
RABL6A Promotes Pancreatic Neuroendocrine Tumor Angiogenesis and Progression In Vivo

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Genotyping
2.3. Mouse Blood Withdrawal and Insulin ELISA
2.4. Islet Isolation
2.5. RNA Isolation and qRT-PCR Analysis
2.6. qRT-PCR Primers
2.7. Analysis of Tumor Burden
2.8. Histopathological Analyses
2.9. Western Blotting and Antibodies
2.10. RNA Interference in pNET Cells
2.11. Statistics
3. Results
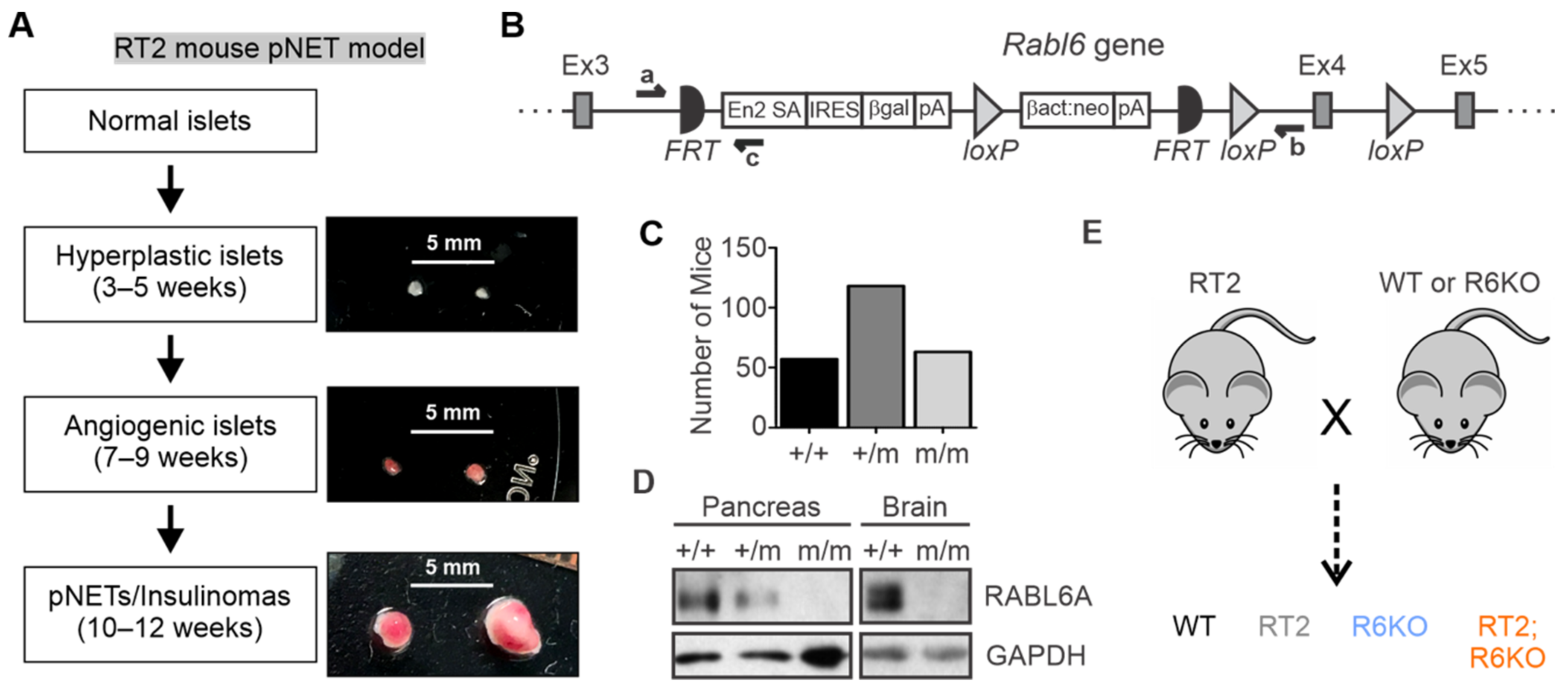
3.1. Description and Generation of the Mouse Models
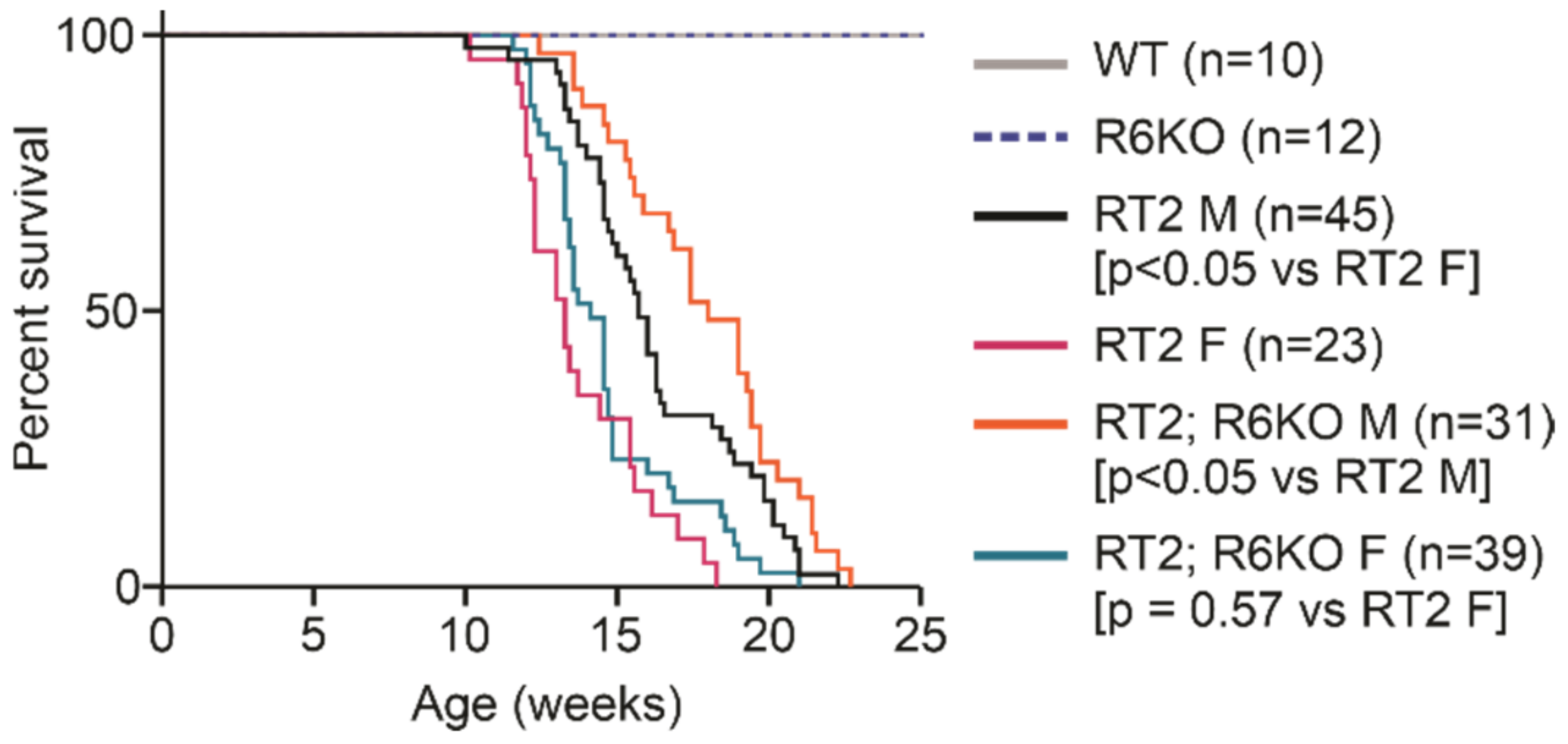
3.2. RABL6A Expression Reduces Survival in RT2 Mice
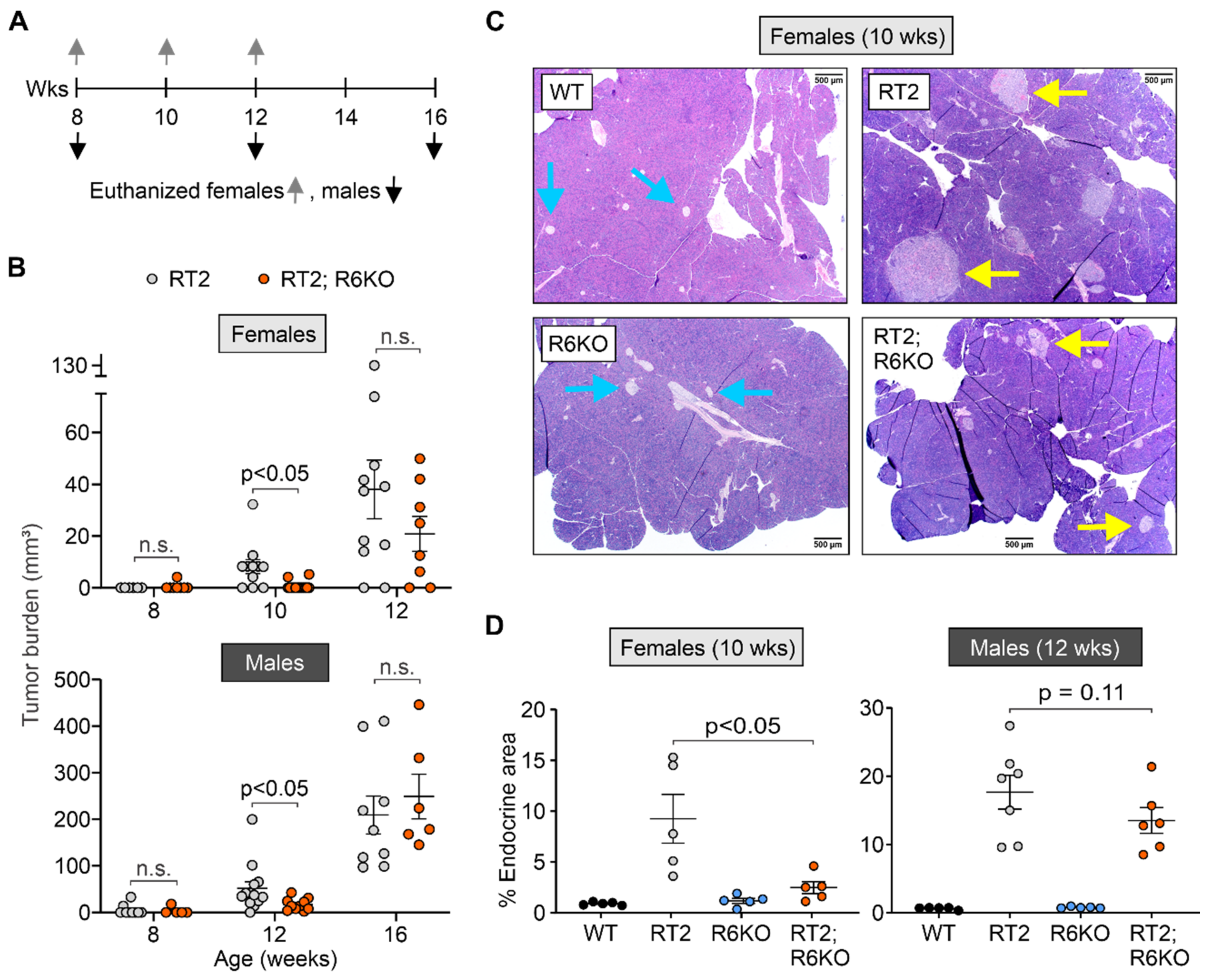
3.3. RABL6A Increases Tumor Burden in RT2 Mice in an Age-Dependent Manner
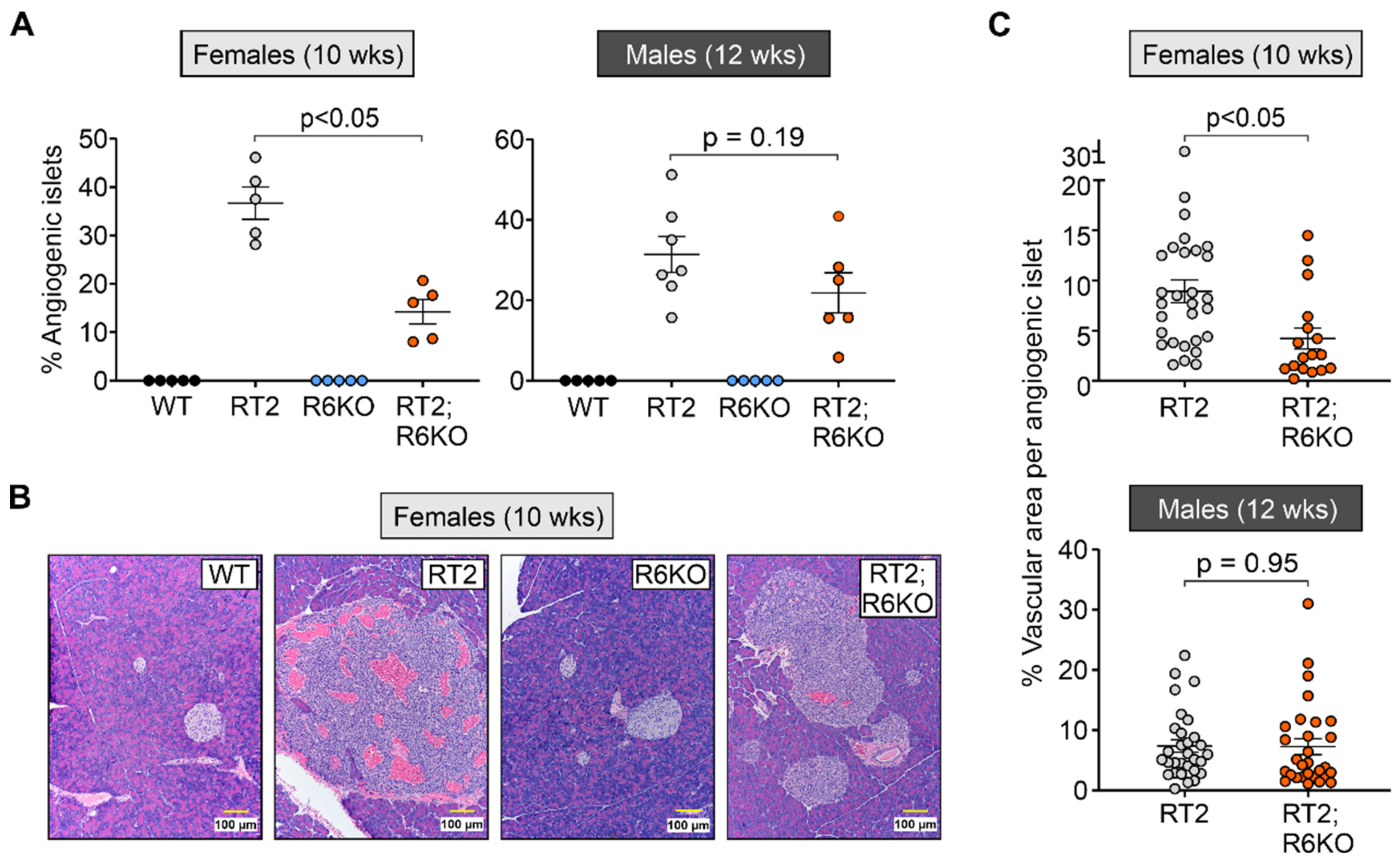
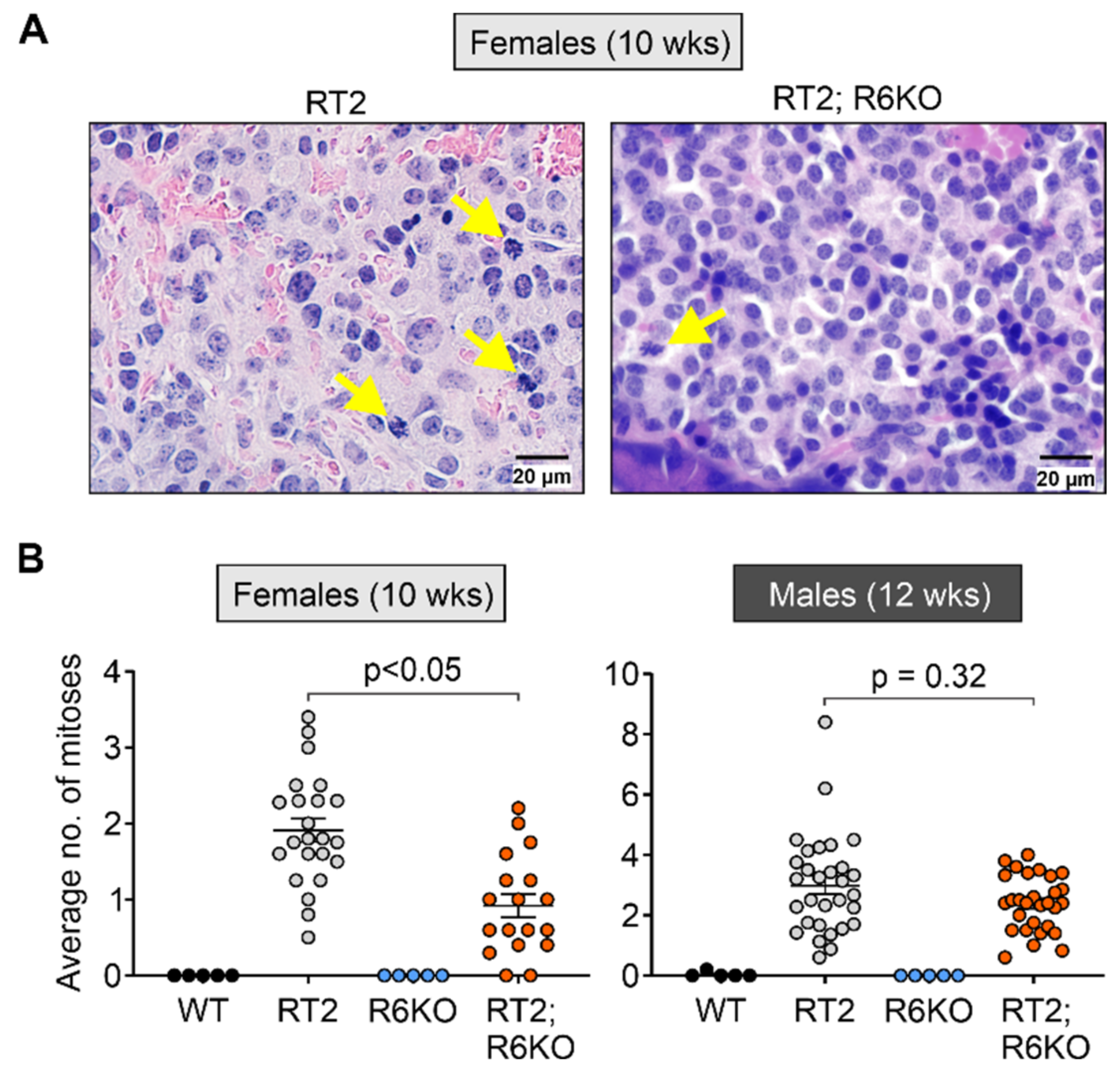
3.4. RABL6A Promotes Angiogenesis during Early Stages of pNET Formation
3.5. RABL6A Drives pNET Cell Mitosis Early in Tumor Development
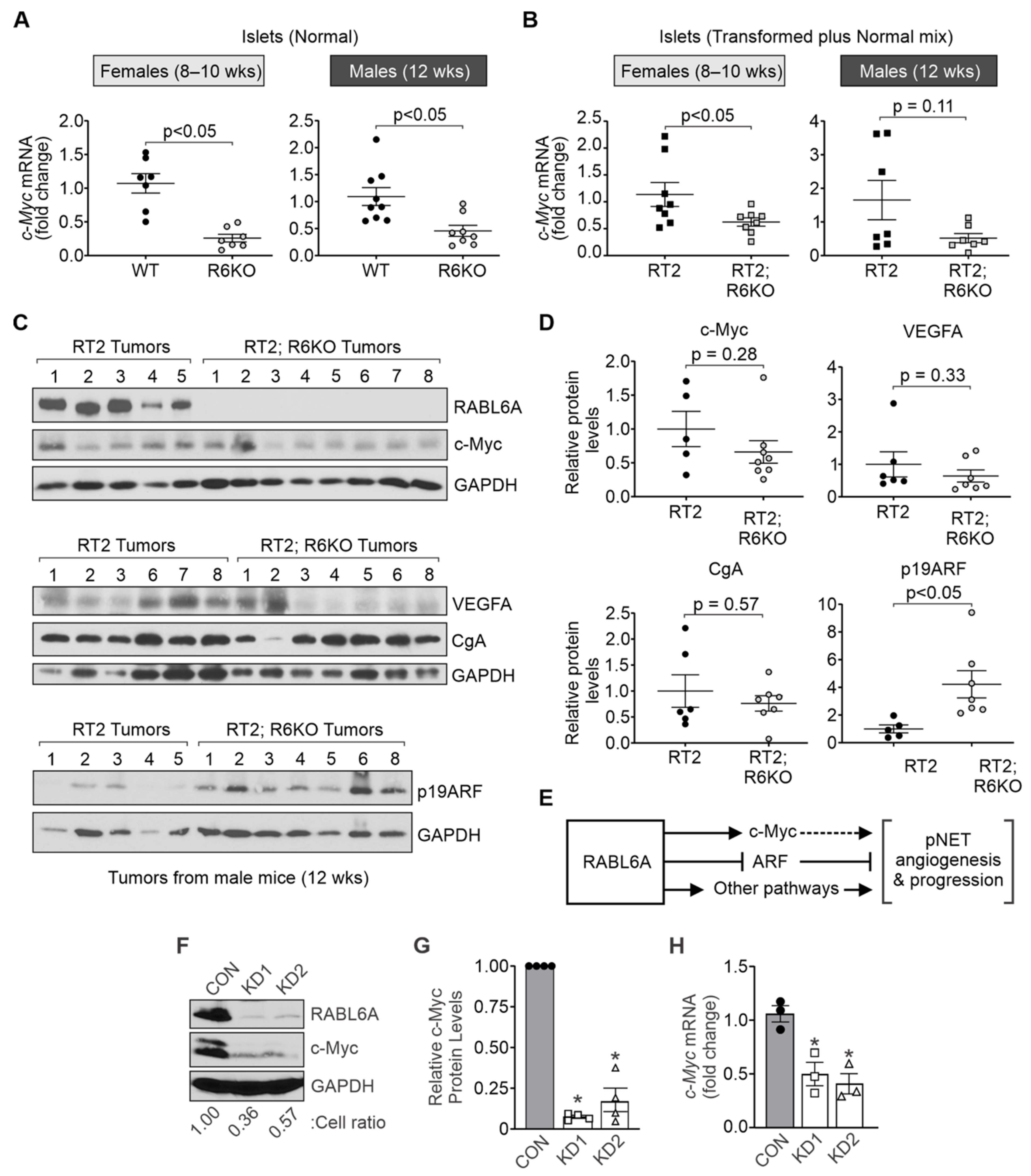
3.6. RABL6A Promotes c-Myc and Limits ARF Expression in Mouse Pancreatic Islets
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Scott, A.T.; Howe, J.R. Evaluation and Management of Neuroendocrine Tumors of the Pancreas. Surg. Clin. N. Am. 2019, 99, 793–814. [Google Scholar] [CrossRef] [PubMed]
- Halfdanarson, T.R.; Rabe, K.G.; Rubin, J.; Petersen, G.M. Pancreatic neuroendocrine tumors (PNETs): Incidence, prognosis and recent trend toward improved survival. Ann. Oncol. 2008, 19, 1727–1733. [Google Scholar] [CrossRef] [PubMed]
- Sun, J. Pancreatic neuroendocrine tumors. Intractable Rare Dis. Res. 2017, 6, 21–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasari, A.; Shen, C.; Halperin, D.M.; Zhao, B.; Zhou, S.; Xu, Y.; Shih, T.; Yao, J.C. Trends in the Incidence, Prevalence, and Survival Outcomes in Patients with Neuroendocrine Tumors in the United States. JAMA Oncol. 2017, 3, 1335–1342. [Google Scholar] [CrossRef]
- Akirov, A.; Larouche, V.; AlShehri, S.; Asa, S.L.; Ezzat, S. Treatment Options for Pancreatic Neuroendocrine Tumors. Cancers 2019, 11, 828. [Google Scholar] [CrossRef] [Green Version]
- Tompkins, V.; Hagen, J.; Zediak, V.P.; Quelle, D.E. Identification of Novel ARF Binding Proteins by Two-Hybrid Screening. Cell Cycle 2006, 5, 642–647. [Google Scholar] [CrossRef] [Green Version]
- Montalbano, J.; Lui, K.; Sheikh, M.S.; Huang, Y. Identification and Characterization of RBEL1 Subfamily of GTPases in the Ras Superfamily Involved in Cell Growth Regulation. J. Biol. Chem. 2009, 284, 18129–18142. [Google Scholar] [CrossRef] [Green Version]
