Gut Microbiome Composition Remains Stable in Individuals with Diabetes-Related Early to Late Stage Chronic Kidney Disease


Abstract
:1. Introduction
2. Materials and Methods
2.1. Participants
2.2. Stool Collection
2.3. DNA Extraction
2.4. DNA Microbiome Profiling and Quality Control
2.5. Data Cleaning, Normalisation and Statistical Analysis
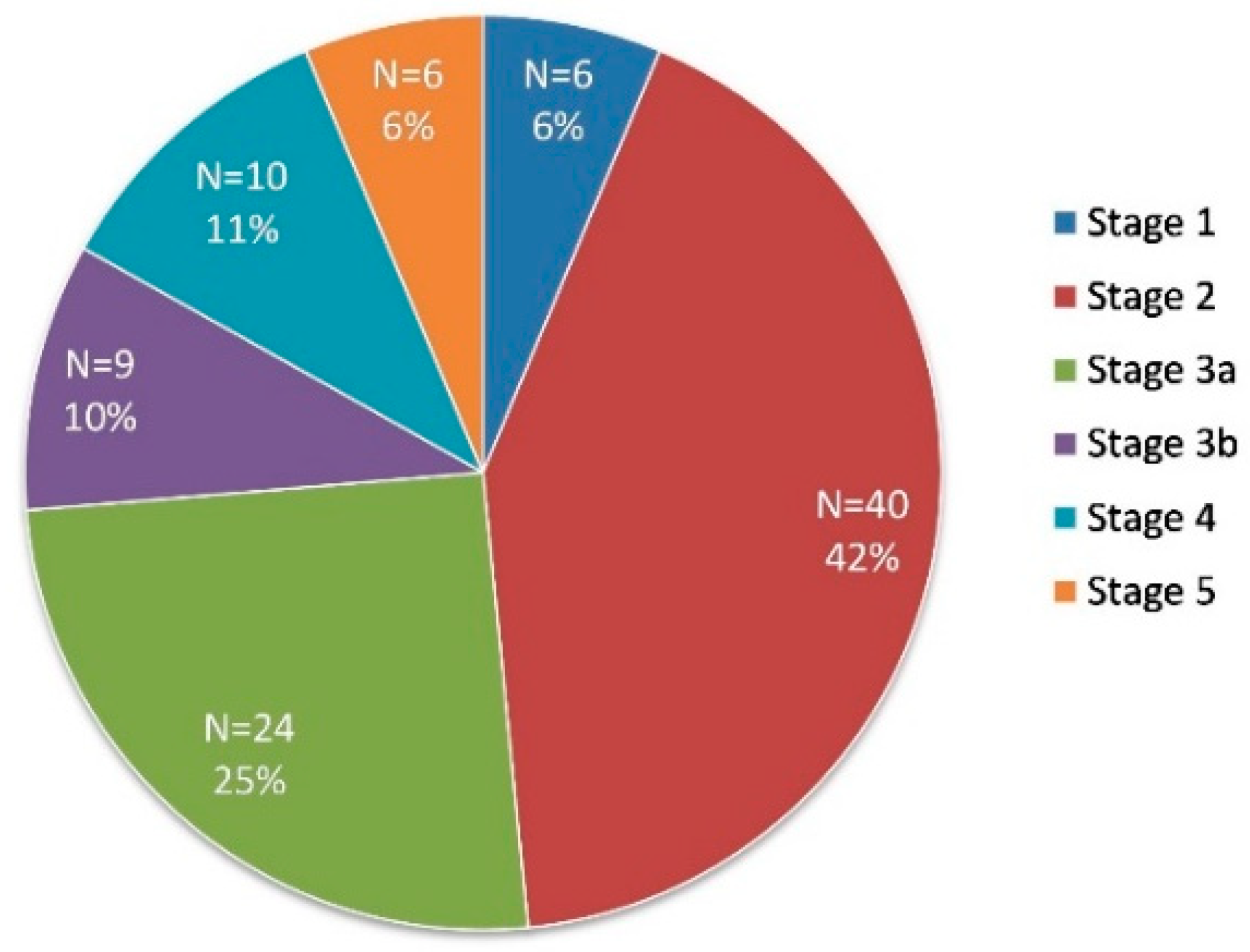
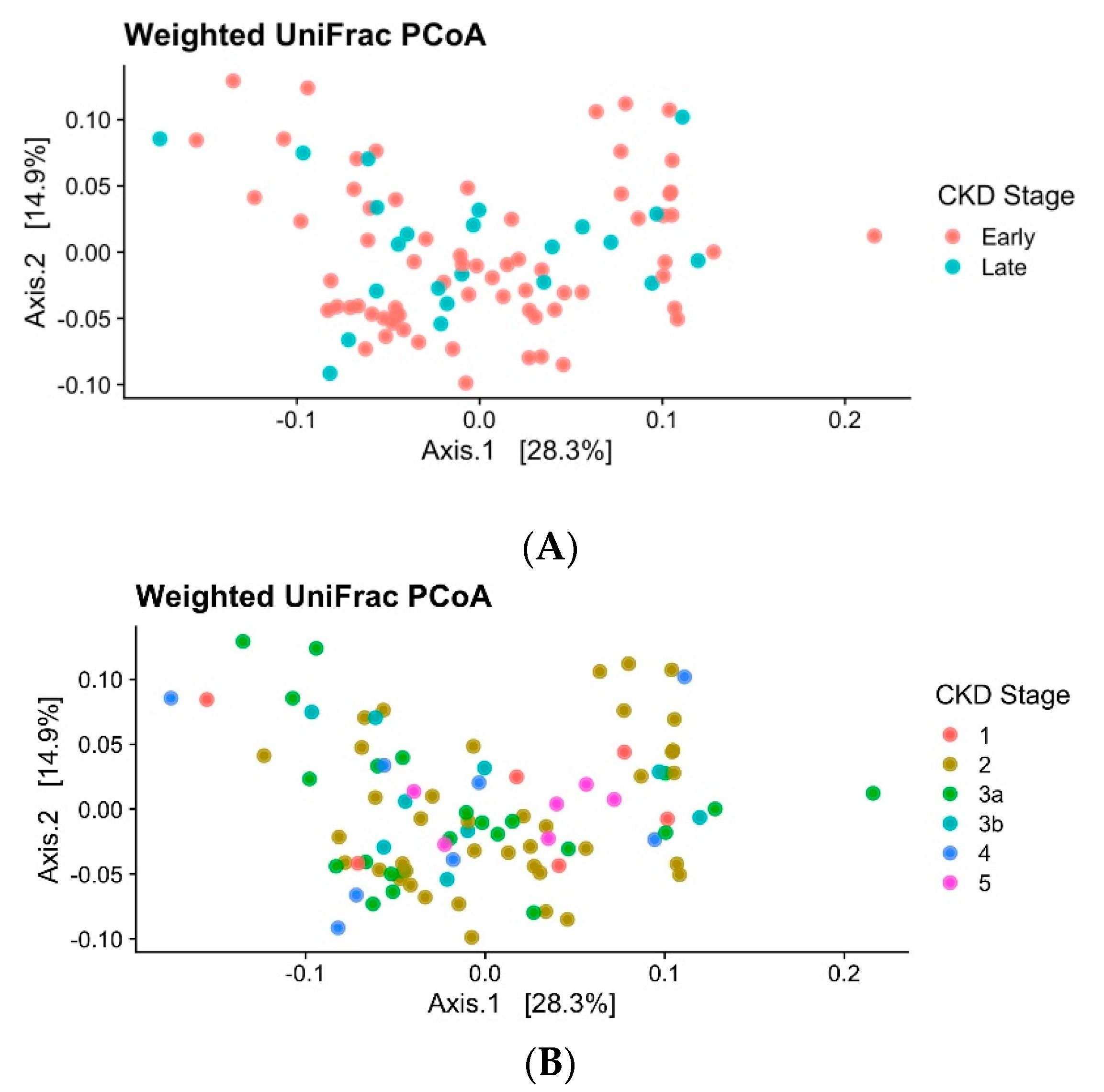
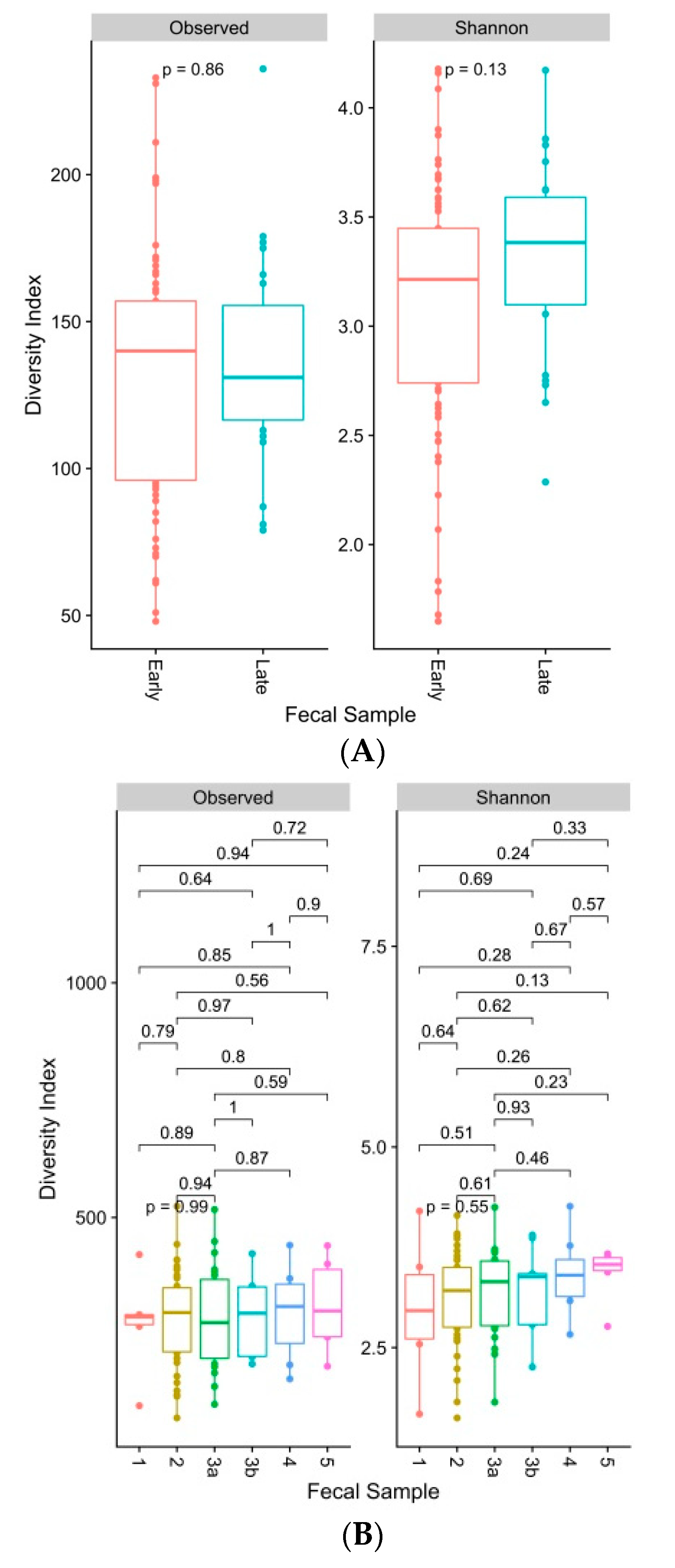
