
β-RA Targets Mitochondrial Metabolism and Adipogenesis, Leading to Therapeutic Benefits against CoQ Deficiency and Age-Related Overweight

 , , and
, , and
Abstract
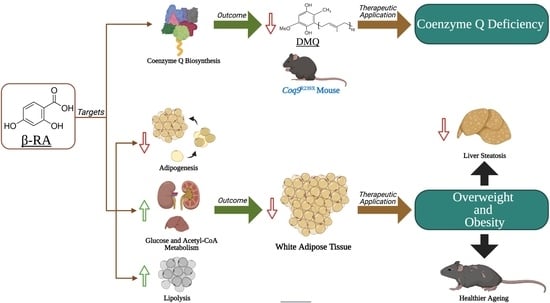
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals and Treatments
2.2. Cell Culture and Cell Assays
2.3. Histology and Immunohistochemistry
2.4. Plasma and Urine Analysis
2.5. Mitochondrial Proteomics Analysis
2.6. Sample Preparation and Western Blot Analysis in Tissues and Cells
2.7. Quantification of CoQ9 and CoQ10 Levels in Mice Tissues and 3T3-L1 Cells
2.8. CoQ-Dependent Respiratory Chain Activities
2.9. Metabolic Assays in Tissues
2.10. Mitochondrial Respiration
2.11. Quantification of β-RA and 4-HB Levels in Mice Tissues
2.12. Statistical Analysis
3. Results
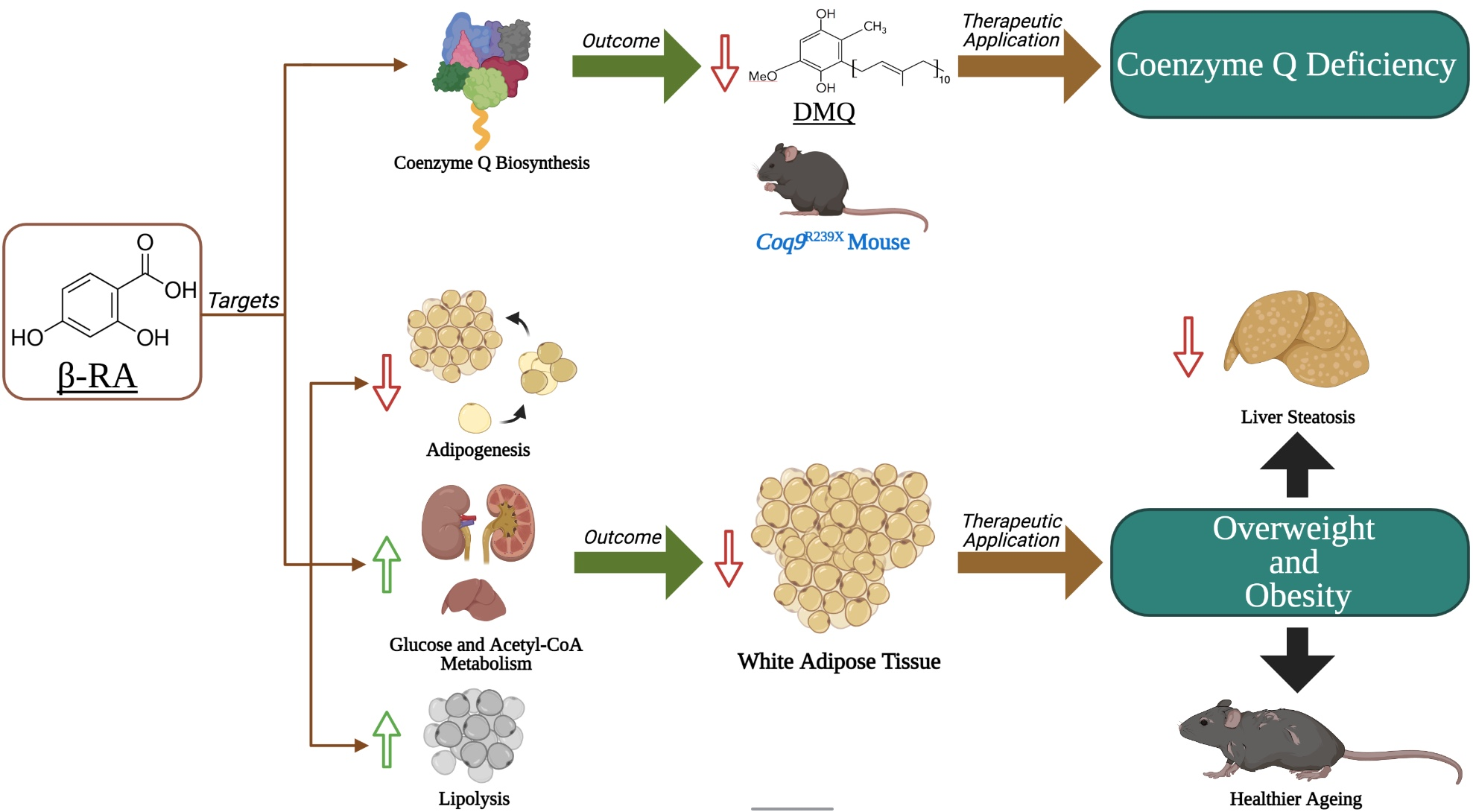
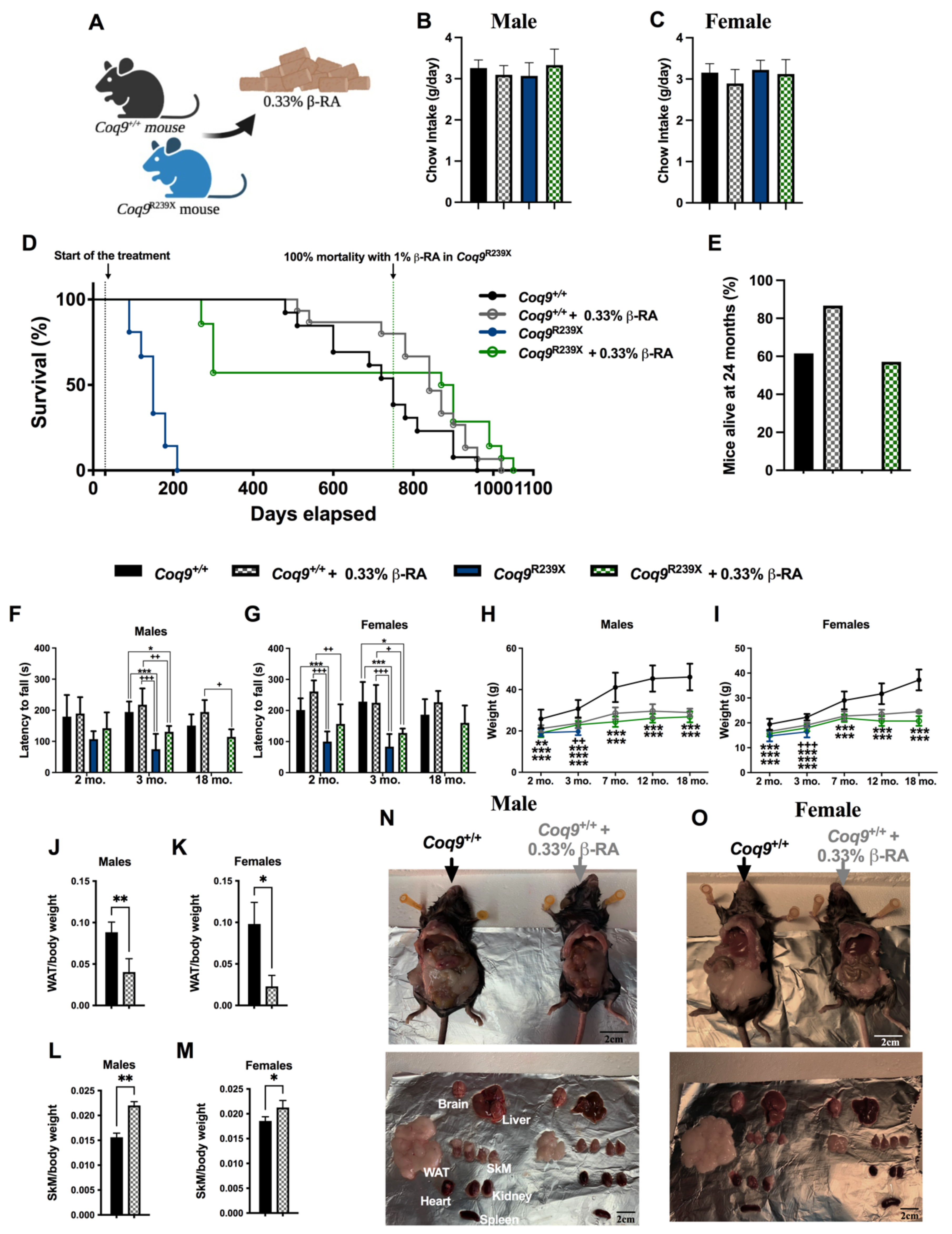
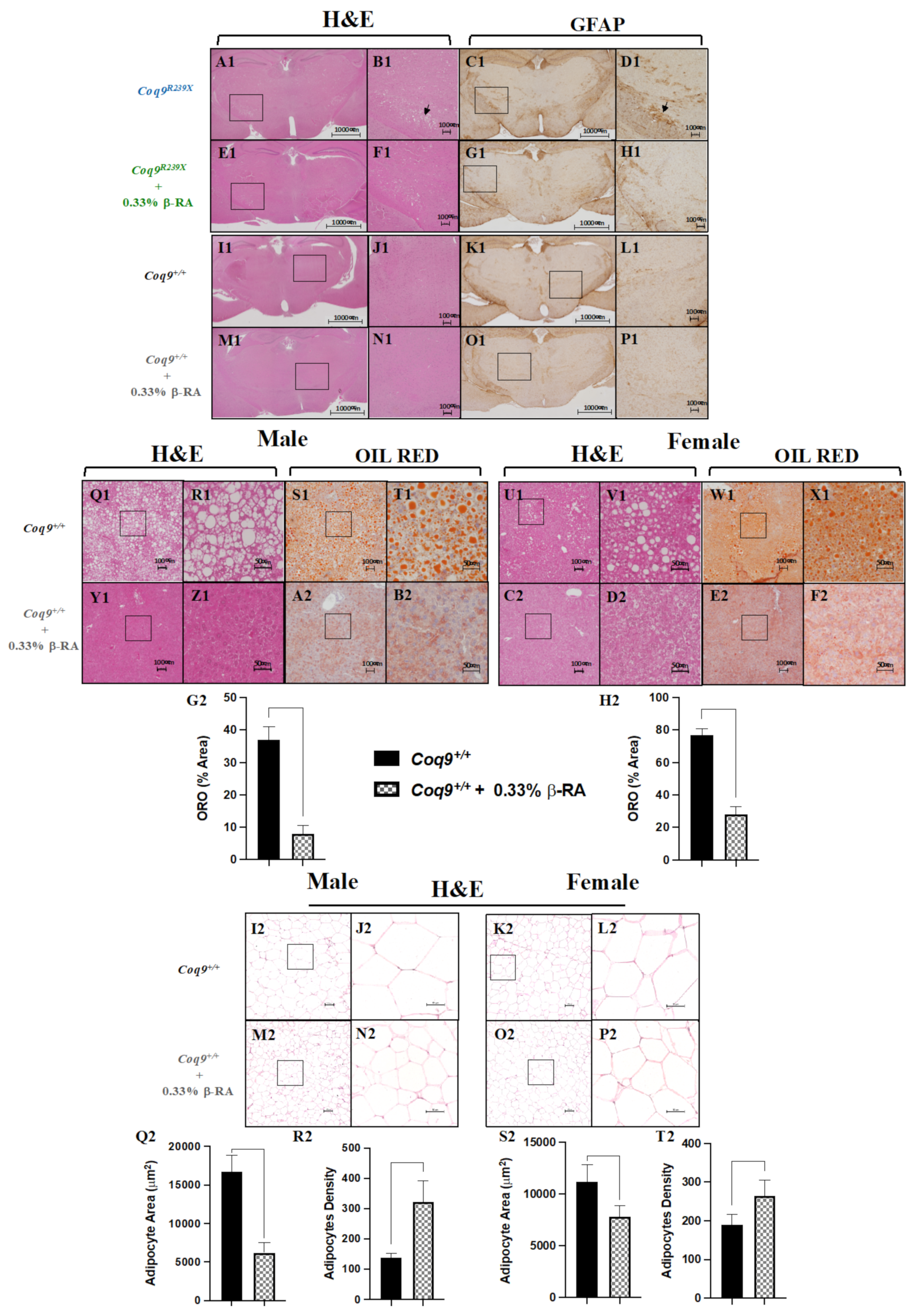
3.1. β-RA Induced Phenotypic and Morphological Benefits against Both Age-Related Obesity and Mitochondrial Encephalopathy due to CoQ Deficiency
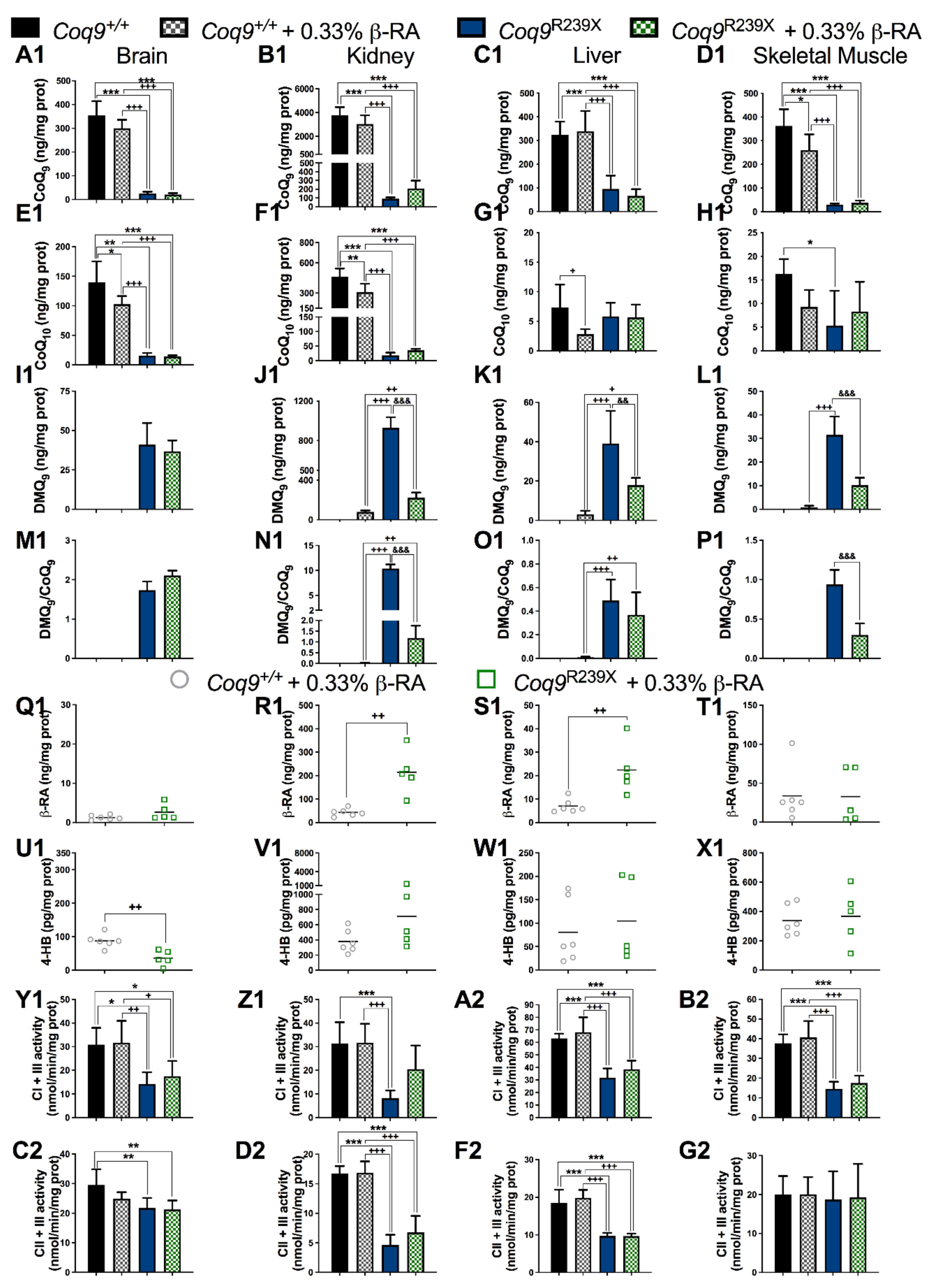
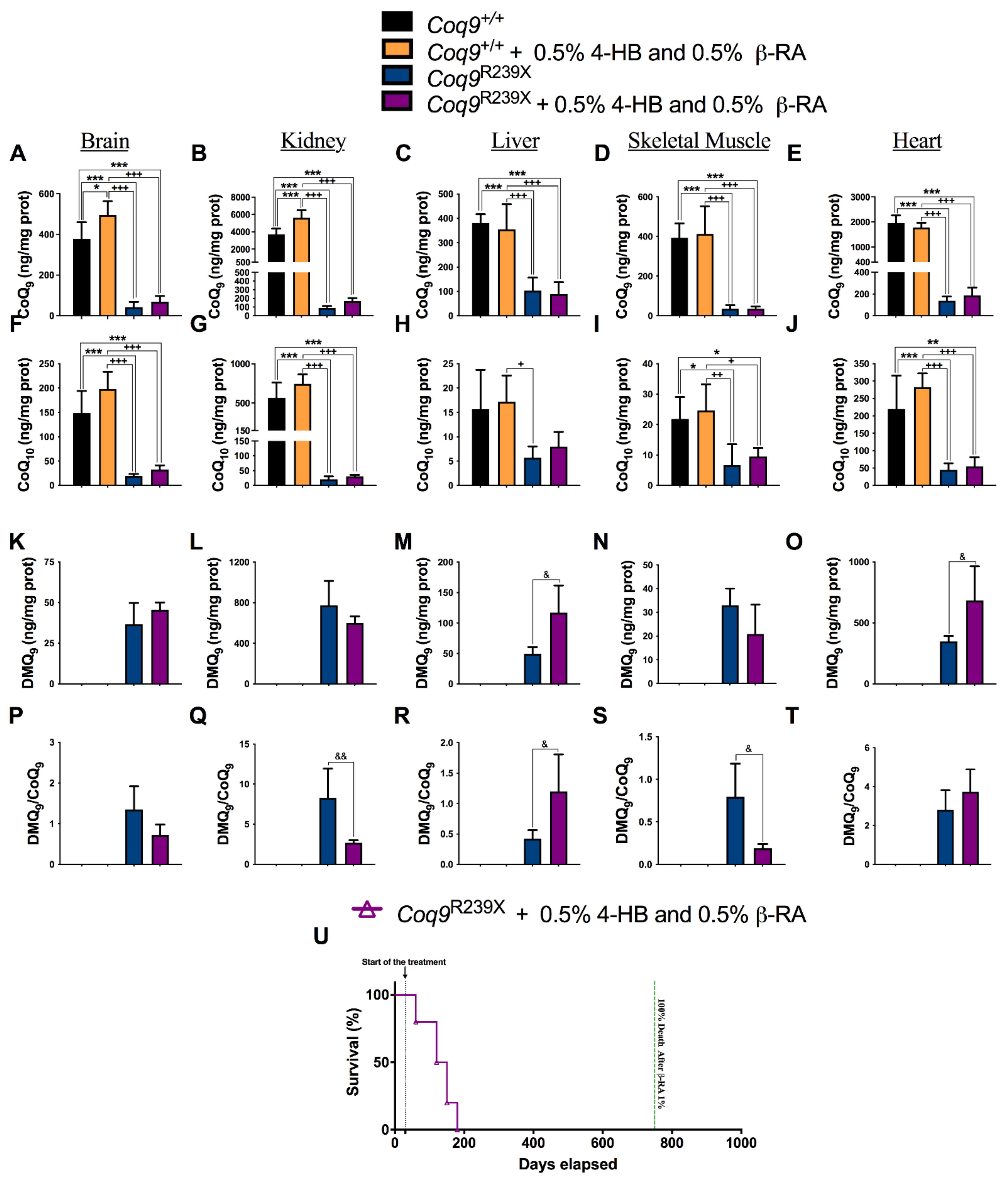
3.2. β-RA Led to Bioenergetics Improvement in Coq9R239X Mice through Its Direct Participation in the CoQ Biosynthetic Pathway
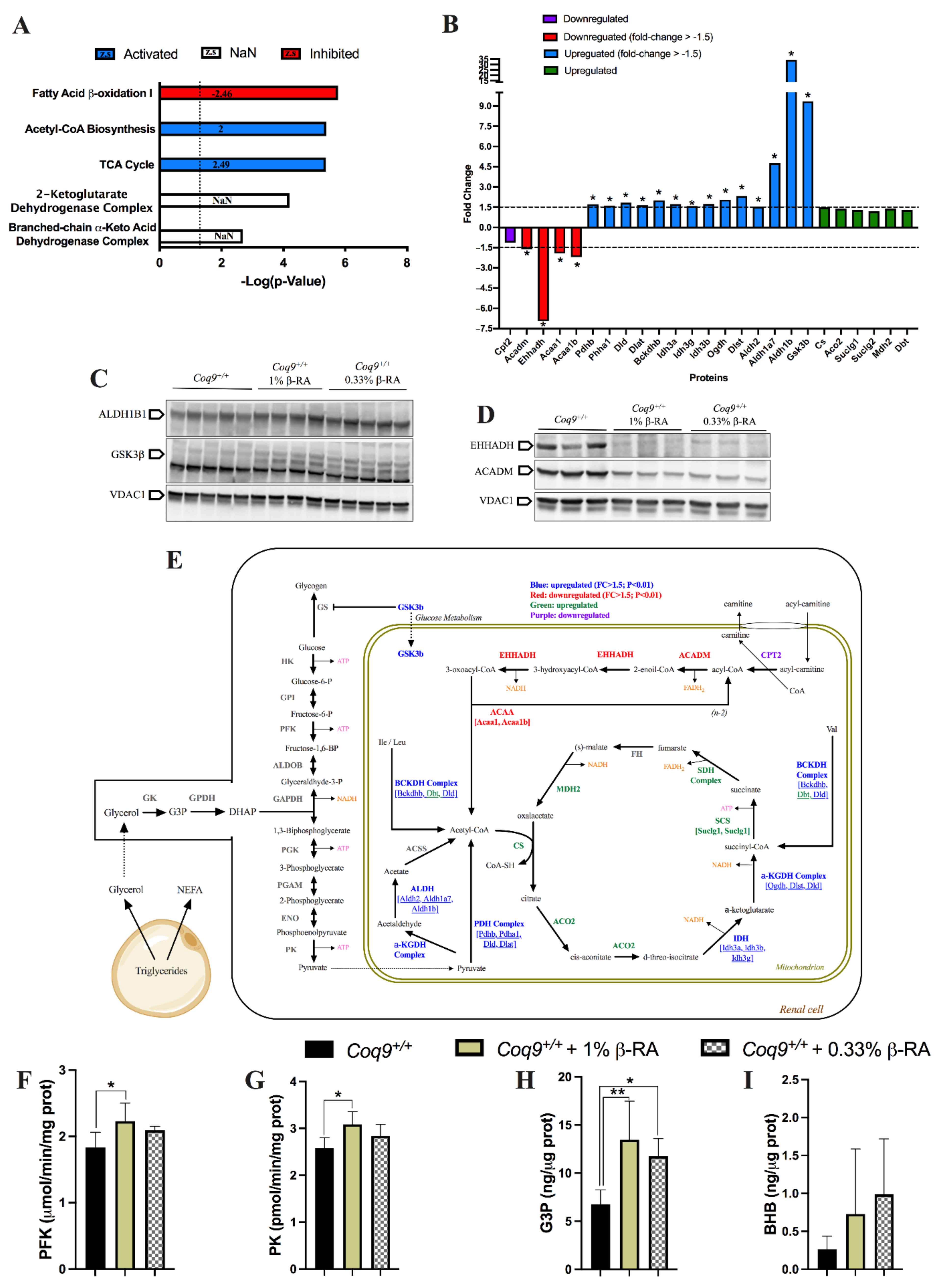
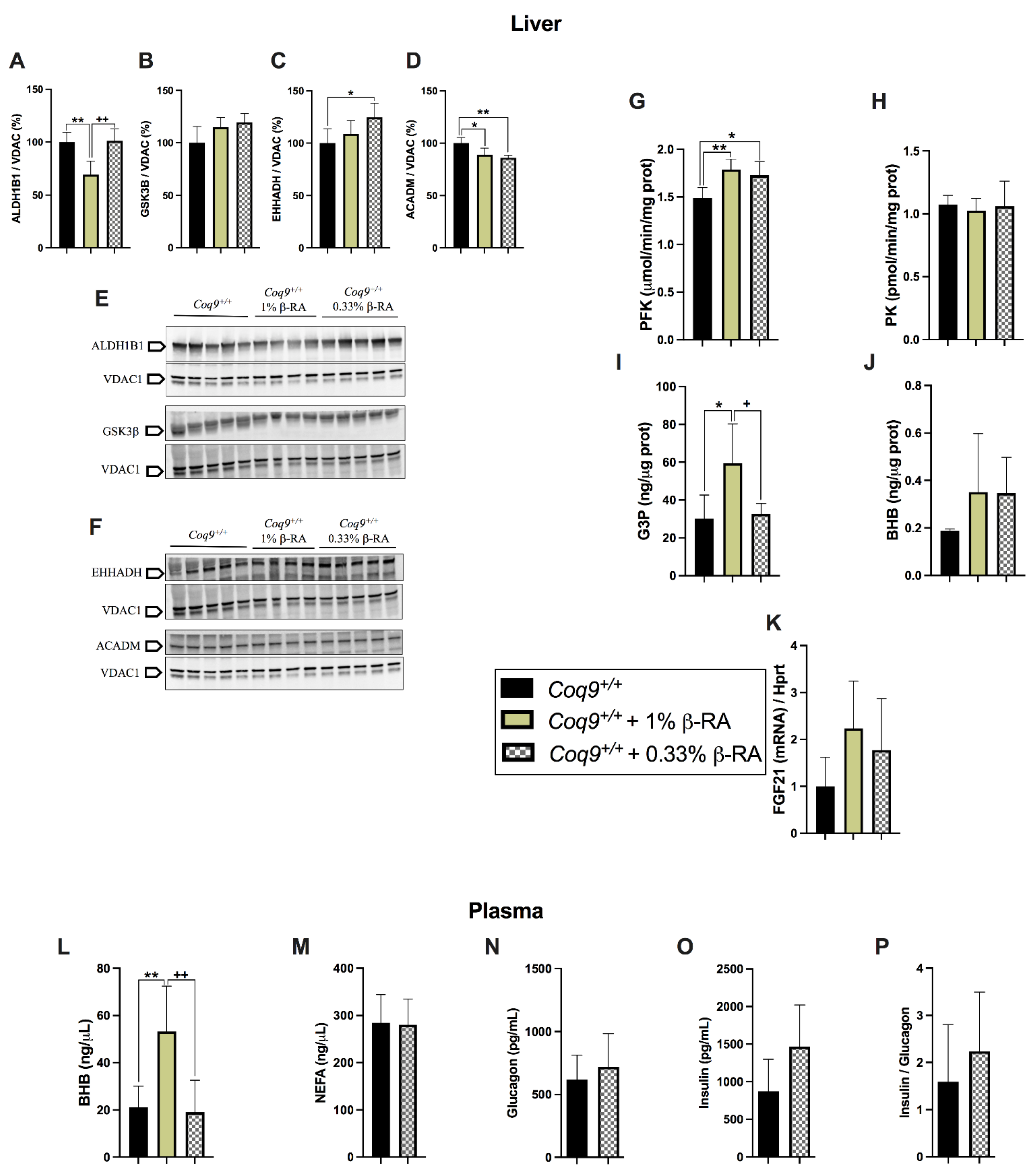
3.3. A Metabolic Switch in Wild-Type Animals Contributed to the Effects of β-RA in Reducing WAT
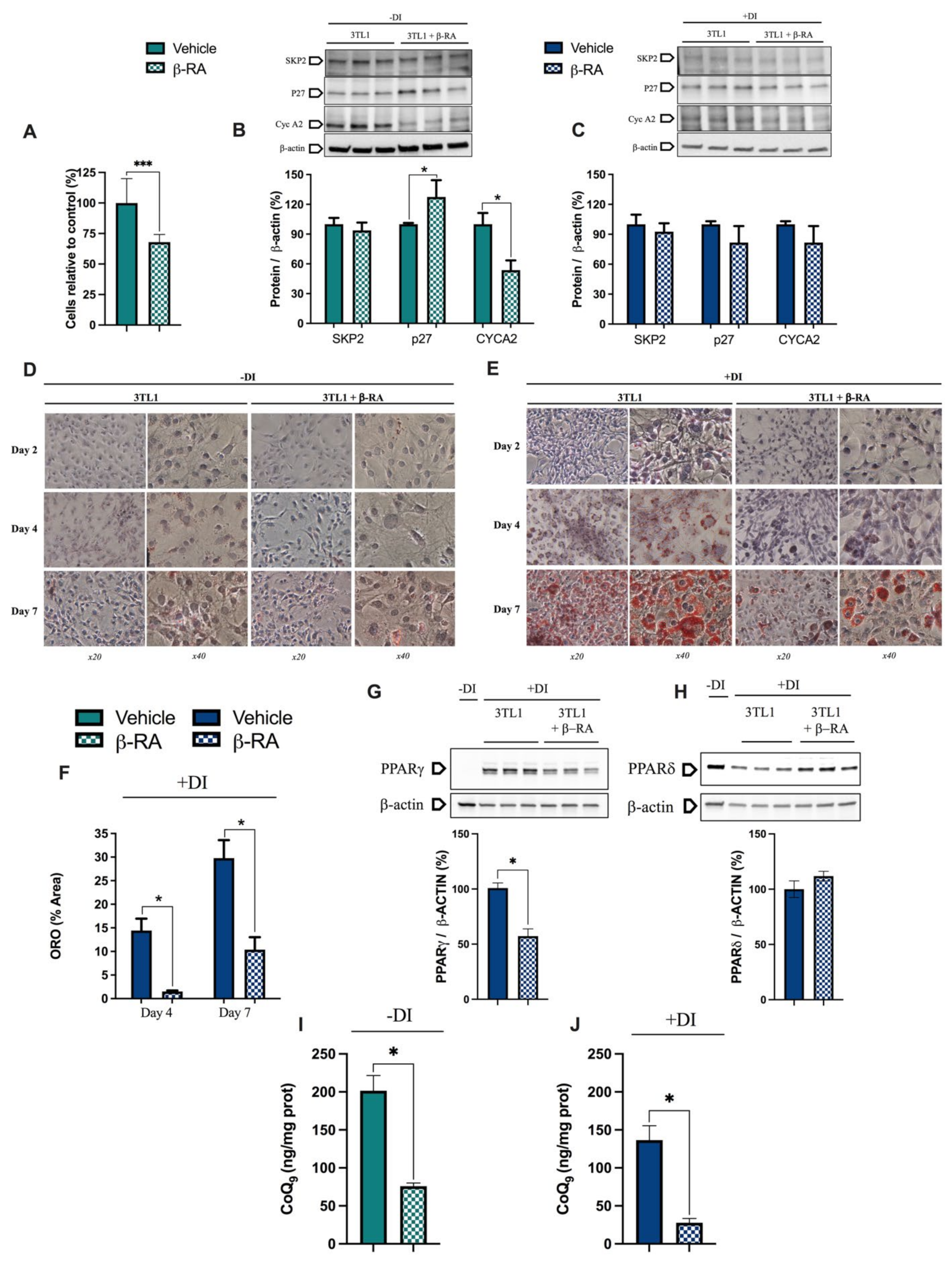
3.4. β-RA Directly Inhibited Adipogenesis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Luna-Sanchez, M.; Diaz-Casado, E.; Barca, E.; Tejada, M.A.; Montilla-Garcia, A.; Cobos, E.J.; Escames, G.; Acuna-Castroviejo, D.; Quinzii, C.M.; Lopez, L.C. The clinical heterogeneity of coenzyme Q10 deficiency results from genotypic differences in the Coq9 gene. EMBO Mol. Med. 2015, 7, 670–687. [Google Scholar] [CrossRef] [PubMed]
