Exploring the Use of Helicogenic Amino Acids for Optimising Single Chain Relaxin-3 Peptide Agonists


 , , and
, , and
Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
2.2. Peptide Synthesis
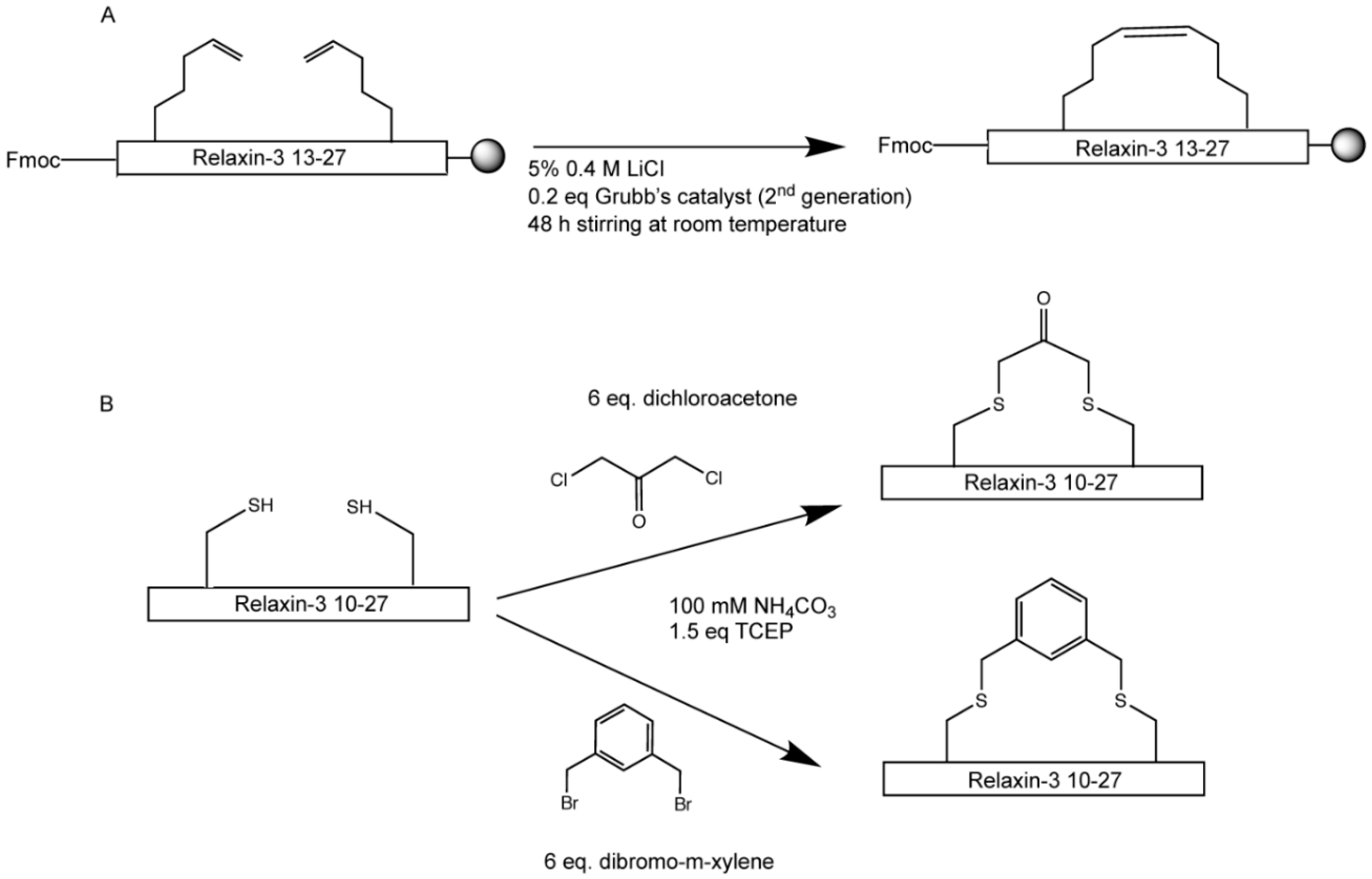
2.3. Hydrocarbon and Halogen Stapling
2.4. NMR Analysis
2.5. Competition Binding Assay
2.6. cAMP Activity Assay
3. Results
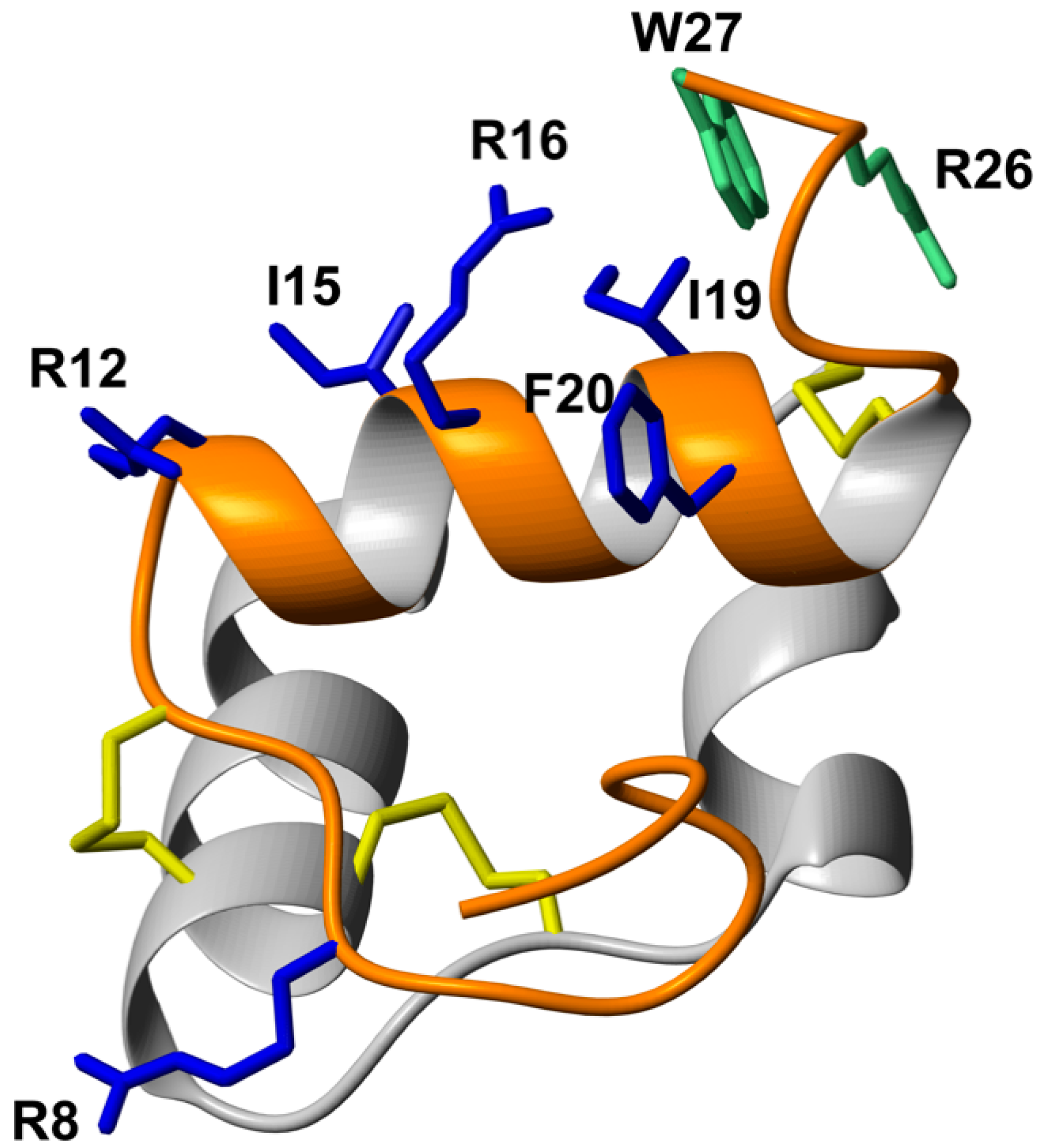
3.1. Peptide Design Rationale and Synthesis
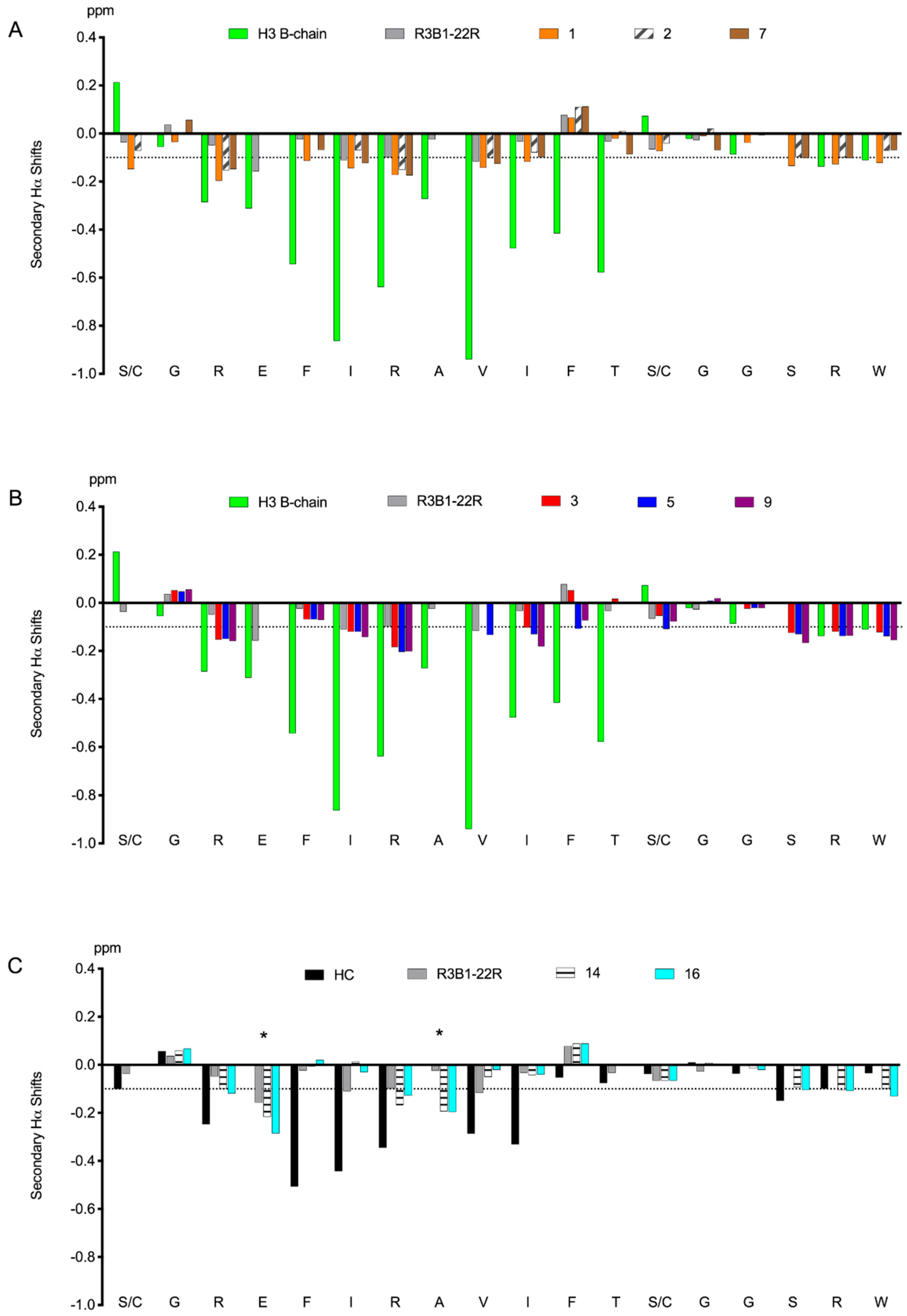
3.2. Helicogenic Amino Acid and Thiol-Based Staples Show Small Improvements in Secondary Structure of Single-Chain Relaxin-3 Agonists
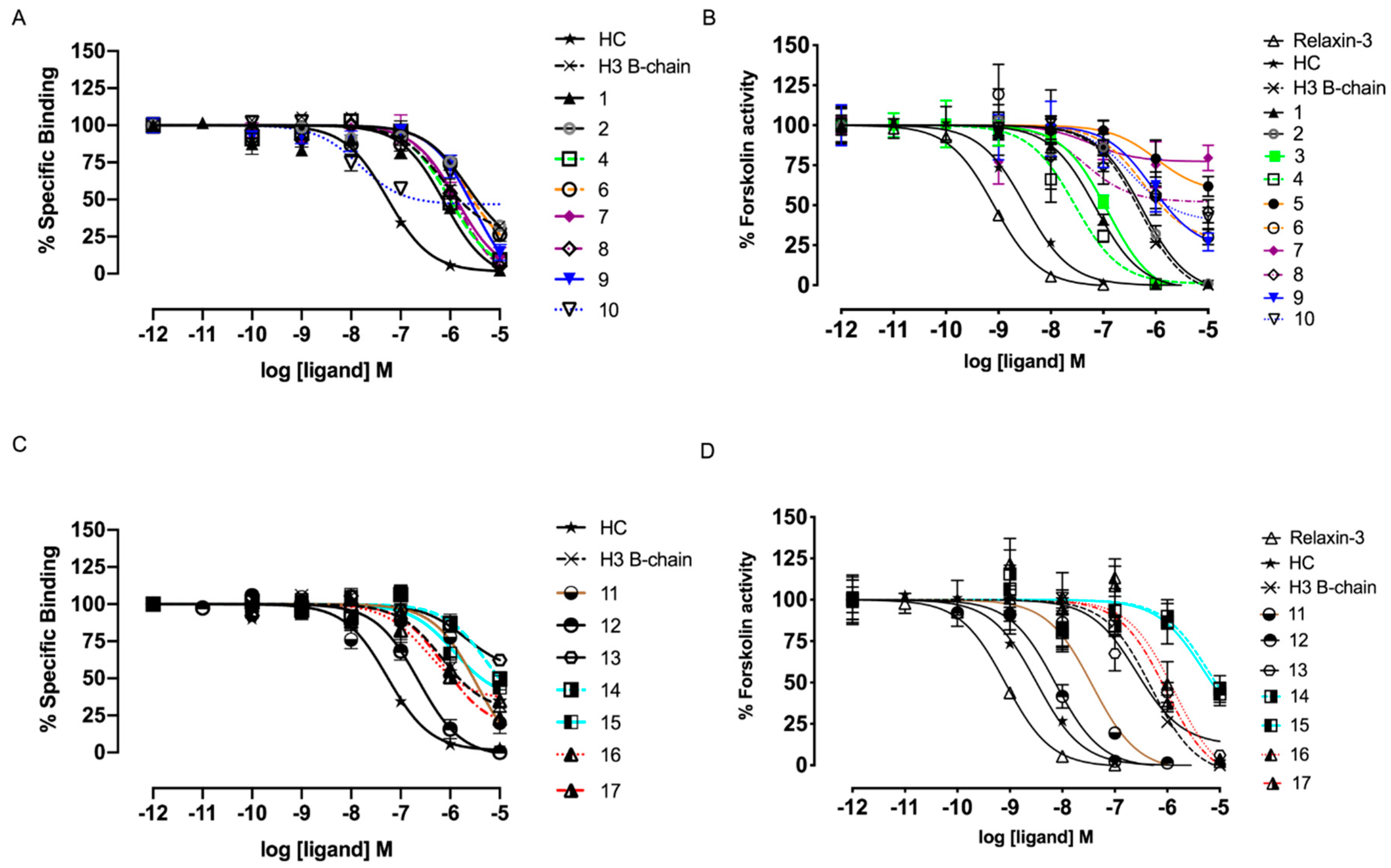
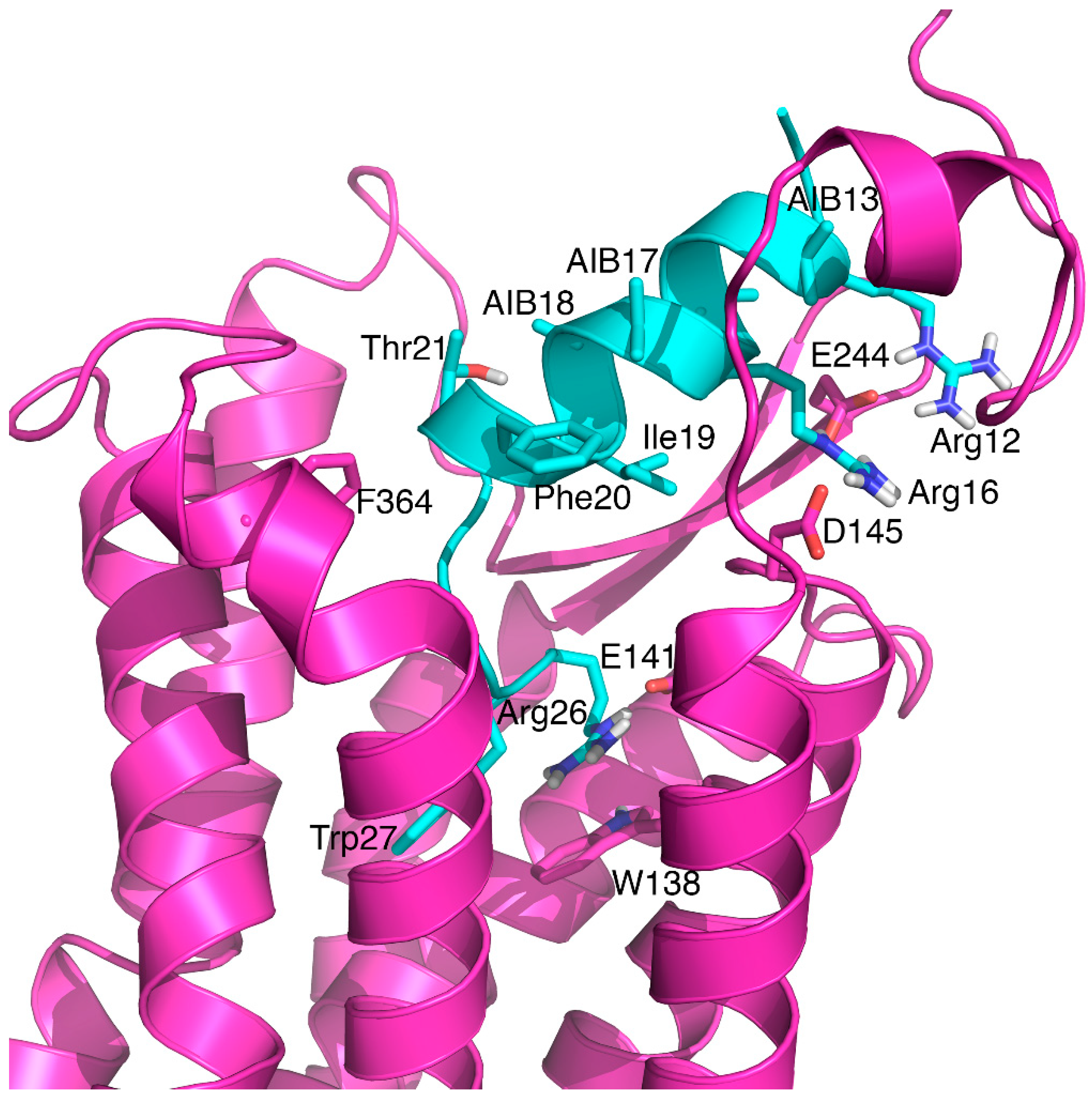
3.3. Aib Residues, but not Cysteine Staples, Can Improve Potency at RXFP3
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ma, S.; Bonaventure, P.; Ferraro, T.; Shen, P.J.; Burazin, T.C.; Bathgate, R.A.; Liu, C.; Tregear, G.W.; Sutton, S.W.; Gundlach, A.L. Relaxin-3 in GABA projection neurons of nucleus incertus suggests widespread influence on forebrain circuits via G-protein-coupled receptor-135 in the rat. Neuroscience 2007, 144, 165–190. [Google Scholar] [CrossRef] [PubMed]
- Sutton, S.W.; Bonaventure, P.; Kuei, C.; Roland, B.; Chen, J.; Nepomuceno, D.; Lovenberg, T.W.; Liu, C. Distribution of G-protein-coupled receptor (GPCR)135 binding sites and receptor mRNA in the rat brain suggests a role for relaxin-3 in neuroendocrine and sensory processing. Neuroendocrinology 2004, 80, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, T.N.; Speed, T.P.; Tregear, G.W.; Bathgate, R.A. Evolution of the relaxin-like peptide family. BMC Evol. Biol. 2005, 5, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGowan, B.M.; Stanley, S.A.; Smith, K.L.; White, N.E.; Connolly, M.M.; Thompson, E.L.; Gardiner, J.V.; Murphy, K.G.; Ghatei, M.A.; Bloom, S.R. Central relaxin-3 administration causes hyperphagia in male Wistar rats. Endocrinology 2005, 146, 3295–3300. [Google Scholar] [CrossRef]
- Ganella, D.E.; Callander, G.E.; Ma, S.; Bye, C.R.; Gundlach, A.L.; Bathgate, R.A. Modulation of feeding by chronic rAAV expression of a relaxin-3 peptide agonist in rat hypothalamus. Gene Ther. 2013, 20, 703–716. [Google Scholar] [CrossRef]