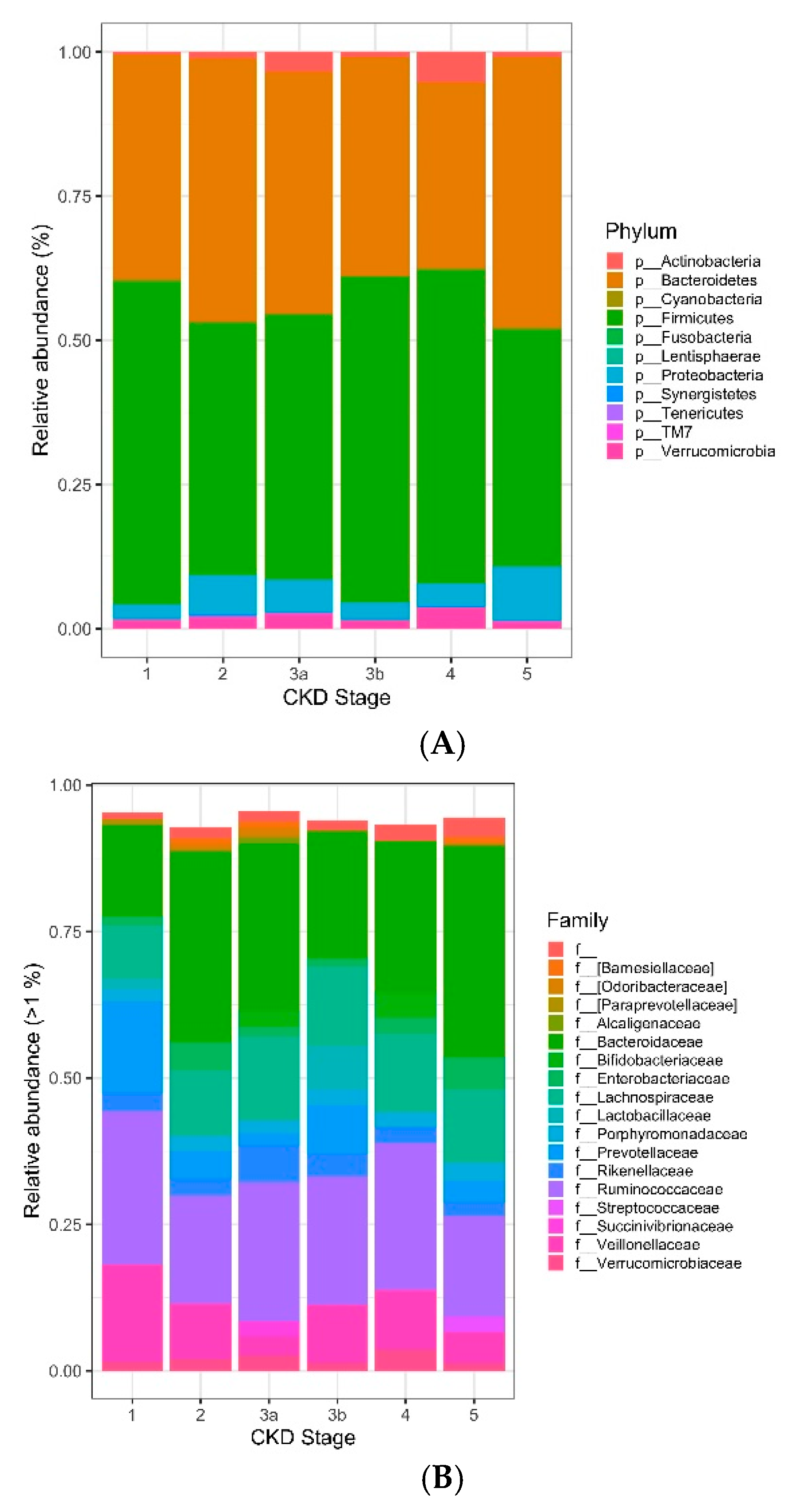
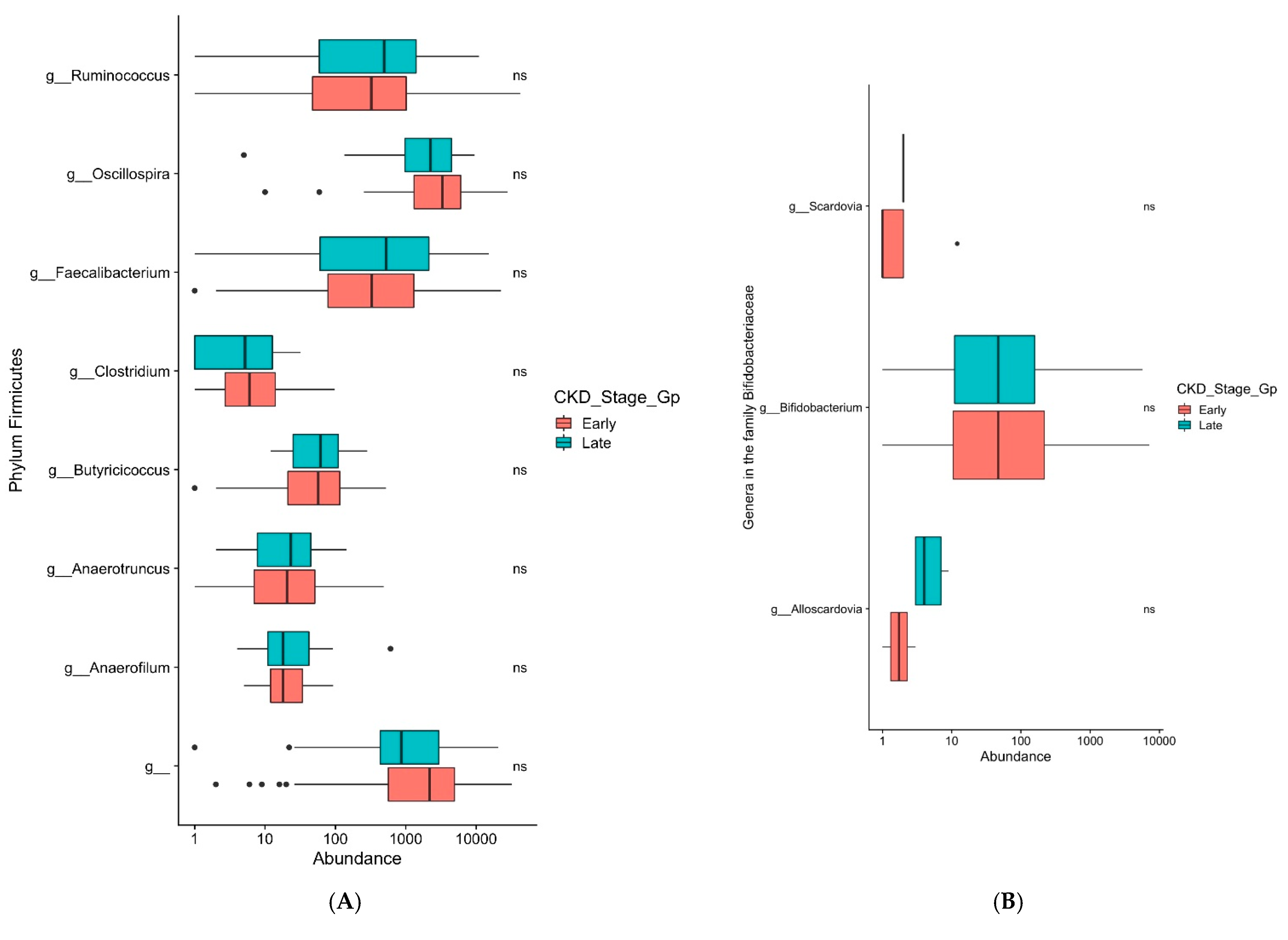
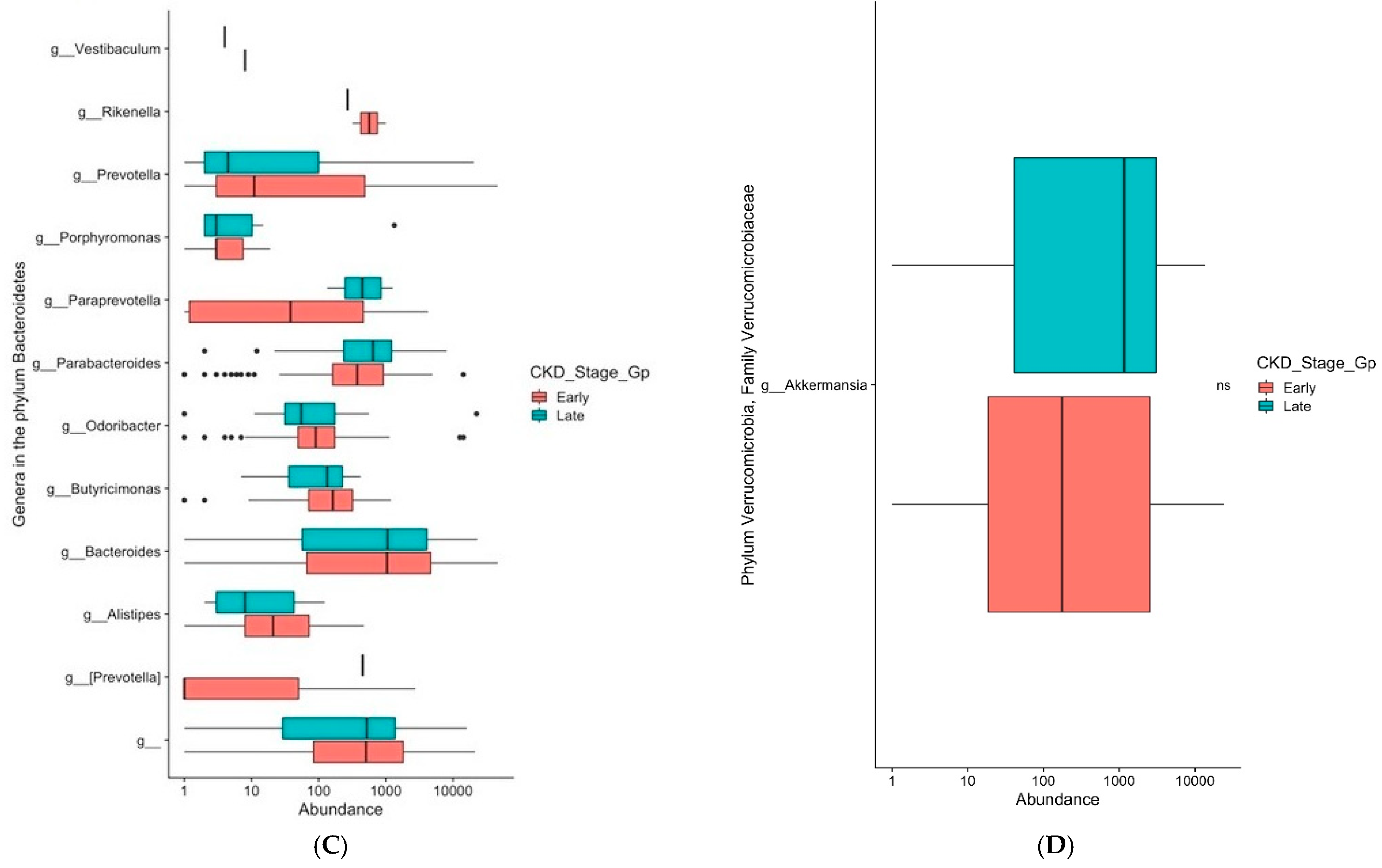
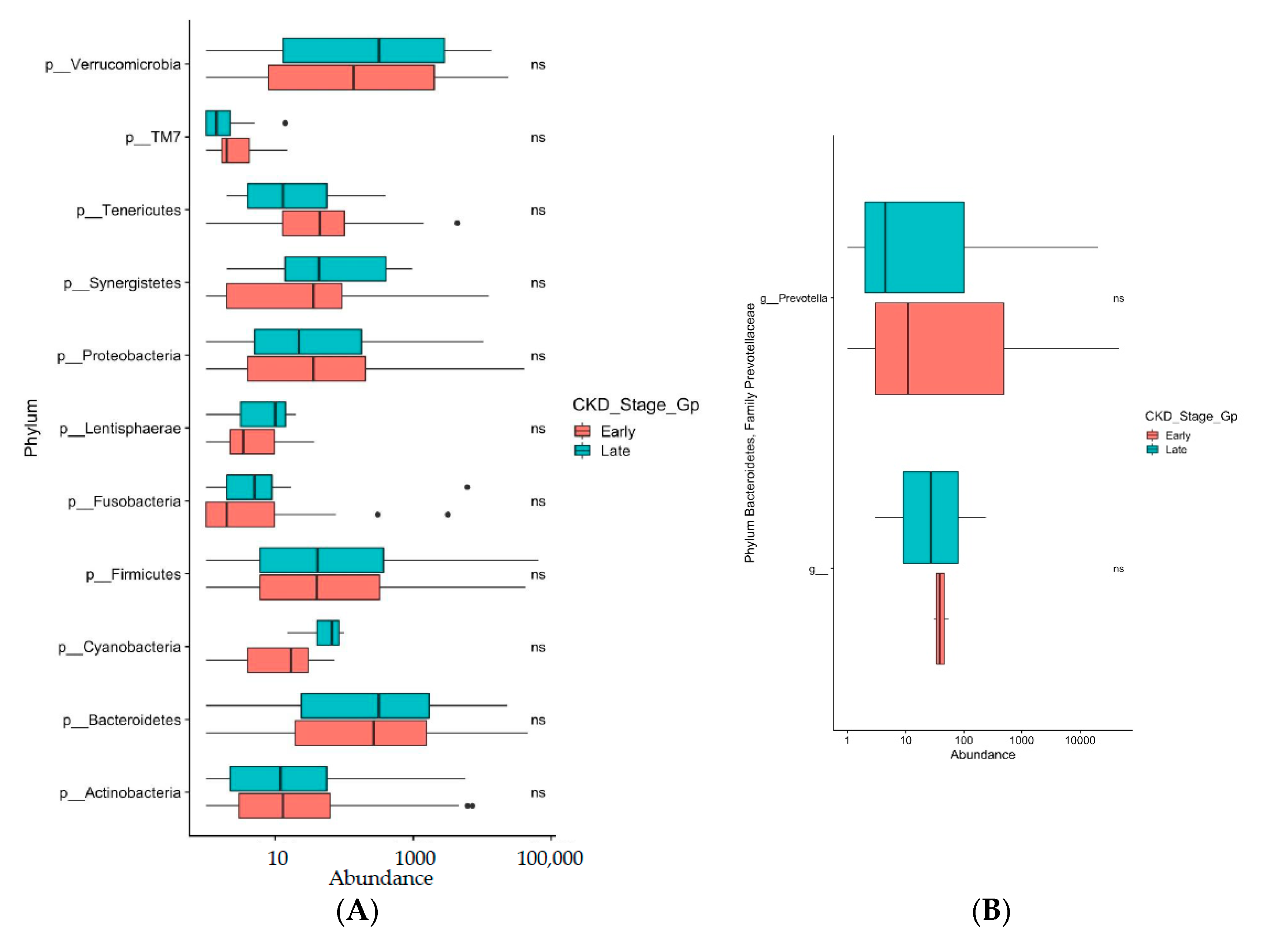
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Hooper, L.V.; Gordon, J.I. Commensal host-bacterial relationships in the gut. Science 2001, 292, 1115–1118. [Google Scholar] [CrossRef]
- Bourlioux, P.; Koletzko, B.; Guarner, F.; Braesco, V. The intestine and its microflora are partners for the protection of the host: Report on