- Muniz, V.P.; Askeland, R.W.; Zhang, X.; Reed, S.M.; Tompkins, V.S.; Hagen, J.; McDowell, B.D.; Button, A.; Smith, B.J.; Weydert, J.A.; et al. RABL6A Promotes Oxaliplatin Resistance in Tumor Cells and Is a New Marker of Survival for Resected Pancreatic Ductal Adenocarcinoma Patients. Genes Cancer 2013, 4, 273–284. [Google Scholar] [CrossRef] [Green Version]
- Lui, K.; An, J.; Montalbano, J.; Shi, J.; Corcoran, C.; He, Q.; Sun, H.; Sheikh, M.S.; Huang, Y. Negative regulation of p53 by Ras superfamily protein RBEL1A. J. Cell Sci. 2013, 126, 2436–2445. [Google Scholar] [CrossRef] [Green Version]
- Hagen, J.; Muniz, V.P.; Falls, K.C.; Reed, S.M.; Taghiyev, A.F.; Quelle, F.; Gourronc, F.A.; Klingelhutz, A.; Major, H.J.; Askeland, R.W.; et al. RABL6A Promotes G1–S Phase Progression and Pancreatic Neuroendocrine Tumor Cell Proliferation in an Rb1-Dependent Manner. Cancer Res. 2014, 74, 6661–6670. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Ji, F.; Sun, J.; Xie, Y.; Xu, Y.; Yue, H. RBEL1 is required for osteosarcoma cell proliferation via inhibiting retinoblastoma 1. Mol. Med. Rep. 2015, 13, 1275–1280. [Google Scholar] [CrossRef] [Green Version]
- Umesalma, S.; Kaemmer, C.A.; Kohlmeyer, J.L.; Letney, B.L.; Schab, A.M.; Reilly, J.A.; Sheehy, R.; Hagen, J.; Tiwari, N.; Zhan, F.; et al. RABL6A inhibits tumor-suppressive PP2A/AKT signaling to drive pancreatic neuroendocrine tumor growth. J. Clin. Investig. 2019, 129, 1641–1653. [Google Scholar] [CrossRef]
- Feng, Y.; Yan, S.; Huang, Y.; Huang, Q.; Wang, F.; Lei, Y. High expression of RABL6 promotes cell proliferation and predicts poor prognosis in esophageal squamous cell carcinoma. BMC Cancer 2020, 20, 602. [Google Scholar] [CrossRef]
- Li, Y.-Y.; Fu, S.; Wang, X.-P.; Wang, H.-Y.; Zeng, M.-S.; Shao, J.-Y. Down-Regulation of C9orf86 in Human Breast Cancer Cells Inhibits Cell Proliferation, Invasion and Tumor Growth and Correlates with Survival of Breast Cancer Patients. PLoS ONE 2013, 8, e71764. [Google Scholar] [CrossRef] [Green Version]
- Kohlmeyer, J.L.; Kaemmer, C.A.; Pulliam, C.; Maharjan, C.K.; Moreno Samayoa, A.; Major, H.J.; Cornick, K.E.; Knepper-Adrian, V.; Khanna, R.; Sieren, J.C.; et al. RABL6A Is an Essential Driver of MPNSTs that Negatively Regulates the RB1 Pathway and Sensitizes Tumor Cells to CDK4/6 Inhibitors. Clin. Cancer Res. 2020, 26, 2997. [Google Scholar] [CrossRef] [Green Version]
- Scott, A.T.; Weitz, M.; Breheny, P.J.; Ear, P.H.; Darbro, B.; Brown, B.J.; Braun, T.A.; Li, G.; Umesalma, S.; Kaemmer, C.A.; et al. Gene Expression Signatures Identify Novel Therapeutics for Metastatic Pancreatic Neuroendocrine Tumors. Clin. Cancer Res. 2020, 26, 2011–2021. [Google Scholar] [CrossRef]