- Desbats, M.A.; Morbidoni, V.; Silic-Benussi, M.; Doimo, M.; Ciminale, V.; Cassina, M.; Sacconi, S.; Hirano, M.; Basso, G.; Pierrel, F.; et al. The COQ2 genotype predicts the severity of coenzyme Q10 deficiency. Hum. Mol. Genet. 2016, 25, 4256–4265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinzii, C.M.; Lopez, L.C.; Von-Moltke, J.; Naini, A.; Krishna, S.; Schuelke, M.; Salviati, L.; Navas, P.; DiMauro, S.; Hirano, M. Respiratory chain dysfunction and oxidative stress correlate with severity of primary CoQ10 deficiency. FASEB J. 2008, 22, 1874–1885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinzii, C.M.; Lopez, L.C.; Gilkerson, R.W.; Dorado, B.; Coku, J.; Naini, A.B.; Lagier-Tourenne, C.; Schuelke, M.; Salviati, L.; Carrozzo, R.; et al. Reactive oxygen species, oxidative stress, and cell death correlate with level of CoQ10 deficiency. FASEB J. 2010, 24, 3733–3743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, L.C.; Quinzii, C.M.; Area, E.; Naini, A.; Rahman, S.; Schuelke, M.; Salviati, L.; DiMauro, S.; Hirano, M. Treatment of CoQ(10) deficient fibroblasts with ubiquinone, CoQ analogs, and vitamin C: Time- and compound-dependent effects. PLoS ONE 2010, 5, e11897. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Corzo, L.; Luna-Sanchez, M.; Doerrier, C.; Garcia, J.A.; Guaras, A.; Acin-Perez, R.; Bullejos-Peregrin, J.; Lopez, A.; Escames, G.; Enriquez, J.A.; et al. Dysfunctional Coq9 protein causes predominant encephalomyopathy associated with CoQ deficiency. Hum. Mol. Genet. 2013, 22, 1233–1248. [Google Scholar] [CrossRef] [Green Version]
- Quinzii, C.M.; Garone, C.; Emmanuele, V.; Tadesse, S.; Krishna, S.; Dorado, B.; Hirano, M. Tissue-specific oxidative stress and loss of mitochondria in CoQ-deficient Pdss2 mutant mice. FASEB J. 2013, 27, 612–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duberley, K.E.; Abramov, A.Y.; Chalasani, A.; Heales, S.J.; Rahman, S.; Hargreaves, I.P. Human neuronal coenzyme Q(10) deficiency results in global loss of mitochondrial respiratory chain activity, increased mitochondrial oxidative stress and reversal of ATP synthase activity: Implications for pathogenesis and treatment. J. Inherit. Metab. Dis. 2013, 36, 63–73. [Google Scholar] [CrossRef]