the Danone Symposium “The Intelligent Intestine”, held in Paris, June 14, 2002. Am. J. Clin. Nutr. 2003, 78, 675–683. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, J.M.; Murphy, K.; Stanton, C.; Ross, R.P.; Kober, O.I.; Juge, N.; Avershina, E.; Rudi, K.; Narbad, A.; Jenmalm, M.C.; et al. The composition of the gut microbiota throughout life, with an emphasis on early life. Microb. Ecol. Heal. Dis. 2015, 26, 26050. [Google Scholar] [CrossRef] [PubMed]
- Aron-Wisnewsky, J.; Clément, K. The gut microbiome, diet, and links to cardiometabolic and chronic disorders. Nat. Rev. Nephrol. 2015, 12, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Huttenhower, C.; Gevers, D.; Knight, R.; Abubucker, S.; Badger, J.H.; Chinwalla, A.T.; Creasy, H.H.; Earl, A.M.; FitzGerald, M.G.; Fulton, R.S.; et al. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Karlsson, F.H.; Tremaroli, V.; Nookaew, I.; Bergström, G.; Behre, C.J.; Fagerberg, B.; Nielsen, J. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 2013, 498, 99–103. [Google Scholar] [CrossRef]
- International Diabetes Federation (IDF). Diabetes Atlas, 7th ed.; International Diabetes Federation: Brussels, Belgium, 2015. [Google Scholar]
- US Renal Data System 2008. Annual Data Report. Atlas of Chronic Kidney Disease and End Stage Renal Disease in the United States; National Institutes of Health, National Institutes of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2008.
