020, 105, 1384–1395. [Google Scholar] [CrossRef]
- Bartekova, M.; Adameova, A.; Gorbe, A.; Ferenczyova, K.; Pechanova, O.; Lazou, A.; Dhalla, N.S.; Ferdinandy, P.; Giricz, Z. Natural and synthetic antioxidants targeting cardiac oxidative stress and redox signaling in cardiometabolic diseases. Free Radic. Biol. Med. 2021, 169, 446–477. [Google Scholar] [CrossRef] [PubMed]
- Seddon, M.; Looi, Y.H.; Shah, A.M. Oxidative stress and redox signalling in cardiac hypertrophy and heart failure. Heart 2007, 93, 903–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreckenberg, R.; Rebelo, M.; Deten, A.; Weber, M.; Rohrbach, S.; Pipicz, M.; Csonka, C.; Ferdinandy, P.; Schluter, K.D. Specific mechanisms underlying right heart failure: The missing upregulation of superoxide dismutase-2 and its decisive role in antioxidant defense. Antioxid. Redox. Signal 2015, 23, 1220–1232. [Google Scholar] [CrossRef]
- Shiomi, T.; Tsutsui, H.; Matsusaka, H.; Murakami, K.; Hayashidani, S.; Ikeuchi, M.; Wen, J.; Kubota, T.; Utsumi, H.; Takeshita, A. Overexpression of glutathione peroxidase prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation 2004, 109, 544–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khaper, N.; Kaur, K.; Li, T.; Farahmand, F.; Singal, P.K. Antioxidant enzyme gene expression in congestive heart failure following myocardial infarction. Mol. Cell. Biochem. 2003, 251, 9–15. [Google Scholar] [CrossRef]
- Yan, Q.; He, B.; Hao, G.; Liu, Z.; Tang, J.; Fu, Q.; Jiang, C.X. KLF9 aggravates ischemic injury in cardiomyocytes through augmenting oxidative stress. Life Sci. 2019, 233, 116641. [Google Scholar] [CrossRef]
- Wang, W.; Li, S.; Wang, H.; Li, B.; Shao, L.; Lai, Y.; Horvath, G.; Wang, Q.; Yamamoto, M.; Janicki, J.S.; et al. Nrf2 enhances myocardial clearance of toxic ubiquitinated proteins. J. Mol. Cell. Cardiol. 2014, 72, 305–315. [Google Scholar] [CrossRef] [Green Version]
- Tian, C.; Gao, L.; Zimmerman, M.C.; Zucker, I.H. Myocardial infarction-induced microRNA-enriched exosomes contribute to cardiac Nrf2 dysregulation in chronic heart failure. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H928–H939. [Google Scholar] [CrossRef]
- Ungvari, Z.; Gupte, S.A.; Recchia, F.A.; Batkai, S.; Pacher, P. Role of oxidative-nitrosative stress and downstream pathways in various forms of cardiomyopathy and heart failure. Curr. Vasc. Pharmacol. 2005, 3, 221–229. [Google Scholar] [CrossRef]
- Schiattarella, G.G.; Altamirano, F.; Tong, D.; French, K.M.; Villalobos, E.; Kim, S.Y.; Luo, X.; Jiang, N.; May, H.I.; Wang, Z.V.; et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature 2019, 568, 351–356. [Google Scholar] [CrossRef]
- Pacher, P.; Schulz, R.; Liaudet, L.; Szabo, C. Nitrosative stress and pharmacological modulation of heart failure. Trends Pharmacol. Sci. 2005, 26, 302–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neri, M.; Fineschi, V.; Di Paolo, M.; Pomara, C.; Riezzo, I.; Turillazzi, E.; Cerretani, D. Cardiac oxidative stress and inflammatory cytokines response after myocardial infarction. Curr. Vasc. Pharmacol. 2015, 13, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Valgimigli, M.; Merli, E.; Malagutti, P.; Soukhomovskaia, O.; Cicchitelli, G.; Antelli, A.; Canistro, D.; Francolini, G.; Macri, G.; Mastrorilli, F.; et al. Hydroxyl radical generation, levels of tumor necrosis factor-alpha, and progression to heart failure after acute myocardial infarction. J. Am. Coll. Cardiol. 2004, 43, 2000–2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari, R.; Guardigli, G.; Mele, D.; Percoco, G.F.; Ceconi, C.; Curello, S. Oxidative stress during myocardial ischemia and heart failure. Curr. Pharm. Des. 2004, 10, 1699–1711. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, F.; Zhou, H.; Hu, Y.; Guo, D.; Fang, X.; Chen, Y. Interplay of TNF-α, soluble TNF receptors and oxidative stress in coronary chronic total occlusion of the oldest patients with coronary heart disease. Cytokine 2020, 125, 154836. [Google Scholar] [CrossRef] [PubMed]