- Kleiner, G.; Barca, E.; Ziosi, M.; Emmanuele, V.; Xu, Y.; Hidalgo-Gutierrez, A.; Qiao, C.; Tadesse, S.; Area-Gomez, E.; Lopez, L.C.; et al. CoQ10 supplementation rescues nephrotic syndrome through normalization of H2S oxidation pathway. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3708–3722. [Google Scholar] [CrossRef] [PubMed]
- Luna-Sanchez, M.; Hidalgo-Gutierrez, A.; Hildebrandt, T.M.; Chaves-Serrano, J.; Barriocanal-Casado, E.; Santos-Fandila, A.; Romero, M.; Sayed, R.K.; Duarte, J.; Prokisch, H.; et al. CoQ deficiency causes disruption of mitochondrial sulfide oxidation, a new pathomechanism associated with this syndrome. EMBO Mol. Med. 2017, 9, 78–95. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Martin, J.M.; Salviati, L.; Trevisson, E.; Montini, G.; DiMauro, S.; Quinzii, C.; Hirano, M.; Rodriguez-Hernandez, A.; Cordero, M.D.; Sanchez-Alcazar, J.A.; et al. Missense mutation of the COQ2 gene causes defects of bioenergetics and de novo pyrimidine synthesis. Hum. Mol. Genet. 2007, 16, 1091–1097. [Google Scholar] [CrossRef]
- Mancuso, M.; Orsucci, D.; Volpi, L.; Calsolaro, V.; Siciliano, G. Coenzyme Q10 in neuromuscular and neurodegenerative disorders. Curr. Drug Targets 2010, 11, 111–121. [Google Scholar] [CrossRef] [Green Version]
- Emmanuele, V.; Lopez, L.C.; Berardo, A.; Naini, A.; Tadesse, S.; Wen, B.; D’Agostino, E.; Solomon, M.; DiMauro, S.; Quinzii, C.; et al. Heterogeneity of coenzyme Q10 deficiency: Patient study and literature review. Arch. Neurol. 2012, 69, 978–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinzii, C.M.; Lopez, L.C.; Naini, A.; DiMauro, S.; Hirano, M. Human CoQ10 deficiencies. Biofactors 2008, 32, 113–118. [Google Scholar] [CrossRef]
- Turunen, M.; Olsson, J.; Dallner, G. Metabolism and function of coenzyme Q. Biochim. Biophys. Acta 2004, 1660, 171–199. [Google Scholar] [CrossRef] [Green Version]