- Bergers, G.; Song, S.; Meyer-Morse, N.; Bergsland, E.; Hanahan, D. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J. Clin. Investig. 2003, 111, 1287–1295. [Google Scholar] [CrossRef] [Green Version]
- Casanovas, O.; Hicklin, D.J.; Bergers, G.; Hanahan, D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 2005, 8, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Chiu, C.W.; Nozawa, H.; Hanahan, D. Survival Benefit With Proapoptotic Molecular and Pathologic Responses From Dual Targeting of Mammalian Target of Rapamycin and Epidermal Growth Factor Receptor in a Preclinical Model of Pancreatic Neuroen-docrine Carcinogenesis. J. Clin. Oncol. 2010, 28, 4425–4433. [Google Scholar] [CrossRef] [Green Version]
- Christofori, G.; Naik, P.; Hanahan, D. A second signal supplied by insulin-like growth factor II in oncogene-induced tumorigenesis. Nat. Cell Biol. 1994, 369, 414–418. [Google Scholar] [CrossRef]
- Hanahan, D. Heritable formation of pancreatic beta-cell tumours in transgenic mice expressing recombinant insulin/simian virus 40 oncogenes. Nature 1985, 315, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Folkman, J. Patterns and Emerging Mechanisms of the Angiogenic Switch during Tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef] [Green Version]
- Inoue, M.; Hager, J.H.; Farrara, N.; Gerber, H.-P.; Hanahan, D. VEGF-A has a critical, nonredundant role in angiogenic switching and pancreatic beta cell carcinogenesis. Cancer Cell 2002, 1, 193–202. [Google Scholar] [CrossRef] [Green Version]
- Olson, P.; Gerald, C.C.; Perry, S.R.; Nolan-Stevaux, O.; Hanahan, D. Imaging guided trials of the angiogenesis inhibitor sunitinib in mouse models predict efficacy in pancreatic neuroendocrine but not ductal carcinoma. Proc. Natl. Acad. Sci. USA 2011, 108, E1275–E1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parangi, S.; Dietrich, W.; Christofori, G.; Lander, E.S.; Hanahan, D. Tumor suppressor loci on mouse chromosomes 9 and 16 are lost at distinct stages of tumorigenesis in a transgenic model of islet cell carcinoma. Cancer Res. 1995, 55, 6071–6076. [Google Scholar]
- Sodir, N.M.; Swigart, L.B.; Karnezis, A.N.; Hanahan, D.; Evan, G.I.; Soucek, L. Endogenous Myc maintains the tumor microenvironment. Genes Dev. 2011, 25, 907–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulanet, D.B.; Hanahan, D. Loss of p19Arf Facilitates the Angiogenic Switch and Tumor Initiation in a Multi-Stage Cancer Model via p53-Dependent and Independent Mechanisms. PLoS ONE 2010, 5, e12454. [Google Scholar] [CrossRef] [Green Version]