- Eleuteri, E.; Magno, F.; Gnemmi, I.; Carbone, M.; Colombo, M.; La Rocca, G.; Anzalone, R.; Genta, F.T.; Zummo, G.; Di Stefano, A.; et al. Role of oxidative stress and nitrosative stress biomarkers in chronic heart failure. Front. Biosci. 2009, 14, 2230–2237. [Google Scholar] [CrossRef] [Green Version]
- Huet, O.; Dupic, L.; Harrois, A.; Duranteau, J. Oxidative stress and endothelial dysfunction during sepsis. Front. Biosci. 2011, 16, 1986–1995. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Davidoff, M.N. Oxidative stress and endothelial dysfunction in heart failure. Congest. Heart Fail. 2002, 8, 165–172. [Google Scholar] [CrossRef]
- Indik, J.H.; Goldman, S.; Gaballa, M.A. Oxidative stress contributes to vascular endothelial dysfunction in heart failure. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H1767–H1770. [Google Scholar] [CrossRef]
- Warnholtz, A.; Wendt, M.; August, M.; Munzel, T. Clinical aspects of reactive oxygen and nitrogen. Biochem. Soc. Symp. 2004, 71, 121–133. [Google Scholar] [CrossRef]
- Dubois-Deruy, E.; Peugnet, V.; Turkieh, A.; Pinet, F. Oxidative stress in cardiovascular diseases. Antioxidants 2020, 9, 864. [Google Scholar] [CrossRef] [PubMed]
- Okonko, D.O.; Shah, A.M. Mitochondrial dysfunction and oxidative stress in CHF. Nat. Rev. Cardiol. 2015, 12, 6–8. [Google Scholar] [CrossRef]
- Anatoliotakis, N.; Deftereos, S.; Bouras, G.; Giannopoulos, G.; Tsounis, D.; Angelidis, C.; Koukis, A.; Stefanadis, C. Myeloperoxidase: Expressing inflammation and oxidative stress in cardiovascular disease. Curr. Top. Med. Chem. 2013, 13, 115–138. [Google Scholar] [CrossRef] [PubMed]
- Heymes, C.; Bendall, J.K.; Ratajczak, P.; Cave, A.C.; Jane-Lise, S.; Hasenfuss, G.; Shah, A.M. Increased myocardial NADPH oxidase activity in human heart failure. J. Am. Coll. Cardiol. 2003, 41, 2164–2171. [Google Scholar] [CrossRef] [Green Version]
- Lauderic, N.; Mialet-Perez, J.; Paolocci, N.; Parini, A.; Di Lisa, F. Monoamine oxidases as sources of oxidants in the heart. J. Mol. Cell. Cardiol. 2014, 73, 34–42. [Google Scholar] [CrossRef] [Green Version]
- Sawyer, D.B.; Colucci, W.S. Mitochondrial oxidative stress in heart failure. Circ. Res. 2000, 86, 119–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayeva, M.; Ardehali, H. Mitochondrial dysfunction and oxidative damage to sarcomeric proteins. Curr. Hypertens. Rep. 2010, 12, 426–432. [Google Scholar] [CrossRef]
- Akhmendov, A.T.; Rybin, V.; Marin-Garcia, J. Mitochondrial oxidative metabolism and uncoupling proteins in the failing heart. Heart Fail. Rev. 2015, 20, 227–249. [Google Scholar] [CrossRef]
- Nickel, A.; Kohlhass, M.; Maack, C. Mitochondrial reactive oxygen species production and elimination. J. Mol. Cell. Cardiol. 2014, 73, 26–33. [Google Scholar] [CrossRef]
- Tsuhima, K.; Bugger, H.; Wende, A.R.; Soto, J.; Jenson, G.A.; Tor, A.R.; McGlauflin, R.; Kenny, H.C.; Zhang, Y.; Souvenir, R.; et al. Mitochondrial reactive oxygen species in lipotoxic hearts induce post-translational medicine modifications of AKAP121, DRP1, and OAP1 that promote mitochondrial fission. Circ. Res. 2018, 122, 58–73. [Google Scholar] [CrossRef]
- Dai, D.F.; Johnson, S.C.; Villarin, J.J.; Chin, M.T.; Nieves-Cintron, M.; Chen, T.; Marcinek, D.J.; Dorn 2nd, G.W.; Kang, Y.J.; Parolla, T.A.; et al. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ. Res. 2011, 108, 837–846. [Google Scholar] [CrossRef] [Green Version]
- Kang, D.; Hamasaki, N. Mitochondrial oxidative stress and mitochondrial DNA. Clin. Chem. Lab. Med. 2003, 41, 1281–1288. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and mitochondrial DNA damage in heart failure. Circ. J. 2008, 72 (Suppl. A), A31–A37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsutsui, H. Mitochondrial oxidative stress and heart failure. Intern. Med. 2006, 45, 809–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Mitochondrial oxidative stress and dysfunction in myocardial remodelling. Cardiovasc. Res. 2009, 81, 449–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murdoch, C.E.; Zhang, M.; Cave, A.C.; Shah, A.M. NADPH oxidase-dependent redox signalling in cardiac hypertrophy, remodelling and failure. Cardiovasc. Res. 2006, 71, 208–215. [Google Scholar] [CrossRef] [Green Version]