- Yubero, D.; Montero, R.; Martin, M.A.; Montoya, J.; Ribes, A.; Grazina, M.; Trevisson, E.; Rodriguez-Aguilera, J.C.; Hargreaves, I.P.; Salviati, L.; et al. Secondary coenzyme Q10 deficiencies in oxidative phosphorylation (OXPHOS) and non-OXPHOS disorders. Mitochondrion 2016, 30, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Fazakerley, D.J.; Chaudhuri, R.; Yang, P.; Maghzal, G.J.; Thomas, K.C.; Krycer, J.R.; Humphrey, S.J.; Parker, B.L.; Fisher-Wellman, K.H.; Meoli, C.C.; et al. Mitochondrial CoQ deficiency is a common driver of mitochondrial oxidants and insulin resistance. eLife 2018, 7, e32111. [Google Scholar] [CrossRef] [PubMed]
- Bentinger, M.; Dallner, G.; Chojnacki, T.; Swiezewska, E. Distribution and breakdown of labeled coenzyme Q10 in rat. Free Radic. Biol. Med. 2003, 34, 563–575. [Google Scholar] [CrossRef]
- Garcia-Corzo, L.; Luna-Sanchez, M.; Doerrier, C.; Ortiz, F.; Escames, G.; Acuna-Castroviejo, D.; Lopez, L.C. Ubiquinol-10 ameliorates mitochondrial encephalopathy associated with CoQ deficiency. Biochim. Biophys. Acta 2014, 1842, 893–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herebian, D.; Lopez, L.C.; Distelmaier, F. Bypassing human CoQ10 deficiency. Mol. Genet. Metab. 2018, 123, 289–291. [Google Scholar] [CrossRef]
- Wang, Y.; Oxer, D.; Hekimi, S. Mitochondrial function and lifespan of mice with controlled ubiquinone biosynthesis. Nat. Commun. 2015, 6, 6393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hidalgo-Gutierrez, A.; Barriocanal-Casado, E.; Bakkali, M.; Diaz-Casado, M.E.; Sanchez-Maldonado, L.; Romero, M.; Sayed, R.K.; Prehn, C.; Escames, G.; Duarte, J.; et al. beta-RA reduces DMQ/CoQ ratio and rescues the encephalopathic phenotype in Coq9 (R239X) mice. EMBO Mol. Med. 2019, 11, e9466. [Google Scholar] [CrossRef]
- Wang, Y.; Smith, C.; Parboosingh, J.S.; Khan, A.; Innes, M.; Hekimi, S. Pathogenicity of two COQ7 mutations and responses to 2,4-dihydroxybenzoate bypass treatment. J. Cell. Mol. Med. 2017, 21, 2329–2343. [Google Scholar] [CrossRef] [PubMed]
- Widmeier, E.; Airik, M.; Hugo, H.; Schapiro, D.; Wedel, J.; Ghosh, C.C.; Nakayama, M.; Schneider, R.; Awad, A.M.; Nag, A.; et al. Treatment with 2,4-Dihydroxybenzoic Acid Prevents FSGS Progression and Renal Fibrosis in Podocyte-Specific Coq6 Knockout Mice. J. Am. Soc. Nephrol. 2019, 30, 393–405. [Google Scholar] [CrossRef] [Green Version]