- Valmadrid, C.T.; Klein, R.; Moss, S.E.; Klein, B.E. The risk of cardiovascular disease mortality associated with microalbuminuria and gross proteinuria in persons with older-onset diabetes mellitus. Arch. Intern. Med. 2000, 160, 1093–1100. [Google Scholar] [CrossRef]
- Carrero, J.J.; Stenvinkel, P. Persistent Inflammation as a Catalyst for Other Risk Factors in Chronic Kidney Disease: A Hypothesis Proposal. Clin. J. Am. Soc. Nephrol. 2009, 4 (Suppl. 1), S49–S55. [Google Scholar] [CrossRef] [Green Version]
- Larsen, N.; Vogensen, F.K.; van den Berg, F.W.J.; Nielsen, D.S.; Andreasen, A.S.; Pedersen, B.K.; Abu Al-Soud, W.; Sørensen, S.J.; Hansen, L.H.; Jakobsen, M. Gut Microbiota in Human Adults with Type 2 Diabetes Differs from Non-Diabetic Adults. PLoS ONE. 2010, 5, e9085. [Google Scholar] [CrossRef]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Hatch, M.; Freel, R.W.; Vaziri, N.D. Intestinal excretion of oxalate in chronic renal failure. J. Am. Soc. Nephrol. 1994, 5, 1339–1343. [Google Scholar] [PubMed]
- Vaziri, N.D.; Freel, R.W.; Hatch, M. Effect of chronic experimental renal insufficiency on urate metabolism. J. Am. Soc. Nephrol. 1995, 6, 1313–1317. [Google Scholar] [PubMed]
- Xu, K.-Y.; Xia, G.-H.; Lu, J.-Q.; Chen, M.-X.; Zhen, X.; Wang, S.; You, C.; Nie, J.; Zhou, H.; Yin, J. Impaired renal function and dysbiosis of gut microbiota contribute to increased trimethylamine-N-oxide in chronic kidney disease patients. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Yacoub, R.; Nugent, M.; Cai, W.; Nadkarni, G.N.; Chaves, L.D.; Abyad, S.; Honan, A.M.; Thomas, S.A.; Zheng, W.; Valiyaparambil, S.A.; et al. Advanced glycation end products dietary restriction effects on bacterial gut microbiota in peritoneal dialysis patients; a randomized open label controlled trial. PLoS ONE 2017, 12, e0184789. [Google Scholar] [CrossRef] [Green Version]
- Sabatino, A.; Regolisti, G.; Cosola, C.; Gesualdo, L.; Fiaccadori, E. Intestinal Microbiota in Type 2 Diabetes and Chronic Kidney Disease. Curr. Diabetes Rep. 2017, 17, 16. [Google Scholar] [CrossRef]
- Felizardo, R.J.F.; Castoldi, A.; Andrade-Oliveira, V.; Câmara, N.O.S. The microbiota and chronic kidney diseases: A double-edged sword. Clin. Transl. Immunol. 2016, 5, e86. [Google Scholar] [CrossRef]
- Wong, J.; Piceno, Y.M.; DeSantis, T.Z.; Pahl, M.; Andersen, G.L.; Vaziri, N.D. Expansion of urease- and uricase-containing, indole- and p-cresol-forming and contraction of short-chain fatty acid-producing intestinal microbiota in ESRD. Am. J. Nephrol. 2014, 39, 230–237. [Google Scholar] [CrossRef] [Green Version]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.-M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef]
- de Andrade, L.S.; Ramos, C.I.; Cuppari, L. The cross-talk between the kidney and the gut: Implications for chronic kidney disease. Nutrire 2017, 42, 27. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Wong, J.; Pahl, M.V.; Piceno, Y.M.; Yuan, J.; DeSantis, T.Z.; Ni, Z.; Nguyen, T.-H.; Andersen, G.L. Chronic kidney disease alters intestinal microbial flora. Kidney Int. 2013, 83, 308–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Castelao, A.; Navarro-Gonzalez, J.F.; Gorriz, J.L.; de Alvaro, F. The Concept and the Epidemiology of Diabetic Nephropathy Have Changed in Recent Years. J. Clin. Med. 2015, 4, 1207–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foundation, N.K. ACR. Available online: https//www.kidney.org/kidneydisease/siemens_hcp_acr (accessed on 29 July 2020).