- Harrison, D.G.; Cai, H.; Landmesser, U.; Griendling, K.K. Interactions of angiotensin II with NAD(P)H oxidase, oxidant stress and cardiovascular disease. J. Renin Angiotensin Aldosterone Syst. 2003, 4, 51–61. [Google Scholar] [CrossRef]
- Cave, A.; Grieve, D.; Johar, S.; Zhang, M.; Shah, A.M. NADPH oxidase-derived reactive oxygen species in cardiac pathophysiology. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2005, 360, 2327–2334. [Google Scholar] [CrossRef] [Green Version]
- Sorescu, D.; Griendling, K.K. Reactive oxygen species, mitochondria, and NAD(P)H oxidases in the development and progression of heart failure. Congest. Heart Fail. 2002, 8, 132–140. [Google Scholar] [CrossRef]
- Nediani, C.; Borchi, E.; Giordano, C.; Baruzzo, S.; Pnziani, V.; Sebastiani, M.; Nassi, P.; Mugelli, A.; d’Amati, G.; Cerbai, E. NADPH oxidase-dependent redox signalling in human heart failure: Relationship between the left and right ventricle. J. Moll. Cell. Cardiol. 2007, 42, 826–834. [Google Scholar] [CrossRef]
- Nabeebaccus, A.; Zhang, M.; Shah, A.M. NADPH oxidase and cardiac remodeling. Heart Fail. Rev. 2011, 16, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Maejima, Y.; Kuroda, J.; Matsushima, S.; Ago, T.; Sadoshima, J. Regulation of myocardial growth and death by NADPH oxidase. J. Moll. Cell. Cardiol. 2011, 50, 408–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuroda, J.; Sadoshima, J. NADPH oxidase and cardiac failure. J. Cardiovasc. Transl. Res. 2010, 3, 314–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joseph, L.C.; Barca, E.; Subramnyam, P.; Komrowski, M.; Pajvani, U.; Colecraft, H.M.; Hirano, M.; Morrow, J.P. Inhibition of NADPH oxidase 2 (NOX2) prevents oxidative stress and mitochondrial abnormalities caused by saturated fat in cardiomyocytes. PLoS ONE 2016, 11, e145750. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, M.D.; Canugovi, C.; Vendrov, A.E.; Hayami, T.; Bowles, D.E.; Krause, K.H.; Madamanchi, N.R.; Runge, M.S. NADPH oxidase 4 regulates inflammation in ischemic heart failure: Role of soluble epoxide hydrolase. Antioxid. Redox. Signal 2019, 31, 39–58. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, J.; Ago, T.; Matshushima, S.; Zhai, P.; Schneider, M.D.; Sadoshima, J. NADPH oxidase 4 (NOX4) is a major source of oxidative stress in the failing heart. Proc. Natl. Acad. Sci. USA 2010, 107, 15565–15570. [Google Scholar] [CrossRef] [Green Version]
- Qin, F. Inhibition of NADPH oxidase reduce myocardial oxidative stress and apoptosis and improves cardiac function in heart failure after myocardial infarction. Free Radic. Biol. Med. 2007, 43, 271–281. [Google Scholar] [CrossRef]
- Liu, Y.; Huang, H.; Xia, W.; Tang, Y.; Li, H.; Huang, C. NADPH oxidase inhibition ameliorates cardiac dysfunction in rabbits with heart failure. Mol. Cell. Biochem. 2010, 343, 143–153. [Google Scholar] [CrossRef]
- Kaludercic, N.; Carpi, A.; Menabo, R.; Di Lisa, F.; Paolocci, N. Monoamine oxidases (MAO) in the pathogenesis of heart failure and ischemia/reperfusion injury. Biochim. Biophys. Acta 2011, 1813, 1323–1332. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, P.; Pimentel, D.R.; Murphy, M.P.; Colucci, W.S.; Parini, A. A new hypertrophic mechanism of serotonin in cardiac myocytes: Receptor-independent ROS generation. FASEB J. 2005, 19, 641–643. [Google Scholar] [CrossRef]
- Mialet-Perez, J.; Santin, Y.; Parini, A. Monoamine oxidase-A, serotonin and norepinephrine: Synergistic players in cardiac physiology and pathology. J. Nueral. Transm. 2018, 125, 1627–1634. [Google Scholar] [CrossRef] [PubMed]
- Santin, Y.; Sicard, P.; Vigneron, F.; Guilbeau-Frugier, C.; Dutuar, M.; Lairez, O.; Couderc, B.; Manni, D.; Korolchuk, V.I.; Lezoualc’h, F.; et al. Oxidative stress by monoamine oxidase-A impairs transcription factor EB activation and autophagosome clearance, leading to cardiomyocyte necrosis and heart failure. Antioxid. Redox. Signal 2016, 25, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Manni, E.; Rigacci, S.; Borchi, E.; Bargelli, V.; Miceli, C.; Giordano, C.; Raimondi, L.; Nediani, C. Monoamine oxidase is overactivated in left and right ventricles from ischemic hearts: An intriguing therapeutic target. Oxid. Med. Cell. Longev. 2016, 2016, 4375418. [Google Scholar] [CrossRef] [PubMed]