- Widmeier, E.; Yu, S.; Nag, A.; Chung, Y.W.; Nakayama, M.; Hugo, H.; Buerger, F.; Schapiro, D.; Choi, W.-I.; Kim, J.-w.; et al. ADCK4 deficiency destabilizes the coenzyme Q complex, which is rescued by 2,4-dihydroxybenzoic acid treatment. J. Am. Soc.Nephrol. 2020, 31, 1191–1211. [Google Scholar] [CrossRef]
- Liu, F.; Lossl, P.; Rabbitts, B.M.; Balaban, R.S.; Heck, A.J.R. The interactome of intact mitochondria by cross-linking mass spectrometry provides evidence for coexisting respiratory supercomplexes. Mol. Cell. Proteom. 2018, 17, 216–232. [Google Scholar] [CrossRef] [Green Version]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Raimundo, N.; Vanharanta, S.; Aaltonen, L.A.; Hovatta, I.; Suomalainen, A. Downregulation of SRF-FOS-JUNB pathway in fumarate hydratase deficiency and in uterine leiomyomas. Oncogene 2009, 28, 1261–1273. [Google Scholar] [CrossRef] [Green Version]
- Barriocanal-Casado, E.; Hidalgo-Gutierrez, A.; Raimundo, N.; Gonzalez-Garcia, P.; Acuna-Castroviejo, D.; Escames, G.; Lopez, L.C. Rapamycin administration is not a valid therapeutic strategy for every case of mitochondrial disease. EBioMedicine 2019, 42, 511–523. [Google Scholar] [CrossRef] [Green Version]
- Rogers, G.W.; Brand, M.D.; Petrosyan, S.; Ashok, D.; Elorza, A.A.; Ferrick, D.A.; Murphy, A.N. High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria. PLoS ONE 2011, 6, e21746. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Regulation of longevity by FGF21: Interaction between energy metabolism and stress responses. Ageing Res. Rev. 2017, 37, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Carriere, A.; Fernandez, Y.; Rigoulet, M.; Penicaud, L.; Casteilla, L. Inhibition of preadipocyte proliferation by mitochondrial reactive oxygen species. FEBS Lett. 2003, 550, 163–167. [Google Scholar] [CrossRef] [Green Version]