- de Avila, C.; Chometton, S.; Lenglos, C.; Calvez, J.; Gundlach, A.L.; Timofeeva, E. Differential effects of relaxin-3 and a selective relaxin-3 receptor agonist on food and water intake and hypothalamic neuronal activity in rats. Behav. Brain Res. 2018, 336, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Ryan, P.J.; Buchler, E.; Shabanpoor, F.; Hossain, M.A.; Wade, J.D.; Lawrence, A.J.; Gundlach, A.L. Central relaxin-3 receptor (RXFP3) activation decreases anxiety- and depressive-like behaviours in the rat. Behav. Brain Res. 2013, 244, 142–151. [Google Scholar] [CrossRef]
- Zhang, C.; Chua, B.E.; Yang, A.; Shabanpoor, F.; Hossain, M.A.; Wade, J.D.; Rosengren, K.J.; Smith, C.M.; Gundlach, A.L. Central relaxin-3 receptor (RXFP3) activation reduces elevated, but not basal, anxiety-like behaviour in C57BL/6J mice. Behav. Brain Res. 2015, 292, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Marwari, S.; Poulsen, A.; Shih, N.; Lakshminarayanan, R.; Kini, R.M.; Johannes, C.W.; Dymock, B.W.; Dawe, G.S. Intranasal administration of a stapled relaxin-3 mimetic has anxiolytic- and antidepressant-like activity in rats. Br. J. Pharmacol. 2019, 176, 3899–3923. [Google Scholar] [CrossRef]
- Ryan, P.J.; Kastman, H.E.; Krstew, E.V.; Rosengren, K.J.; Hossain, M.A.; Churilov, L.; Wade, J.D.; Gundlach, A.L.; Lawrence, A.J. Relaxin-3/RXFP3 system regulates alcohol-seeking. Proc. Natl. Acad. Sci. USA 2013, 110, 20789–20794. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.M.; Chua, B.E.; Zhang, C.; Walker, A.W.; Haidar, M.; Hawkes, D.; Shabanpoor, F.; Hossain, M.A.; Wade, J.D.; Rosengren, K.J.; et al. Central injection of relaxin-3 receptor (RXFP3) antagonist peptides reduces motivated food seeking and consumption in C57BL/6J mice. Behav. Brain Res. 2014, 268, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.C.; Kastman, H.E.; Krstew, E.V.; Gundlach, A.L.; Lawrence, A.J. Central amygdala relaxin-3/relaxin family peptide receptor 3 signalling modulates alcohol seeking in rats. Br. J. Pharmacol. 2017, 174, 3359–3369. [Google Scholar] [CrossRef] [PubMed]
- Olucha-Bordonau, F.E.; Albert-Gasco, H.; Ros-Bernal, F.; Rytova, V.; Ong-Palsson, E.K.E.; Ma, S.; Sanchez-Perez, A.M.; Gundlach, A.L. Modulation of forebrain function by nucleus incertus and relaxin-3/RXFP3 signaling. CNS Neurosci. Ther. 2018, 24, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Rosengren, K.J.; Lin, F.; Bathgate, R.A.; Tregear, G.W.; Daly, N.L.; Wade, J.D.; Craik, D.J. Solution structure and novel insights into the determinants of the receptor specificity of human relaxin-3. J. Biol. Chem. 