- Kaludercic, N.; Takimoto, E.; Nagayama, T.; Feng, N.; Laim, E.W.; Bedja, D.; Chen, K.; Gabrielson, K.L.; Blakely, R.D.; Shih, J.C.; et al. Monoamine oxidase A–mediated enhanced catabolism of norepinephrine contributes to adverse remodeling and pump failure in hearts with pressure overload. Circ. Res. 2010, 106, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Duicu, O.M.; Lighezan, R.; Sturza, A.; Ceausu, R.A.; Borza, C.; Vaduva, A.; Noveanu, L.; Gaspar, M.; Ionac, A.; Feier, H.; et al. Monoamine oxidase as potential contributors to oxidative stress in diabetes: Time for study in patients undergoing heart surgery. Biomed. Res. Int. 2015, 2015, 515437. [Google Scholar] [CrossRef] [PubMed]
- Belch, J.J.; Bridges, A.B.; Scott, N.; Chopra, M. Oxygen free radicals and congestive heart failure. Br. Heart J. 1991, 65, 245–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Tian, J.; Sun, Y.; Xu, T.R.; Chi, R.F.; Zhang, X.L.; Zhang, Y.A.; Qin, F.Z.; Zhang, W.F. Activation of NADPH oxidase mediates increased endoplasmic reticulum stress and left ventricular remodeling after myocardial infarction in rabbits. Biochim. Biophys. Acta 2015, 1852, 805–815. [Google Scholar] [CrossRef] [Green Version]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2181–H2190. [Google Scholar] [CrossRef] [Green Version]
- Adameova, A.; Shah, A.K.; Dhalla, N.S. Role of oxidative stress in the genesis of ventricular arrhythmias. Int. J. Mol. Sci. 2020, 21, 4200. [Google Scholar] [CrossRef]
- Luczak, E.D.; Anderson, M.E. CaMKII oxidative activation and the pathogenesis of cardiac disease. J. Moll. Cell. Cardiol. 2014, 73, 112–116. [Google Scholar] [CrossRef] [Green Version]
- Shah, A.K.; Bhullar, S.K.; Elimban, V.; Dhalla, N.S. Oxidative stress as a mechanism for functional alterations in cardiac hypertrophy and heart failure. Antioxidants 2021, 10, 931. [Google Scholar] [CrossRef] [PubMed]
- Ziolo, M.T.; Houser, S.R. Abnormal Ca2+ cycling in failing ventricular myocytes: Role of NOS1-mediated nitroso-redox balance. Antioxid. Redox. Signal. 2014, 21, 2044–2059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhary, G.; Dudley Jr, S.C. Heart failure, oxidative stress, and ion channel modulation. Congest. Heart Fail. 2002, 8, 148–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bode, D.; Wen, Y.; Hegemann, N.; Primessnig, U.; Parwani, A.; Boldt, L.H.; Pieske, B.M.; Heinzel, F.R.; Hohendanner, F. Oxidative stress and inflammatory modulation of Ca2+ handling in metabolic HFpEF-related left atrial cardiomyopathy. Antioxidants 2020, 9, 860. [Google Scholar] [CrossRef] [PubMed]
- Qin, F.; Yan, C.; Patel, R.; Liu, W.; Dong, E. Vitamins C and E attenuates apoptosis, β-adrenergic receptor desensitization, and sarcoplasmic reticular Ca2+ ATPase downregulation after myocardial infarction. Free Radic. Biol. Med. 2006, 40, 1827–1842. [Google Scholar] [CrossRef]
- Liu, W.; Chen, P.; Deng, J.; Lv, J.; Liu, J. Resveratrol and polydatin as modulators of Ca2+ mobilization in the cardiovascular system. Ann. New York Acad. Sci. 2017, 1403, 82–91. [Google Scholar] [CrossRef]
- Lacerda, D.; Turck, P.; Campos-Carraro, C.; Hickmann, A.; Ortiz, V.; Bianchi, S.; Bello-Klein, A.; de Castro, A.L.; Bassani, V.L.; da Rosa Araujo, A.S. Pterostilbene improves cardiac function in a rat model of right heart failure through modulation of calcium handling proteins and oxidative stress. Appl. Physiol. Nutr. Metab. 2020, 45, 987–995. [Google Scholar] [CrossRef]
- Wu, T.; Yao, H.; Zhang, B.; Zhou, S.; Hou, P.; Chen, K. κ Opioid receptor agonist inhibits myocardial injury in heart failure rats through activating Nrf2/HO-1 pathway and regulating Ca2+-SERCA2a. Oxid. Med. Cell. Longev. 2021, 2021, 7328437. [Google Scholar] [CrossRef]