- Wagner, G.; Lindroos-Christensen, J.; Einwallner, E.; Husa, J.; Zapf, T.C.; Lipp, K.; Rauscher, S.; Groger, M.; Spittler, A.; Loewe, R.; et al. HO-1 inhibits preadipocyte proliferation and differentiation at the onset of obesity via ROS dependent activation of Akt2. Sci. Rep. 2017, 7, 40881. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Olivier, P.; Diehl, J.A.; Fero, M.; Roussel, M.F.; Roberts, J.M.; Sherr, C.J. The p21(Cip1) and p27(Kip1) CDK ‘inhibitors’ are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J. 1999, 18, 1571–1583. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, B.S.; Nam, H.; Morrison, R.F. Curcumin Inhibits 3T3-L1 Preadipocyte Proliferation by Mechanisms Involving Post-transcriptional p27 Regulation. Biochem. Biophys. Rep. 2016, 5, 16–21. [Google Scholar] [CrossRef] [Green Version]
- Lehrke, M.; Lazar, M.A. The many faces of PPARgamma. Cell 2005, 123, 993–999. [Google Scholar] [CrossRef] [Green Version]
- Schoonjans, K.; Staels, B.; Auwerx, J. The peroxisome proliferator activated receptors (PPARS) and their effects on lipid metabolism and adipocyte differentiation. Biochim. Biophys. Acta 1996, 1302, 93–109. [Google Scholar] [CrossRef]
- Pierrel, F. Impact of Chemical Analogs of 4-Hydroxybenzoic Acid on Coenzyme Q Biosynthesis: From Inhibition to Bypass of Coenzyme Q Deficiency. Front. Physiol. 2017, 8, 436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Lin, Y.; Kang, T.; Huang, B.; Xu, W.; Garcia-Barrio, M.; Olatinwo, M.; Matthews, R.; Chen, Y.E.; Thompson, W.E. Mitochondrial dysfunction and adipogenic reduction by prohibitin silencing in 3T3-L1 cells. PLoS ONE 2012, 7, e34315. [Google Scholar] [CrossRef] [Green Version]
- Hawley, S.A.; Fullerton, M.D.; Ross, F.A.; Schertzer, J.D.; Chevtzoff, C.; Walker, K.J.; Peggie, M.W.; Zibrova, D.; Green, K.A.; Mustard, K.J.; et al. The ancient drug salicylate directly activates AMP-activated protein kinase. Science 2012, 336, 918–922. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.; Park, J.; Kim, H.L.; Youn, D.H.; Kang, J.; Lim, S.; Jeong, M.Y.; Sethi, G.; Park, S.J.; Ahn, K.S.; et al. Vanillic acid attenuates testosterone-induced benign prostatic hyperplasia in rats and inhibits proliferation of prostatic epithelial cells. Oncotarget 2017, 8, 87194–87208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [Green Version]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.Y.; Vasta, V.; Hahn, S.; Gangoiti, J.A.; Opheim, E.; Sedensky, M.M.; Morgan, P.G. The role of DMQ(9) in the long-lived mutant clk-1. Mech. Ageing Dev. 2011, 132, 331–339. [Google Scholar] [CrossRef] [Green Version]