2006, 281, 5845–5851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Eriste, E.; Sutton, S.; Chen, J.; Roland, B.; Kuei, C.; Farmer, N.; Jornvall, H.; Sillard, R.; Lovenberg, T.W. Identification of relaxin-3/INSL7 as an endogenous ligand for the orphan G-protein-coupled receptor GPCR135. J. Biol. Chem. 2003, 278, 50754–50764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudo, S.; Kumagai, J.; Nishi, S.; Layfield, S.; Ferraro, T.; Bathgate, R.A.; Hsueh, A.J. H3 relaxin is a specific ligand for LGR7 and activates the receptor by interacting with both the ectodomain and the exoloop 2. J. Biol. Chem. 2003, 278, 7855–7862. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Chen, J.; Sutton, S.; Roland, B.; Kuei, C.; Farmer, N.; Sillard, R.; Lovenberg, T.W. Identification of relaxin-3/INSL7 as a ligand for GPCR142. J. Biol. Chem. 2003, 278, 50765–50770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Chen, J.; Kuei, C.; Sutton, S.; Nepomuceno, D.; Bonaventure, P.; Lovenberg, T.W. Relaxin-3/insulin-like peptide 5 chimeric peptide, a selective ligand for G protein-coupled receptor (GPCR)135 and GPCR142 over leucine-rich repeat-containing G protein-coupled receptor 7. Mol. Pharmacol. 2005, 67, 231–240. [Google Scholar] [CrossRef]
- Hossain, M.A.; Rosengren, K.J.; Haugaard-Jonsson, L.M.; Zhang, S.; Layfield, S.; Ferraro, T.; Daly, N.L.; Tregear, G.W.; Wade, J.D.; Bathgate, R.A. The A-chain of human relaxin family peptides has distinct roles in the binding and activation of the different relaxin family peptide receptors. J. Biol. Chem. 2008, 283, 17287–17297. [Google Scholar] [CrossRef] [Green Version]
- Kuei, C.; Sutton, S.; Bonaventure, P.; Pudiak, C.; Shelton, J.; Zhu, J.; Nepomuceno, D.; Wu, J.; Chen, J.; Kamme, F.; et al. R3(BDelta23 27)R/I5 chimeric peptide, a selective antagonist for GPCR135 and GPCR142 over relaxin receptor LGR7: In vitro and in vivo characterization. J. Biol. Chem. 2007, 282, 25425–25435. [Google Scholar] [CrossRef] [Green Version]
- Haugaard-Kedström, L.M.; Shabanpoor, F.; Hossain, M.A.; Clark, R.J.; Ryan, P.J.; Craik, D.J.; Gundlach, A.L.; Wade, J.D.; Bathgate, R.A.D.; Rosengren, K.J. Design, synthesis, and characterization of a single-chain peptide antagonist for the relaxin-3 receptor RXFP3. J. Am. Chem. Soc. 2011, 133, 4965–4974. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.L.L.; Scott, D.J.; Hossain, M.A.; Kaas, Q.; Rosengren, K.J.; Bathgate, R.A.D. Distinct but overlapping binding sites of agonist and antagonist at the relaxin family peptide 3 (RXFP3) receptor. J. Biol. Chem. 2018, 293, 15777–15789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bathgate, R.A.; Oh, M.H.; Ling, W.J.; Kaas, Q.; Hossain, M.A.; Gooley, P.R.; Rosengren, K.J. Elucidation of relaxin-3 binding interactions in the extracellular loops of RXFP3. Front. Endocrinol. 2013, 4, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, M.J.; Shao, X.X.; Wang, J.H.; Wei, D.; Liu, Y.L.; Xu, Z.G.; Guo, Z.Y. Identification of hydrophobic interactions between relaxin-3 and its receptor RXFP3: Implication for a conformational change in the B-chain C-terminus during receptor binding. Amino Acids 2016, 48, 2227–2236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.J.; Wang, X.Y.; Guo, Y.Q.; Luo, X.; Gao, X.J.; Shao, X.X.; Liu, Y.L.; Xu, Z.G.; Guo, Z.Y. The highly conserved negatively charged Glu141 and Asp145 of the G-protein-coupled receptor RXFP3 interact with the highly conserved positively charged arginine residues of relaxin-3. Amino Acids 2014, 46, 1393–1402. [Google Scholar] [CrossRef]
- Hossain, M.A.; Bathgate, R.A.; Rosengren, K.J.; Shabanpoor, F.; Zhang, S.; Lin, F.; Tregear, G.W.; Wade, J.D. The structural and functional role of the B-chain C-terminal arginine in the relaxin-3 peptide antagonist, R3(BDelta23-27)R/I5. Chem. Biol. Drug Des. 2009, 73, 46–52. [Google Scholar] [CrossRef]
- Haugaard-Kedström, L.M.; Lee, H.S.; Jones, M.V.; Song, A.; Rathod, V.; Hossain, M.A.; Bathgate, R.A.D.; Rosengren, K.J. Binding conformation and determinants of a single-chain peptide antagonist at the relaxin-3 receptor RXFP3. J. Biol. Chem. 2018, 293, 15765–15776. [Google Scholar] [CrossRef] [Green Version]
- Hojo, K.; Hossain, M.A.; Tailhades, J.; Shabanpoor, F.; Wong, L.L.; Ong-Palsson, E.E.; Kastman, H.E.; Ma, S.K.; Gundlach, A.L.; Rosengren, K.J.; et al. Development of a single-chain peptide agonist of the relaxin-3 receptor using hydrocarbon stapling. J. Med. Chem. 2016, 59, 7445–7456. [Google Scholar] [CrossRef]
- Jayakody, T.; Marwari, S.; Lakshminarayanan, R.; Tan, F.C.; Johannes, C.W.; Dymock, B.W.; Poulsen, A.; Herr, D.R.; Dawe, G.S. Hydrocarbon stapled B chain analogues of relaxin-3 retain biological activity. Peptides 2016, 84, 44–57. [Google Scholar] [CrossRef] [Green Version]