- Luo, J.; Xuan, Y.T.; Gu, Y.; Prabhu, S.D. Prolonged oxidative stress inverts the cardiac force-frequency relation: Role of altered calcium handling and myofilament calcium responsiveness. J. Mol. Cell. Cardiol. 2006, 40, 64–75. [Google Scholar] [CrossRef]
- Ahmed, M.; Gladden, J.D.; Litovsky, S.H.; Llyod, S.G.; Gupta, H.; Inusah, S.; Denny, T., Jr.; Powell, P.; McGiffin, D.C.; Dell’ltalia, L.J. Increased oxidative stress and cardiomyocyte myofibrillar degeneration in patients with chronic isolated mitral regurgitation and ejection fraction >60%. J. Am. Coll. Cardiol. 2010, 55, 671–679. [Google Scholar] [CrossRef] [Green Version]
- Canton, M.; Menazza, S.; Sheeran, F.L.; de Laureto, P.P.; Di Lisa, F.; Pepe, S. Oxidation of myofibrillar proteins in human heart. J. Am. Coll. Cardiol. 2011, 57, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.G.; Ravi, R.; Stull, L.B.; Murphy, A.M. Chronic xanthine oxidase inhibition prevents myofibrillar protein oxidation and preserves cardiac function in a transgenic mouse model of cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H1512–H1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, Q.; Ren, B.; Elimban, V.; Tappia, P.S.; Takeda, N.; Dhalla, N.S. Modification of sarcolemmal Na+-K+-ATPase and Na+/Ca2+ exchanger in heart failure by blockade of renin-angiotensin system. Am. J. Heart Physiol. Circ. Physiol. 2005, 288, H2637–H2646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Pogwizd, S.M.; Prabhu, S.D.; Zhou, L. Inhibiting Na+-K+ ATPase can impair mitochondrial energetics and induce abnormal Ca2+ cycling and automaticity in guinea pig cardiomyocytes. PLoS ONE 2014, 9, e93928. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, M.; Panagia, V.; Paolillo, G.; Majumder, S.; Ou, C.; Dhalla, N.S. Inhibiton of cardiac phosphatidylethanolamine N-methylation by oxygen free radicals. Biochim. Biophys. Acta 1990, 1021, 33–38. [Google Scholar] [CrossRef]
- Tappia, P.S.; Dent, M.R.; Dhalla, N.S. Oxidative stress and redox regulation of phospholipase D in myocardial disease. Free Radic. Biol. Med. 2006, 41, 349–361. [Google Scholar] [CrossRef]
- Kohlhaas, M.; Maack, C. Interplay of defective excitation-contraction coupling, energy starvation, and oxidative stress in heart failure. Trends. Cardiovasc. Med. 2011, 21, 69–73. [Google Scholar] [CrossRef]
- Nojiri, H.; Shimizu, T.; Funakoshi, M.; Yamaguchi, O.; Zhou, H.; Kawakami, S.; Ohta, Y.; Sami, M.; Ishikawa, H.; Kurosawa, H.; et al. Oxidative stress causes heart failure with impaired mitochondrial respiration. J. Biol. Chem. 2006, 281, 33789–33801. [Google Scholar] [CrossRef] [Green Version]
- Shaheen, M.; Cheema, Y.; Shahbaz, A.U.; Bhattacharya, S.K.; Weber, K.T. Intracellular calcium overloading and oxidative stress in cardiomyocyte necrosis via a mitochondriocentric signal-transducer-effector pathway. Exp. Clin. Cardiol. 2011, 16, 109–115. [Google Scholar]
- Dietl, A.; Maack, C. Targeting mitochondrial calcium handling and reactive oxygen species in heart failure. Curr. Heart Fail. Rep. 2017, 14, 338–349. [Google Scholar] [CrossRef]
- Cortassa, S.; Juhaszova, M.; Aon, M.A.; Zorov, D.B.; Sollott, S.J. Mitochondrial Ca2+, redox environment and ROS emission in heart failure: Two sides of the same coin? J. Mol. Cell. Cardiol. 2021, 151, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Pavez-Giani, M.G.; Sanchez-Aguilera, P.I.; Bomer, n.; Miyamota, S.; Booij, H.G.; Giraldo, P.; Oberdorf-Maass, S.U.; Nijholt, K.T.; Yurista, S.R.; Milting, H.; et al. ATPase inhibitory factor-1 disrputs mitochondrial Ca2+ handling and promotes pathological cardiac hypertrophy through CaMKIIδ. Int. J. Mol. Sci. 2021, 22, 4427. [Google Scholar] [CrossRef] [PubMed]
- Bertero, E.; Nickel, A.; Kohlhaas, M.; Hohl, M.; Sequeira, V.; Brune, C.; Schwemmlein, J.; Abeber, M.; Schuh, K.; Kutschka, I.; et al. Loss of mitochondrial Ca2+ uniporter limits inotropic reserve and provides trigger and substrate for arrhythmias in Barth syndrome cardiomyopathy. Circulation 2021, 144, 1694–1713. [Google Scholar] [CrossRef] [PubMed]