- Kang, C.; Xie, L.; Gunasekar, S.K.; Mishra, A.; Zhang, Y.; Pai, S.; Gao, Y.; Kumar, A.; Norris, A.W.; Stephens, S.B.; et al. SWELL1 is a glucose sensor regulating β-cell excitability and systemic glycaemia. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Ling, J.; Gallie, D.R. RNase activity requires formation of disulfide bonds and is regulated by the redox state. Plant Mol. Biol. 2004, 55, 83–96. [Google Scholar] [CrossRef]
- Meyerholz, D.K.; Beck, A.P. Principles and approaches for reproducible scoring of tissue stains in research. Lab. Investig. 2018, 98, 844–855. [Google Scholar] [CrossRef]
- Townsend, C.M.; Ishizuka, J.; Thompson, J.C. Studies of Growth Regulation in a Neuroendocrine Cell Line. Acta Oncol. 1993, 32, 125–130. [Google Scholar] [CrossRef] [Green Version]
- Folkman, J.; Watson, K.; Ingber, D.; Hanahan, D. Induction of angiogenesis during the transition from hyperplasia to neoplasia. Nat. Cell Biol. 1989, 339, 58–61. [Google Scholar] [CrossRef]
- Rindi, G.; Klimstra, D.S.; Abedi-Ardekani, B.; Asa, S.L.; Bosman, F.T.; Brambilla, E.; Busam, K.J.; De Krijger, R.R.; Dietel, M.; El-Naggar, A.K.; et al. A common classification framework for neuroendocrine neoplasms: An International Agency for Research on Cancer (IARC) and World Health Organization (WHO) expert consensus proposal. Mod. Pathol. 2018, 31, 1770–1786. [Google Scholar] [CrossRef]
- Pelengaris, S.; Khan, M.; Evan, I.G. Suppression of Myc-Induced Apoptosis in β Cells Exposes Multiple Oncogenic Properties of Myc and Triggers Carcinogenic Progression. Cell 2002, 109, 321–334. [Google Scholar] [CrossRef] [Green Version]
- Baudino, T.A.; McKay, C.; Pendeville-Samain, H.; Nilsson, J.A.; MacLean, K.H.; White, E.L.; Davis, A.C.; Ihle, J.N.; Cleveland, J.L. c-Myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev. 2002, 16, 2530–2543. [Google Scholar] [CrossRef] [Green Version]
- Raymond, E.; Dahan, L.; Raoul, J.-L.; Bang, Y.-J.; Borbath, I.; Lombard-Bohas, C.; Valle, J.; Metrakos, P.; Smith, D.; Vinik, A.; et al. Sunitinib Malate for the Treatment of Pancreatic Neuroendocrine Tumors. N. Engl. J. Med. 2011, 364, 501–513. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.C.; Shah, M.H.; Ito, T.; Bohas, C.L.; Wolin, E.M.; Van Cutsem, E.; Hobday, T.J.; Okusaka, T.; Capdevila, J.; De Vries, E.G.; et al. Everolimus for Advanced Pancreatic Neuroendocrine Tumors. N. Engl. J. Med. 2011, 364, 514–523. [Google Scholar] [CrossRef] [Green Version]