- Jo, H.; Meinhardt, N.; Wu, Y.; Kulkarni, S.; Hu, X.; Low, K.E.; Davies, P.L.; DeGrado, W.F.; Greenbaum, D.C. Development of alpha-helical calpain probes by mimicking a natural protein-protein interaction. J. Am. Chem. Soc. 2012, 134, 17704–17713. [Google Scholar] [CrossRef] [Green Version]
- Assem, N.; Ferreira, D.J.; Wolan, D.W.; Dawson, P.E. Acetone-Linked Peptides: A Convergent Approach for Peptide Macrocyclization and Labeling. Angew. Chem. Int. Ed. Engl. 2015, 54, 8665–8668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Findeisen, F.; Campiglio, M.; Jo, H.; Abderemane-Ali, F.; Rumpf, C.H.; Pope, L.; Rossen, N.D.; Flucher, B.E.; DeGrado, W.F.; Minor, D.L., Jr. Stapled Voltage-Gated Calcium Channel (CaV) alpha-Interaction Domain (AID) Peptides Act As Selective Protein-Protein Interaction Inhibitors of CaV Function. ACS Chem. Neurosci. 2017, 8, 1313–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahalakshmi, R.; Balaram, P. Non-protein amino acids in the design of secondary structure scaffolds. Methods Mol. Biol. 2006, 340, 71–94. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.A.; Haugaard-Kedström, L.M.; Rosengren, K.J.; Bathgate, R.A.; Wade, J.D. Chemically synthesized dicarba H2 relaxin analogues retain strong RXFP1 receptor activity but show an unexpected loss of in vitro serum stability. Org. Biomol. Chem. 2015, 13, 10895–10903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, R.L.J. The Computer Aided Resonance Assignment Tutorial; Cantina Verlag: Goldau, Switzerland, 2004. [Google Scholar]
- Wishart, D.S.; Bigam, C.G.; Holm, A.; Hodges, R.S.; Sykes, B.D. 1H, 13C and 15N random coil NMR chemical shifts of the common amino acids. I. Investigations of nearest-neighbor effects. J. Biomol. NMR 1995, 5, 67–81. [Google Scholar] [CrossRef]
- Haugaard-Kedström, L.M.; Wong, L.L.; Bathgate, R.A.; Rosengren, K.J. Synthesis and pharmacological characterization of a europium-labelled single-chain antagonist for binding studies of the relaxin-3 receptor RXFP3. Amino Acids 2015, 47, 1267–1271. [Google Scholar] [CrossRef]
- Van der Westhuizen, E.T.; Sexton, P.M.; Bathgate, R.A.; Summers, R.J. Responses of GPCR135 to human gene 3 (H3) relaxin in CHO-K1 cells determined by microphysiometry. Ann. N. Y. Acad. Sci. 2005, 1041, 332–337. [Google Scholar] [CrossRef]
- Shabanpoor, F.; Akhter Hossain, M.; Ryan, P.J.; Belgi, A.; Layfield, S.; Kocan, M.; Zhang, S.; Samuel, C.S.; Gundlach, A.L.; Bathgate, R.A.; et al. Minimization of human relaxin-3 leading to high-affinity analogues with increased selectivity for relaxin-family peptide 3 receptor (RXFP3) over RXFP1. J. Med. Chem. 2012, 55, 1671–1681. [Google Scholar] [CrossRef]