- Donoso, P.; Sanchez, G.; Bull, R.; Hidalgo, C. Modulation of cardiac ryanodine receptor activity by ROS and RNS. Front. Biosci. 2011, 16, 553–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokuhisa, T.; Yano, M.; Obayashi, M.; Noma, T.; Mochizuki, M.; Oda, T.; Okuda, S.; Doi, M.; Liu, J.; Ikeda, Y.; et al. AT1 receptor antagonist restores cardiac ryanodine receptor function, rendering isoproterenol-induced failing heart less susceptible to Ca2+-leak induced by oxidative stress. Circ. J. 2006, 70, 777–786. [Google Scholar] [CrossRef] [Green Version]
- Terentyev, D.; Gyorke, I.; Belevych, A.E.; Terentyeva, R.; Sridhar, A.; Nishijima, Y.; de Blanco, E.C.; Khanna, S.; Sen, C.K.; Cardounel, A.J.; et al. Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ. Res. 2008, 103, 1466–1472. [Google Scholar] [CrossRef] [Green Version]
- Yano, M.; Okuda, S.; Oda, T.; Tokuhisa, T.; Tateishi, H.; Mochizuki, M.; Noma, T.; Doi, M.; Kobayashi, S.; Yamamoto, T.; et al. Correction of defective interdomain interaction within ryanodine receptor by antioxidant is a new therapeutic strategy against heart failure. Circulation 2005, 112, 3633–3643. [Google Scholar] [CrossRef] [Green Version]
- Koitabashi, N.; Arai, M.; Tomaru, K.; Takizawa, T.; Watanabe, A.; Niwano, K.; Yokoyama, T.; Wuytack, F.; Periasamy, M.; Nagai, R.; et al. Carvedilol effectively blocks oxidative stress-mediated downregulation of sarcoplasmic reticulum Ca2+-ATPase 2 gene transcription through modification of SP1 binding. Biochem. Biophys. Res. Commun. 2005, 328, 116–124. [Google Scholar] [CrossRef]
- Kaneko, M.; Elimban, V.; Dhalla, N.S. Mechanism for depression of heart sarcolemmal Ca2+ pump by oxygen free radicals. Am. J. Physiol. 1989, 257, H804–H811. [Google Scholar] [CrossRef]
- Kaneko, M.; Beamish, R.E.; Dhalla, N.S. Depression of heart sarcolemmal Ca2+-pump activity by oxygen free radicals. Am. J. Physiol. 1989, 256, H368–H374. [Google Scholar] [CrossRef]
- Kaneko, M.; Lee, S.L.; Wolf, C.M.; Dhalla, N.S. Reduction of calcium channel antagonist binding sites by oxygen free radicals in rat heart. J. Mol. Cell. Cardiol. 1989, 21, 935–943. [Google Scholar] [CrossRef]
- Kaneko, M.; Singal, P.K.; Dhalla, N.S. Alterations in heart sarcolemmal Ca2+ ATPase and Ca2+-binding activities due to oxygen free radicals. Basic Res. Cardiol. 1990, 85, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, T.; Dhalla, N.S. Relationship between mechanical dysfunction and depression of sarcolemmal Ca2+-pump activity in hearts perfused with oxygen free radicals. Mol. Cell. Biochem. 1996, 160–161, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Mochizuki, S.; Saini, H.K.; Elimban, V.; Dhalla, N.S. Modification of alterations in cardiac function and sarcoplasmic reticulum by vanadate in ischemic-reperfused rat hearts. J. Appl. Physiol. 2005, 99, 999–1005. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, S.; Kaneko, M.; Chapman, D.C.; Dhalla, N.S. Alterations in cardiac contractile proteins due to oxygen free radicals. Biochim. Biophys. Acta 1991, 1074, 95–100. [Google Scholar] [CrossRef] [Green Version]
- Makazan, Z.; Saini, H.K.; Dhalla, N.S. Role of oxidative stress in alterations of mitochondria function in ischemic-reperfused hearts. Am. J. Physiol. Heart. Circ. Physiol. 2007, 292, H1986–H1994. [Google Scholar] [CrossRef]
- Kato, K.; Shao, Q.; Elimban, V. Mechanism of depression in cardiac sarcolemmal Na+-K+ ATPase by hypochlorous acid. Am. J. Physiol. 1998, 275, C826–C831. [Google Scholar] [CrossRef]
Figure 1.
Mechanisms of oxidative stress induced defects in the function of subcellular organelles.

Figure 2.
Role of Ca2+-handling abnormalities in oxidative stress induced cardiac dysfunction in heart disease. SL, sarcolemma; SR, sarcoplasmic reticulum.
Figure 2.
Role of Ca2+-handling abnormalities in oxidative stress induced cardiac dysfunction in heart disease. SL, sarcolemma; SR, sarcoplasmic reticulum.


{kind=link}
{kind=link}
Table 1.
Modification of Na+-K+ ATPase and Na+-Ca2+ exchange activities as well as malondialdehyde (MDA) and sulfhydryl (SH-group) content of cardiac sarcolemma upon incubation for 30 min with or without different oxyradical generating systems.