- Freyer, C.; Stranneheim, H.; Naess, K.; Mourier, A.; Felser, A.; Maffezzini, C.; Lesko, N.; Bruhn, H.; Engvall, M.; Wibom, R.; et al. Rescue of primary ubiquinone deficiency due to a novel COQ7 defect using 2,4-dihydroxybensoic acid. J. Med. Genet. 2015, 52, 779–783. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Garcia, P.; Hidalgo-Gutierrez, A.; Mascaraque, C.; Barriocanal-Casado, E.; Bakkali, M.; Ziosi, M.; Abdihankyzy, U.B.; Sanchez-Hernandez, S.; Escames, G.; Prokisch, H.; et al. Coenzyme Q10 modulates sulfide metabolism and links the mitochondrial respiratory chain to pathways associated to one carbon metabolism. Hum. Mol. Genet. 2020, 29, 3296–3311. [Google Scholar] [CrossRef] [PubMed]
- Aranaz, P.; Navarro-Herrera, D.; Zabala, M.; Migueliz, I.; Romo-Hualde, A.; Lopez-Yoldi, M.; Martinez, J.A.; Vizmanos, J.L.; Milagro, F.I.; Gonzalez-Navarro, C.J. Phenolic Compounds Inhibit 3T3-L1 Adipogenesis Depending on the Stage of Differentiation and Their Binding Affinity to PPARgamma. Molecules 2019, 24, 1045. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.L.; Yen, G.C. Effects of flavonoids and phenolic acids on the inhibition of adipogenesis in 3T3-L1 adipocytes. J. Agric. Food Chem. 2007, 55, 8404–8410. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.X.; Williams, K.J.; He, C.H.; Weng, E.; Khong, S.; Rose, T.E.; Kwon, O.; Bensinger, S.J.; Marbois, B.N.; Clarke, C.F. Resveratrol and para-coumarate serve as ring precursors for coenzyme Q biosynthesis. J. Lipid Res. 2015, 56, 909–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, E.D.; Spiegelman, B.M. Molecular regulation of adipogenesis. Annu. Rev. Cell Dev. Biol. 2000, 16, 145–171. [Google Scholar] [CrossRef]
- Jo, J.; Gavrilova, O.; Pack, S.; Jou, W.; Mullen, S.; Sumner, A.E.; Cushman, S.W.; Periwal, V. Hypertrophy and/or Hyperplasia: Dynamics of Adipose Tissue Growth. PLoS Comput. Biol. 2009, 5, e1000324. [Google Scholar] [CrossRef] [PubMed]