- Wada, S.; Tsuda, H.; Okada, T.; Urata, H. Cellular uptake of Aib-containing amphipathic helix peptide. Bioorg. Med. Chem. Lett. 2011, 21, 5688–5691. [Google Scholar] [CrossRef]
- Fairlie, D.P.; de Dantas Araujo, A. Review stapling peptides using cysteine crosslinking. Biopolymers 2016, 106, 843–852. [Google Scholar] [CrossRef]
- Lee, H.S.; Postan, M.; Song, A.; Clark, R.J.; Bathgate, R.A.D.; Haugaard-Kedstrom, L.M.; Rosengren, K.J. Development of Relaxin-3 Agonists and Antagonists Based on Grafted Disulfide-Stabilized Scaffolds. Front. Chem. 2020, 8, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakagami, K.; Masuda, T.; Kawano, K.; Futaki, S. Importance of Net Hydrophobicity in the Cellular Uptake of All-Hydrocarbon Stapled Peptides. Mol. Pharm. 2018, 15, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Yuen, T.Y.; Brown, C.J.; Tan, Y.S.; Johannes, C.W. Synthesis of Chiral Alkenyl Cyclopropane Amino Acids for Incorporation into Stapled Peptides. J. Org. Chem. 2020, 85, 1556–1566. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Jiang, Y.; Li, J.; Wang, D.; Zhao, H.; Li, Z. Effect of Stapling Architecture on Physiochemical Properties and Cell Permeability of Stapled α-Helical Peptides: A Comparative Study. ChemBioChem 2017, 18, 2087–2093. [Google Scholar] [CrossRef]
- Zhang, Q.; Yang, M.; Sorensen, K.K.; Madsen, C.S.; Boesen, J.T.; An, Y.; Peng, S.H.; Wei, Y.; Wang, Q.; Jensen, K.J.; et al. A brain-targeting lipidated peptide for neutralizing RNA-mediated toxicity in Polyglutamine Diseases. Sci. Rep. 2017, 7, 12077. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Perlas, C.; Varese, M.; Guardiola, S.; Garcia, J.; Sanchez-Navarro, M.; Giralt, E.; Teixido, M. From venoms to BBB-shuttles. MiniCTX3: A molecular vector derived from scorpion venom. Chem. Commun. 2018, 54, 12738–12741. [Google Scholar] [CrossRef]
- Oller-Salvia, B.; Sanchez-Navarro, M.; Ciudad, S.; Guiu, M.; Arranz-Gibert, P.; Garcia, C.; Gomis, R.R.; Cecchelli, R.; Garcia, J.; Giralt, E.; et al. MiniAp-4: A Venom-Inspired Peptidomimetic for Brain Delivery. Angew. Chem. Int. Ed. Engl. 2016, 55, 572–575. [Google Scholar] [CrossRef] [Green Version]