- Strosberg, J.R.; Halfdanarson, T.R.; Bellizzi, A.M.; Chan, J.A.; Dillon, J.S.; Heaney, A.P.; Kunz, P.L.; O’Dorisio, T.M.; Salem, R.; Segelov, E.; et al. The North American Neuroendocrine Tumor Society Consensus Guidelines for Surveillance and Medical Man-agement of Midgut Neuroendocrine Tumors. Pancreas 2017, 46, 707–714. [Google Scholar] [CrossRef]
- Jiao, Y.; Shi, C.; Edil, B.H.; De Wilde, R.F.; Klimstra, D.S.; Maitra, A.; Schulick, R.D.; Tang, L.H.; Wolfgang, C.L.; Choti, M.A.; et al. DAXX/ATRX, MEN1, and mTOR Pathway Genes Are Frequently Altered in Pancreatic Neuroendocrine Tumors. Science 2011, 331, 1199–1203. [Google Scholar] [CrossRef] [Green Version]
- Missiaglia, E.; Dalai, I.; Barbi, S.; Beghelli, S.; Falconi, M.; della Peruta, M.; Ouemonti, L.; Capurso, G.; Di Florio, A.; della Fave, G.; et al. Pancreatic Endocrine Tumors: Expression Profiling Evidences a Role for AKT-mTOR Pathway. J. Clin. Oncol. 2010, 28, 245–255. [Google Scholar] [CrossRef] [Green Version]
- Aparicio-Gallego, G.; Blanco, M.; Figueroa, A.; García-Campelo, R.; Valladares-Ayerbes, M.; Grande-Pulido, E.; Antón-Aparicio, L. New Insights into Molecular Mechanisms of Sunitinib-Associated Side Effects. Mol. Cancer Ther. 2011, 10, 2215–2223. [Google Scholar] [CrossRef] [Green Version]
- Chang, T.-M.; Shan, Y.-S.; Chu, P.-Y.; Jiang, S.S.; Hung, W.-C.; Chen, Y.-L.; Tu, H.-C.; Lin, H.Y.; Tsai, H.-J.; Chen, L.-T. The regulatory role of aberrant Phosphatase and Tensin Homologue and Liver Kinase B1 on AKT/mTOR/c-Myc axis in pancreatic neuroendocrine tumors. Oncotarget 2017, 8, 98068–98083. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, E.V.; Ravitz, M.J.; Chen, L.; Lynch, M. Growth controls connect: Interactions between c-myc and the tuberous sclerosis complex-mTOR pathway. Cell Cycle 2009, 8, 1344–1351. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Signal transduction to hypoxia-inducible factor 1. Biochem. Pharmacol. 2002, 64, 993–998. [Google Scholar] [CrossRef]
- Ngo, C.V.; Gee, M.; Akhtar, N.; Yu, D.; Volpert, O.; Auerbach, R.; Thomas-Tikhonenko, A. An in vivo function for the transforming Myc protein: Elicitation of the angiogenic phenotype. Cell Growth Differ. Mol. Boil. J. Am. Assoc. Cancer Res. 2000, 11, 201–210. [Google Scholar]
- Janz, A. Activation of the Myc oncoprotein leads to increased turnover of thrombospondin-1 mRNA. Nucleic Acids Res. 2000, 28, 2268–2275. [Google Scholar] [CrossRef] [Green Version]
- Shchors, K.; Shchors, E.; Rostker, F.; Lawlor, E.R.; Brown-Swigart, L.; Evan, G.I. The Myc-dependent angiogenic switch in tumors is mediated by interleukin 1beta. Genes Dev. 2006, 20, 2527–2538. [Google Scholar] [CrossRef] [Green Version]