Table 1.
Modification of Na+-K+ ATPase and Na+-Ca2+ exchange activities as well as malondialdehyde (MDA) and sulfhydryl (SH-group) content of cardiac sarcolemma upon incubation for 30 min with or without different oxyradical generating systems.
| Parameters | Control | X + XO | 0.5 mM H2O2 | 0.1 mM H2O2 + 0.05 mM Fe2+ |
|---|---|---|---|---|
| Na+-K+ ATPase (µmol Pi/mg/h) | 14.27 ± 1.07 | 6.81 ± 1.03 * | 8.64 ± 1.04 * | 6.98 ± 0.13 * |
| Na+-Ca2+ exchange (nmol/mg/2 s) | 4.59 ± 0.08 | 1.97 ± 0.13 * | 2.97 ± 0.11 * | 3.02 ± 0.14 * |
| MDA content (nmol/mg protein) | 61.67 ± 3.67 | 81.85 ± 2.54 * | 78.77 ± 3.23 * | 88.26 ± 3.07 * |
| SH-group content (nmol/mg protein) | 72.61 ± 2.37 | 37.59 ± 4.41 * | 42.36 ± 2.75 * | 40.86 ± 3.54 * |
The data are taken from our papers (Kaneko et al. [119], and Kaneko et al. [120]). X + XO, 2 mM xanthine plus 0.03 U xanthine oxidase. The mixture of X+XO was used to generate superoxide radicals and the mixture of low concentrations of H2O2 and Fe2+ was used to generate hydroxyl radicals. * p < 0.05 vs. respective control.
Table 2.
Modification of ATP-dependent Ca2+ accumulation, Mg2+ ATPase and Ca2+-stimulated ATPase activities in cardiac sarcolemma upon incubation for 30 min with different oxyradical generating systems in the absence or presence of their scavengers.
Table 2.
Modification of ATP-dependent Ca2+ accumulation, Mg2+ ATPase and Ca2+-stimulated ATPase activities in cardiac sarcolemma upon incubation for 30 min with different oxyradical generating systems in the absence or presence of their scavengers.
| Parameters | ATP-Dependent Ca2+ Accumulation (nmol Ca2+/mg/5 min) | Mg2+ ATPase (µmol/mg/h) | Ca2+-Stimulated ATPase (µmol/mg/h) |
|---|---|---|---|
| Control | 27.0 ± 1.7 | 195 ± 3 | 13.6 ± 0.7 |
| X + XO treated | 9.5 ± 0.8 * | 176 ± 2 * | 2.6 ± 0.5 * |
| X + XO + 80 µg/mL SOD | 21.8 ± 0.8 † | 192 ± 2 † | 10.1 ± 0.4 † |
| 0.5 mM H2O2 treated | 4.7 ± 1.3 * | 165 ± 6 * | 2.9 ± 0.4 * |
| 0.5 mM H2O2 + 10 µg/mL catalase | 20.6 ± 1.1 † | 190 ± 5 † | 8.4 ± 0.3 † |
| 0.1 mM H2O2 + 0.2 mM Fe2+ treated | 6.7 ± 0.5 * | 169 ± 4 * | 4.0 ± 0.3 * |
| 0.1 mM H2O2 + 0.2 mM Fe2+ + 20 mM mannitol | 17.2 ± 0.8 † | 184 ± 2 † | 8.0 ± 0.5 † |
The data are taken from our paper (Kaneko et al. [120]). X + XO, 2 mM xanthine oxidase plus 0.03 U/mL xanthine oxidase. The mixture of X + XO was used to generate superoxide radicals whereas that with low concentration of H2O2 plus Fe2+ was used for generating hydroxyl radicals. SOD, superoxide dismutase. * p < 0.05 vs. respective control, † p < 0.05 vs. respective oxyradical treated.
Table 3.
Modification of Ca2+-channels, ATP receptors, Ca2+-binding and Ca2+-ecto ATPase in cardiac sarcolemma upon incubation for 30 min with oxyradical generating systems.
Table 3.
Modification of Ca2+-channels, ATP receptors, Ca2+-binding and Ca2+-ecto ATPase in cardiac sarcolemma upon incubation for 30 min with oxyradical generating systems.