- Fuster, C.; Varese, M.; Garcia, J.; Giralt, E.; Sanchez-Navarro, M.; Teixido, M. Expanding the MiniAp-4 BBB-shuttle family: Evaluation of proline cis-trans ratio as tool to fine-tune transport. J. Pept. Sci. 2019, 25, e3172. [Google Scholar] [CrossRef]
- Diaz-Perlas, C.; Sanchez-Navarro, M.; Oller-Salvia, B.; Moreno, M.; Teixido, M.; Giralt, E. Phage display as a tool to discover blood-brain barrier (BBB)-shuttle peptides: Panning against a human BBB cellular model. Biopolymers 2017, 108, e22928. [Google Scholar] [CrossRef] [Green Version]
- Urich, E.; Schmucki, R.; Ruderisch, N.; Kitas, E.; Certa, U.; Jacobsen, H.; Schweitzer, C.; Bergadano, A.; Ebeling, M.; Loetscher, H.; et al. Cargo Delivery into the Brain by in vivo identified Transport Peptides. Sci. Rep. 2015, 5, 14104. [Google Scholar] [CrossRef]
- Prades, R.; Oller-Salvia, B.; Schwarzmaier, S.M.; Selva, J.; Moros, M.; Balbi, M.; Grazu, V.; de La Fuente, J.M.; Egea, G.; Plesnila, N.; et al. Applying the retro-enantio approach to obtain a peptide capable of overcoming the blood-brain barrier. Angew. Chem. Int. Ed. Engl. 2015, 54, 3967–3972. [Google Scholar] [CrossRef] [PubMed]
- Oller-Salvia, B.; Sanchez-Navarro, M.; Giralt, E.; Teixido, M. Blood-brain barrier shuttle peptides: An emerging paradigm for brain delivery. Chem. Soc. Rev. 2016, 45, 4690–4707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Sequence | Analogue |
|---|---|---|
| H3 B-chain | RAAPYGVRLSGREFIRAVIFTSGGSRW | |
| R3B 1-22R C | RAAPYGVRLSGREFIRAVIFTSR | |
| Ac-R3 B10-27 13,17 Pa b | Ac-SGR(Pa)FIR(Pa)VIFTSGGSRW | Ac-R3B10-27 [13/17 HC] |
| Aib variants | ||
| Ac-R3 B10-27 13,17 Aib | Ac-SGR(Aib)FIR(Aib)VIFTSGGSRW | 1 |
| Ac-R3 B10-27 13,14,17 Aib | Ac-SGR(Aib)(Aib)IR(Aib)VIFTSGGSRW | 2 |
| R3 B10-27 13,17,18 Aib | SGR(Aib)FIR(Aib)(Aib)IFTSGGSRW | 3 |
| Ac-R3 B10-27 13,17,18 Aib | Ac-SGR(Aib)FIR(Aib)(Aib)IFTSGGSRW | 4 |
| R3 B10-27 13,17,21 Aib | SGR(Aib)FIR(Aib)VIF(Aib)SGGSRW | 5 |
| Ac-R3 B10-27 13,17,21 Aib | Ac-SGR(Aib)FIR(Aib)VIF(Aib)SGGSRW | 6 |
| R3 B10-27 13,17,22 Aib | SGR(Aib)FIR(Aib)VIFT(Aib)GGSRW | 7 |
| AC-R3 B10-27 13,17,22 Aib | Ac-SGR(Aib)FIR(Aib)VIFT(Aib)GGSRW | 8 |
| R3 B10-27 13,17,18,21 Aib | SGR(Aib)FIR(Aib)(Aib)IF(Aib)SGGSRW | 9 |
| AC-R3 B10-27 13,17,18,21 Aib | Ac-SGR(Aib)FIR(Aib)(Aib)IF(Aib)SGGSRW | 10 |
| Hydrocarbon variants | ||
| Ac-R3 B10-27 13,17 Pa a | Ac-SGR(Pa)FIR(Pa)VIFTSGGSRW | 11 |
| 4K-R3 B10-27 13,17 Pa b | KKKKSGR(Pa)FIR(Pa)VIFTSGGSRW | 12 |
| Ac-R3 B10-27 13,17 Pg a | Ac-SGR(Pg)FIR(Pg)VIFTSGGSRW | 13 |