- Karar, J.; Maity, A. PI3K/AKT/mTOR Pathway in Angiogenesis. Front. Mol. Neurosci. 2011, 4, 51. [Google Scholar] [CrossRef] [Green Version]
- Xie, F.; Bao, X.; Yu, J.; Chen, W.; Wang, L.; Zhang, Z.; Xu, Q. Disruption and inactivation of the PP2A complex promotes the proliferation and angiogenesis of hemangioma endothelial cells through activating AKT and ERK. Oncotarget 2015, 6, 25660–25676. [Google Scholar] [CrossRef] [Green Version]
- Kuo, M.-L.; Besten, W.D.; Bertwistle, D.; Roussel, M.F.; Sherr, C.J. N-terminal polyubiquitination and degradation of the Arf tumor suppressor. Genes Dev. 2004, 18, 1862–1874. [Google Scholar] [CrossRef] [Green Version]
- Besten, W.D.; Kuo, M.-L.; Williams, R.T.; Sherr, C.J. Myeloid Leukemia-Associated Nucleophosmin Mutants Perturb p53-Dependent and Independent Activities of the Arf Tumor Suppressor Protein. Cell Cycle 2005, 4, 1593–1598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, E.; Bonetti, P.; Denchi, E.L.; Martinelli, P.; Zamponi, R.; Marine, J.-C.; Helin, K.; Falini, B.; Pelicci, P.G. Nucleophosmin Is Required for DNA Integrity and p19Arf Protein Stability. Mol. Cell. Biol. 2005, 25, 8874–8886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, J.; Seong, D.; Lee, S.R.; Oh, D.-B.; Song, J. Post-Translational Regulation of ARF: Perspective in Cancer. Biomolecules 2020, 10, 1143. [Google Scholar] [CrossRef] [PubMed]
- Rodway, H.; Llanos, S.; Rowe, J.; Peters, G. Stability of nucleolar versus non-nucleolar forms of human p14ARF. Oncogene 2004, 23, 6186–6192. [Google Scholar] [CrossRef] [Green Version]
- Ulanet, D.B.; Ludwig, D.L.; Kahn, C.R.; Hanahan, D. Insulin receptor functionally enhances multistage tumor progression and conveys intrinsic resistance to IGF-1R targeted therapy. Proc. Natl. Acad. Sci. USA 2010, 107, 10791–10798. [Google Scholar] [CrossRef] [Green Version]
- Reidy-Lagunes, D.L.; Vakiani, E.; Ba, M.F.S.; Hollywood, E.M.; Tang, L.H.; Solit, D.B.; Pietanza, M.C.; Capanu, M.; Saltz, L.B. A phase 2 study of the insulin-like growth factor-1 receptor inhibitor MK-0646 in patients with metastatic, well-differentiated neuroendocrine tumors. Cancer 2012, 118, 4795–4800. [Google Scholar] [CrossRef] [Green Version]
- Tang, L.H.; Contractor, T.; Clausen, R.; Klimstra, D.S.; Du, Y.-C.N.; Allen, P.J.; Brennan, M.F.; Levine, A.J.; Harris, C.R. Attenuation of the Retinoblastoma Pathway in Pancreatic Neuroendocrine Tumors Due to Increased Cdk4/Cdk6. Clin. Cancer Res. 2012, 18, 4612–4620. [Google Scholar] [CrossRef] [Green Version]