| Parameters | Control | X+XO | 1 mM H2O2 | 0.1 mM H2O2 + 0.2 mM Fe2+ |
|---|---|---|---|---|
| A. Ca2+-channel binding | ||||
| Kd (nM) | 0.231 ± 0.011 | 0.252 ± 0.011 | 0.254 ± 0.018 | 0.267 ± 0.017 |
| Bmax (fmol/mg) | 199 ± 12 | 139 ± 7.0 * | 142 ± 8.0 * | 157 ± 9.0 * |
| B. ATP-binding | ||||
| Low affinity (1.25 mM Ca2+) | 97.8 ± 4.3 | 147.2 ± 6.1 * | 141.3 ± 5.4 * | 41.4 ± 4.9 * |
| High affinity (50 µM Ca2+) | 7.95 ± 0.32 | 12.08 ± 0.68 * | 13.92 ± 0.66 * | 4.08 ± 0.24 * |
| C. Ca2+-ecto ATPase | ||||
| (µmol Pi/mg/h) | 44.3 ± 1.1 | 57.7 ± 1.4 * | 57.0 ± 1.2 * | 31. 4 ± 1.3 * |
The data are taken from our papers (Kaneko et al. [121] and Kaneko et al. [122]). X + XO, 2 mM xanthine plus 0.03 U xanthine oxidase. The mixture of X + XO was used to generate superoxide radicals whereas the mixture of H2O2 plus Fe2+ mixture was used for the generation of hydroxyl radicals. * p < 0.05 vs. respective control.
Table 4.
Modification of some biochemical activities of cardiac sarcoplasmic reticulum, myofibrils and mitochondria upon incubation for 30 min with some oxyradical generating systems.
Table 4.
Modification of some biochemical activities of cardiac sarcoplasmic reticulum, myofibrils and mitochondria upon incubation for 30 min with some oxyradical generating systems.
| Parameters | Control | X+XO | 1 mM H2O2 |
|---|---|---|---|
| A. Sarcoplasmic reticulum: | |||
| Ca2+-release (nmol Ca2+/mg/15 s) | 8.5 ± 1.4 | 4.2 ± 0.8 * | 3.9 ± 0.7 * |
| Ca2+-uptake (nmol Ca2+/mg/min) | 29.6 ± 2.4 | 15.9 ± 1.6 * | 12.7 ± 1.5 * |
| Ca2+-pump ATPase (µmol Pi/mg/h) | 14.7 ± 1.3 | 6.4 ± 0.8 * | 5.7 ± 0.9 * |
| B. Myofibrils: | |||
| Mg2+-ATPase (µmol/mg/h) | 2.53 ± 0.13 | 4.97 ± 0.16 * | 5.46 ± 0.18 * |
| Ca2+-stimulated ATPase (µmol/mg/h) | 10.29 ± 0.17 | 6.48 ± 0.18 * | 5.92 ± 0.38 * |
| Sulfhydryl group content (nmol/mg protein) | 67.0 ± 1.3 | 54.2 ± 1.6 * | 47.6 ± 2.06 * |
| C. Mitochondria | |||
| State 3 respiration (O/mg/min) | 293 ± 7.0 | 138 ± 7.0 * | 106 ± 4.0 * |
| RCI (State 3 to state 4 ratio) | 5.36 ± 0.13 | 2.66 ± 0.23 * | 1.89 ± 0.07 * |
| ADP to O ratio (nmol ADP/ng atom O) | 3.00 ± 0.15 | 2.55 ± 0.07 * | 2.37 ± 0.03 * |
Table 5.
Modification of ATPase activities in cardiac sarcolemma and myofibrils upon incubation with HOCl for 30 min in the presence or absence of L-methionine.
Table 5.
Modification of ATPase activities in cardiac sarcolemma and myofibrils upon incubation with HOCl for 30 min in the presence or absence of L-methionine.
| Parameters | Control | 0.1 mM HOCl | HOCl Plus 10 mM L-methionine |
|---|---|---|---|
| A. Sarcolemma: | |||
| Na+-K+ ATPase (µmol Pi/mg/h) | 18.86 ± 2.03 | 2.16 ± 1.05 * | 13.31 ± 2.44 † |
| MDA content (nmol/mg protein) | 51.64 ± 3.97 | 67.33 ± 3.97 * | 48.2 ± 3.59 † |
| Sulfhydryl group content (nmol/mg protein) | 64.84 ± 6.36 | 28.67 ± 4.40 * | 55.86 ± 5.72 † |
| B. Myofibrill ATPase (µmol/mg/h) | |||
| Mg2+ ATPase | 2.80 ± 0.12 | 9.51 ± 0.16 * | 3.79 ± 0.16 † |
| Ca2+-stimulated ATPase | 10.96 ± 0.15 | 5.73 ± 0.31 * | 11.64 ± 0.12 † |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Dhalla, N.S.; Elimban, V.; Bartekova, M.; Adameova, A. Involvement of Oxidative Stress in the Development of Subcellular Defects and Heart Disease. Biomedicines 2022, 10, 393. https://doi.org/10.3390/biomedicines10020393
AMA Style
Dhalla NS, Elimban V, Bartekova M, Adameova A. Involvement of Oxidative Stress in the Development of Subcellular Defects and Heart Disease. Biomedicines. 2022; 10(2):393. https://doi.org/10.3390/biomedicines10020393
Chicago/Turabian StyleDhalla, Naranjan S., Vijayan Elimban, Monika Bartekova, and Adriana Adameova. 2022. "Involvement of Oxidative Stress in the Development of Subcellular Defects and Heart Disease" Biomedicines 10, no. 2: 393. https://doi.org/10.3390/biomedicines10020393
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.