| R3 B10-27 13,17 DCA b | SGRCFIRCVIFTSGGSRW | 14 |
| AC-R3 B10-27 13,17 DCA b | Ac-SGRCFIRCVIFTSGGSRW | 15 |
| R3 B10-27 13,17 DBX b | SGRCFIRCVIFTSGGSRW | 16 |
| AC-R3 B10-27 13,17 DBX b | Ac-SGRCFIRCVIFTSGGSRW | 17 |
| Peptide | Analogue | pKi [logM] c | pEC50 [logM] |
|---|---|---|---|
| R3B1-27 | H3 B-chain | 5.91 ± 0.21 d | 5.93 ± 0.02 d,i |
| R3 B10-27 13,17 Pa b | Ac-R3B10-27 [13/17 HC] | 7.38 ± 0.03 | 8.48 ± 0.06 i |
| Aib variants | |||
| Ac-R3 B10-27 13,17 Aib | 1 | 6.25 ± 0.12 e | 7.17 ± 0.11 d,g |
| Ac-R3 B10-27 13,14,17 Aib | 2 | 5.83 ± 0.18 d | 6.26 ± 0.06 d |
| R3 B10-27 13,17,18 Aib | 3 | ND | 7.04 ± 0.04 d,g |
| Ac-R3 B10-27 13,17,18 Aib | 4 | 6.12 ± 0.11 d | 7.48 ± 0.20 e,f |
| R3 B10-27 13,17,21 Aib | 5 | ND | 5.99 d |
| Ac-R3 B10-27 13,17,21 Aib | 6 | 5.77 ± 0.28 d | 5.73 ± 0.04 d |
| R3 B10-27 13,17,22 Aib | 7 | 5.95 ± 0.05 d | <5 |
| AC-R3 B10-27 13,17,22 Aib | 8 | 6.02 ± 0.08 d | <5 |
| R3 B10-27 13,17,18,21 Aib | 9 | 5.70 ± 0.19 d | 5.75 ± 0.27 d |
| AC-R3 B10-27 13,17,18,21 Aib | 10 | <5 | 5.49 ± 0.09 d |
| Stapled variants | |||
| Ac-R3 B10-27 13,17 Pa a | 11 | 6.03 ± 0.45 d | 7.38 ± 0.16 e,f |
| 4K-R3 B10-27 13,17 Pa b | 12 | 6.74 ± 0.16 h | 8.19 ± 0.14 f |
| Ac-R3 B10-27 13,17 Pg a | 13 | <5 | 6.51 ± 0.27 d |
| R3 B10-27 13,17 DCA b | 14 | <5 | 5.21 ± 0.75 d |
| AC-R3 B10-27 13,17 DCA b | 15 | 5.32 ± 0.21 d | 5.32 ± 0.76 d |
| R3 B10-27 13,17 DBX b | 16 | 6.00 ± 0.44 d | 5.98 ± 0.07 d |
| AC-R3 B10-27 13,17 DBX b | 17 | 5.98 ± 0.04 d | 6.07 ± 0.03 d |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.S.; Wang, S.H.; Daniel, J.T.; Hossain, M.A.; Clark, R.J.; Bathgate, R.A.D.; Rosengren, K.J. Exploring the Use of Helicogenic Amino Acids for Optimising Single Chain Relaxin-3 Peptide Agonists. Biomedicines 2020, 8, 415. https://doi.org/10.3390/biomedicines8100415
Lee HS, Wang SH, Daniel JT, Hossain MA, Clark RJ, Bathgate RAD, Rosengren KJ. Exploring the Use of Helicogenic Amino Acids for Optimising Single Chain Relaxin-3 Peptide Agonists. Biomedicines. 2020; 8(10):415. https://doi.org/10.3390/biomedicines8100415
Chicago/Turabian StyleLee, Han Siean, Shu Hui Wang, James T. Daniel, Mohammed Akhter Hossain, Richard J. Clark, Ross A. D. Bathgate, and K. Johan Rosengren. 2020. "Exploring the Use of Helicogenic Amino Acids for Optimising Single Chain Relaxin-3 Peptide Agonists" Biomedicines 8, no. 10: 415. https://doi.org/10.3390/biomedicines8100415