- Muscarella, P.; Melvin, W.S.; Fisher, E.W.; Foor, J.; Ellison, E.C.; Herman, J.G.; Schirmer, W.J.; Hitchcock, C.L.; Deyoung, B.R.; Weghorst, C.M. Genetic alterations in gastrinomas and nonfunctioning pancreatic neuroendocrine tumors: An analysis of p16/MTS1 tumor suppressor gene inactivation. Cancer Res. 1998, 58, 237–240. [Google Scholar]
- Serrano, J.; Goebel, S.U.; Peghini, P.L.; Lubensky, I.A.; Gibril, F.; Jensen, R.T. Alterations in the p16INK4a/CDKN2A tumor suppressor gene in gastrinomas. J. Clin. Endocrinol. Metab. 2000, 85, 4146–4156. [Google Scholar] [CrossRef]
- Soler, A.; Figueiredo, A.M.; Castel, P.; Martin, L.; Monelli, E.; Angulo-Urarte, A.; Milà-Guasch, M.; Viñals, F.; Baselga, J.; Casanovas, O.; et al. Therapeutic Benefit of Selective Inhibition of p110α PI3-Kinase in Pancreatic Neuroendocrine Tumors. Clin. Cancer Res. 2016, 22, 5805–5817. [Google Scholar] [CrossRef] [Green Version]
- Crabtree, J.S.; Scacheri, P.C.; Ward, J.M.; McNally, S.R.; Swain, G.P.; Montagna, C.; Hager, J.H.; Hanahan, D.; Edlund, H.; Magnuson, M.A.; et al. Of Mice and MEN1: Insulinomas in a Conditional Mouse Knockout. Mol. Cell. Biol. 2003, 23, 6075–6085. [Google Scholar] [CrossRef] [Green Version]
- Bertolino, P.; Tong, W.-M.; Herrera, P.L.; Casse, H.; Zhang, C.X.; Wang, Z.-Q. Pancreatic beta-cell-specific ablation of the multiple endocrine neoplasia type 1 (MEN1) gene causes full penetrance of insulinoma development in mice. Cancer Res. 2003, 63, 4836–4841. [Google Scholar]
- Jensen, R.T.; Berna, M.J.; Bingham, D.B.; Norton, J.A. Inherited pancreatic endocrine tumor syndromes: Advances in molecular pathogenesis, diagnosis, management, and controversies. Cancer 2008, 113, 1807–1843. [Google Scholar] [CrossRef] [Green Version]
- Scarpa, A.; Chang, D.K.; Nones, K.; Corbo, V.; Patch, A.-M.; Bailey, P.; Lawlor, R.T.; Johns, A.L.; Miller, D.K.; Mafficini, A.; et al. Whole-genome landscape of pancreatic neuroendocrine tumours. Nat. Cell Biol. 2017, 543, 65–71. [Google Scholar] [CrossRef]
- Wong, C.; Tang, L.H.; Davidson, C.; Vosburgh, E.; Chen, W.; Foran, D.J.; Notterman, D.A.; Levine, A.J.; Xu, E.Y. Two well-differentiated pancreatic neuroendocrine tumor mouse models. Cell Death Differ. 2020, 27, 269–283. [Google Scholar] [CrossRef] [Green Version]
- Sennino, B.; Ishiguro-Oonuma, T.; Wei, Y.; Naylor, R.M.; Williamson, C.W.; Bhagwandin, V.; Tabruyn, S.P.; You, W.-K.; Chapman, H.A.; Christensen, J.G.; et al. Suppression of Tumor Invasion and Metastasis by Concurrent Inhibition of c-Met and VEGF Signaling in Pancreatic Neuroendocrine Tumors. Cancer Discov. 2012, 2, 270–287. [Google Scholar] [CrossRef] [Green Version]