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C.-Y. Chronic Kidney Disease and the Risks of Death, Cardiovascular Events, and Hospitalization. N. Engl. J. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: A fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 2014, 30, 614–620. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [Green Version]
- McMurdie, P.J.; Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE. 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Shannon, C.E. A Mathematical Theory of Communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef] [Green Version]
- Bauer, D.F. Constructing Confidence Sets Using Rank Statistics. J. Am. Stat. Assoc. 1972, 67, 687. [Google Scholar] [CrossRef]
- Lozupone, C.; Knight, R. UniFrac: A New Phylogenetic Method for Comparing Microbial Communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozupone, C.; Lladser, M.E.; Knights, D.; Stombaugh, J.; Knight, R. UniFrac: An effective distance metric for microbial community comparison. ISME J. 2011, 5, 169–172. [Google Scholar] [CrossRef] [Green Version]
- Oksanen, J.; Kindt, R.; Legendre, P.; O’Hara, B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Eduard Szoecs, E.; Wagner, H. Vegan: Community Ecology Package. R package version 1.13-12. 2008. Available online: https://cran.r-project.org/web/packages/vegan/index.html (accessed on 29 December 2020).
- Gurung, M.; Li, Z.; You, H.; Rodrigues, R.; Jump, D.B.; Morgun, A.; Shulzhenko, N. Role of gut microbiota in type 2 diabetes pathophysiology. EBioMedicine 2020, 51, 102590. [Google Scholar] [CrossRef] [Green Version]
- Sircana, A.; Framarin, L.; Leone, N.; Berrutti, M.; Castellino, F.; Parente, R.; De Michieli, F.; Paschetta, E.; Musso, G. Altered Gut Microbiota in Type 2 Diabetes: Just a Coincidence? Curr. Diabetes Rep. 2018, 18, 98. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Yuan, J.; Nazertehrani, S.; Ni, Z.; Liu, S. Chronic Kidney Disease Causes Disruption of Gastric and Small Intestinal Epithelial Tight Junction. Am. J. Nephrol. 2013, 38, 99–103. [Google Scholar] [CrossRef] [Green Version]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. What is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef] [Green Version]
- Bammens, B.; Verbeke, K.; Vanrenterghem, Y.; Evenepoel, P. Evidence for impaired assimilation of protein in chronic renal failure. Kidney Int. 2003, 64, 2196–2203. [Google Scholar] [CrossRef]
- Moraes, C.; Fouque, D.; Amaral, A.C.; Mafra, D. Trimethylamine N-Oxide from Gut Microbiota in Chronic Kidney Disease Patients: Focus on Diet. J. Ren. Nutr. 2015, 25, 459–465. [Google Scholar] [CrossRef] [Green Version]
- Montemurno, E.; Cosola, C.; Dalfino, G.; Daidone, G.; De Angelis, M.; Gobbetti, M.; Gesualdo, L. What would you like to eat, Mr CKD Microbiota? A Mediterranean Diet, please! Kidney Blood Press. Res. 2014, 39, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Gradisteanu, G.; Stoica, R.; Petcu, L.; Picu, A.; Suceveanu, A.; Salmen, T.; Stefan, D.; Serafinceanu, C.; Chifiriuc, M.C.; Stoian, A.P. Microbiota signatures in type-2 diabetic patients with chronic kidney disease—A Pilot Study. J. Mind Med Sci. 2019, 6, 130–136. [Google Scholar] [CrossRef]
- Li, F.; Wang, M.; Wang, J.; Li, R.; Zhang, Y. Alterations to the Gut Microbiota and Their Correlation with Inflammatory Factors in Chronic Kidney Disease. Front. Cell. Infect. Microbiol. 2019, 9, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yacoub, R.; Nadkarni, G.N.; McSkimming, D.I.; Chaves, L.D.; Abyad, S.; Bryniarski, M.A.; Honan, A.M.; Thomas, S.A.; Gowda, M.; He, J.C.; et al. Fecal microbiota analysis of polycystic kidney disease patients according to renal function: A pilot study. Exp. Biol. Med. 2019, 244, 505–513. [Google Scholar] [CrossRef]
- Precup, G.; Vodnar, D.-C. Gut Prevotella as a possible biomarker of diet and its eubiotic versus dysbiotic roles: A comprehensive literature review. Br. J. Nutr. 2019, 122, 131–140. [Google Scholar] [CrossRef]
- den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy me-tabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [Green Version]
- Peng, L.; Li, Z.R.; Green, R.S.; Holzman, I.R.; Lin, J. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J. Nutr. 2009, 139, 1619–1625. [Google Scholar] [CrossRef]
- Keku, T.O.; Dulal, S.; Deveaux, A.; Jovov, B.; Han, X. The gastrointestinal microbiota and colorectal cancer. Am. J. Physiol. Liver Physiol. 2015, 308, G351–G363. [Google Scholar] [CrossRef] [Green Version]
- Staples, A.; Wong, C. Risk factors for progression of chronic kidney disease. Curr. Opin. Pediatr. 2010, 22, 161–169. [Google Scholar] [CrossRef] [Green Version]