- Sennino, B.; Ishiguro-Oonuma, T.; Schriver, B.J.; Christensen, J.G.; McDonald, D.M. Inhibition of c-Met Reduces Lymphatic Metastasis in RIP-Tag2 Transgenic Mice. Cancer Res. 2013, 73, 3692–3703. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyses | Females | Males | |||||
|---|---|---|---|---|---|---|---|
| RT2 | RT2; R6KO | p-Value | RT2 | RT2; R6KO | p-Value | ||
| Survival (wks) | Median survival | 13.3 | 14.1 | 0.57 | 15.7 | 18.0 | <0.05 |
| pNET formation | Tumor burden (mm3) | 8.2 ± 2.7 | 0.7 ± 0.5 | <0.05 | 52.0 ± 14.2 | 16.1 ± 3.8 | <0.05 |
| % Endocrine area | 9.3 ± 2.4 | 2.5 ± 0.6 | <0.05 | 17.7 ± 2.5 | 13.5 ± 1.9 | 0.11 | |
| Islet transformation | % Angiogenic islets | 36.7 ± 3.3 | 14.2 ± 2.5 | <0.05 | 31.4 ± 4.5 | 21.9 ± 5.0 | 0.19 |
| % Vasculature area | 8.9 ± 1.1 | 4.2 ± 1.0 | <0.05 | 7.4 ± 1.0 | 7.3 ± 1.3 | 0.95 | |
| Mitoses (per field of view at 600×) | 1.9 ± 0.2 | 0.9 ± 0.2 | <0.05 | 3.0 ± 0.3 | 2.4 ± 0.2 | 0.32 | |
| c-Myc expression | c-Myc mRNA | 1.1 ± 0.2 | 0.6 ± 0.1 | <0.05 | 1.7 ± 0.6 | 0.5 ± 0.1 | 0.11 |
| c-Myc protein | ND | ND | ND | 1.0 ± 0.6 | 0.7 ± 0.2 | 0.28 | |
| VEGFA expression | Vegfa mRNA | 1.2 ± 0.2 | 1.05 ± 0.2 | 0.78 | 1.1 ± 0.2 | 1.8 ± 0.3 | 0.05 |
| VEGFA protein | ND | ND | ND | 1.0 ± 0.4 | 0.6 ± 0.2 | 0.33 | |
| ARF expression | Arf mRNA | 1.9 ± 0.8 | 1.8 ± 0.8 | 0.61 | 1.1 ± 0.2 | 0.8 ± 0.4 | 0.26 |
| p19ARF protein | ND | ND | ND | 1.0 ± 0.3 | 4.2 ± 1.0 | <0.05 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maharjan, C.K.; Umesalma, S.; Kaemmer, C.A.; Muniz, V.P.; Bauchle, C.; Mott, S.L.; Zamba, K.D.; Breheny, P.; Leidinger, M.R.; Darbro, B.W.; et al. RABL6A Promotes Pancreatic Neuroendocrine Tumor Angiogenesis and Progression In Vivo. Biomedicines 2021, 9, 633. https://doi.org/10.3390/biomedicines9060633
Maharjan CK, Umesalma S, Kaemmer CA, Muniz VP, Bauchle C, Mott SL, Zamba KD, Breheny P, Leidinger MR, Darbro BW, et al. RABL6A Promotes Pancreatic Neuroendocrine Tumor Angiogenesis and Progression In Vivo. Biomedicines. 2021; 9(6):633. https://doi.org/10.3390/biomedicines9060633
Chicago/Turabian StyleMaharjan, Chandra K., Shaikamjad Umesalma, Courtney A. Kaemmer, Viviane P. Muniz, Casey Bauchle, Sarah L. Mott, K. D. Zamba, Patrick Breheny, Mariah R. Leidinger, Benjamin W. Darbro, and et al. 2021. "RABL6A Promotes Pancreatic Neuroendocrine Tumor Angiogenesis and Progression In Vivo" Biomedicines 9, no. 6: 633. https://doi.org/10.3390/biomedicines9060633