- Wing, M.R.; Ramezani, A.; Gill, H.S.; Devaney, J.M.; Raj, D.S. Epigenetics of progression of chronic kidney disease: Fact or fantasy? Semin. Nephrol. 2013, 33, 363–374. [Google Scholar] [CrossRef] [Green Version]
- Sabatino, A.; Regolisti, G.; Brusasco, I.; Cabassi, A.; Morabito, S.; Fiaccadori, E. Alterations of intestinal barrier and microbiota in chronic kidney disease. Nephrol. Dial. Transplant. 2014, 30, 924–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hand, T.W.; Vujkovic-Cvijin, I.; Ridaura, V.K.; Belkaid, Y. Linking the Microbiota, Chronic Disease, and the Immune System. Trends Endocrinol. Metab. 2016, 27, 831–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, M.; Campbell, K.L.; Johnson, D.W.; Stanton, T.; Vesey, D.A.; Coombes, J.S.; Weston, K.S.; Hawley, C.M.; McWhinney, B.C.; Ungerer, J.P.; et al. Protein-bound uremic toxins, inflammation and oxidative stress: A cross-sectional study in stage 3–4 chronic kidney disease. Arch. Med Res. 2014, 45, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Darisipudi, M.N.; Knauf, F. An update on the role of the inflammasomes in the pathogenesis of kidney diseases. Pediatr. Nephrol. 2015, 31, 535–544. [Google Scholar] [CrossRef]
- Turin, T.C.; Tonelli, M.; Manns, B.J.; Ahmed, S.B.; Ravani, P.; James, M.; Hemmelgarn, B.R. Lifetime risk of ESRD. J. Am. Soc. Nephrol. 2012, 23, 1569–1578. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Cycle | Initial | Disassociate | Anneal | Extension | Finish |
|---|---|---|---|---|---|---|
| 16S: V3–V4 | 29 | 95 °C for 7 min | 94 °C for 30 s | 50 °C for 60 s | 72 °C for 60 s | 72 °C for 7 min |
| Target | 341F-806R | |||||
| Forward Primer(341F) | CCTAYGGGRBGCASCAG | |||||
| ReversePrimer(806R) | GGACTACNNGGGTATCTAAT | |||||
| Patient Characteristics | Mean Early CKD (Group 1) | SD | Mean Late CKD (Group 2) | SD | p-Value (Wilcoxon Signed Rank Test) | p-Value (Welch Two Sample t-Test) |
|---|---|---|---|---|---|---|
| Age (years) | 66.24 | 10.22 | 72.68 | 10.21 | 0.01 | 0.01 |
| Male (%) | 80.70 | 19.30 | 0.09 | |||
| Type of Diabetes | 0.83 | 0.42 | 0.92 | 0.28 | 0.29 | 0.22 |
| Type 1 (%) | 86.67 | 13.33 | ||||
| Type 2 (%) | 70.89 | 29.11 | ||||
| Latent autoimmune diabetes in adults (LADA) (%) | 100.00 | 0.00 | ||||
| Duration of Diabetes (years) | 18.81 | 11.63 | 21.00 | 10.99 | 0.32 | 0.41 |
| Diabetic Retinopathy (%) | 0.39 | 0.49 | 0.40 | 0.50 | 0.90 | 0.90 |
| Cardiovascular Disease (%) | 57.89 | 42.11 | 0.09 | |||
| Stroke/Transient Ischaemic Attack (TIA%) | 0.13 | 0.34 | 0.12 | 0.33 | 0.92 | 0.91 |
| Anxiety | 6.77 | 4.22 | 8.32 | 4.98 | 0.30 | 0.17 |
| Depression | 5.64 | 3.67 | 7.00 | 3.74 | 0.11 | 0.12 |
| Smoking quantity pack_year | 9.83 | 13.12 | 6.60 | 11.70 | 0.22 | 0.26 |
| Smoking status (%) | 0.51 | 0.61 | 0.32 | 0.56 | 0.14 | 0.15 |
| Non-smoker (%) | 67.86 | 32.14 | ||||
| Ex-smoker (%) | 82.35 | 17.65 | ||||
| Current-smoker (%) | 80.00 | 20.00 | ||||
| Body Mass Index (BMI)(kg/m2) | 29.64 | 7.46 | 30.12 | 5.23 | 0.40 | 0.73 |
| Systolic blood pressure (SBP) (mmHg) | 127.83 | 27.37 | 135.04 | 29.32 | 0.74 | 0.29 |
| Diastolic blood pressure (DBP) (mmHg) | 73.30 | 15.91 | 71.72 | 9.86 | 0.21 | 0.57 |
| Haemoglobin (Hb) (g/L) | 132.66 | 16.92 | 117.72 | 12.72 | 0.00 | 0.00 |
| Estimated glomerular filteration rate (eGFR) (mL/min/1.73 m2) | 67.51 | 13.68 | 24.48 | 10.68 | 0.00 | 0.00 |
| Glycated haemoglobin (HbA1c) (%) | 7.68 | 1.95 | 6.86 | 3.35 | 0.62 | 0.25 |
| Urine Albumin/Creatinine ratio (ACR) | 3.37 | 10.52 | 33.65 | 122.81 | 0.97 | 0.23 |
| Urine Protein/Creatinine ratio (PCR) | 0.02 | 0.03 | 0.06 | 0.16 | 0.99 | 0.15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lecamwasam, A.; Nelson, T.M.; Rivera, L.; Ekinci, E.I.; Saffery, R.; Dwyer, K.M. Gut Microbiome Composition Remains Stable in Individuals with Diabetes-Related Early to Late Stage Chronic Kidney Disease. Biomedicines 2021, 9, 19. https://doi.org/10.3390/biomedicines9010019
Lecamwasam A, Nelson TM, Rivera L, Ekinci EI, Saffery R, Dwyer KM. Gut Microbiome Composition Remains Stable in Individuals with Diabetes-Related Early to Late Stage Chronic Kidney Disease. Biomedicines. 2021; 9(1):19. https://doi.org/10.3390/biomedicines9010019
Chicago/Turabian StyleLecamwasam, Ashani, Tiffanie M. Nelson, Leni Rivera, Elif I. Ekinci, Richard Saffery, and Karen M. Dwyer. 2021. "Gut Microbiome Composition Remains Stable in Individuals with Diabetes-Related Early to Late Stage Chronic Kidney Disease" Biomedicines 9, no. 1: 19. https://doi.org/10.3390/biomedicines9010019