- Tchoukalova, Y.D.; Votruba, S.B.; Tchkonia, T.; Giorgadze, N.; Kirkland, J.L.; Jensen, M.D. Regional differences in cellular mechanisms of adipose tissue gain with overfeeding. Proc. Natl. Acad. Sci. USA 2010, 107, 18226–18231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landgraf, K.; Rockstroh, D.; Wagner, I.V.; Weise, S.; Tauscher, R.; Schwartze, J.T.; Loffler, D.; Buhligen, U.; Wojan, M.; Till, H.; et al. Evidence of early alterations in adipose tissue biology and function and its association with obesity-related inflammation and insulin resistance in children. Diabetes 2015, 64, 1249–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerich, J.E.; Meyer, C.; Woerle, H.J.; Stumvoll, M. Renal gluconeogenesis: Its importance in human glucose homeostasis. Diabetes Care 2001, 24, 382–391. [Google Scholar] [CrossRef] [Green Version]
- Legouis, D.; Faivre, A.; Cippa, P.E.; de Seigneux, S. Renal gluconeogenesis: An underestimated role of the kidney in systemic glucose metabolism. Nephrol. Dial. Transplant. 2020, gfaa302. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Cooper, D.E.; Cluntun, A.A.; Warmoes, M.O.; Zhao, S.; Reid, M.A.; Liu, J.; Lund, P.J.; Lopes, M.; Garcia, B.A.; et al. Acetate Production from Glucose and Coupling to Mitochondrial Metabolism in Mammals. Cell 2018, 175, 502–513. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Tu, B.P. Acetyl-CoA and the regulation of metabolism: Mechanisms and consequences. Curr. Opin. Cell Biol. 2015, 33, 125–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.; Zhang, M.; Plec, A.A.; Estill, S.J.; Cai, L.; Repa, J.J.; McKnight, S.L.; Tu, B.P. ACSS2 promotes systemic fat storage and utilization through selective regulation of genes involved in lipid metabolism. Proc. Natl. Acad. Sci. USA 2018, 115, E9499–E9506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fletcher, J.A.; Deja, S.; Satapati, S.; Fu, X.; Burgess, S.C.; Browning, J.D. Impaired ketogenesis and increased acetyl-CoA oxidation promote hyperglycemia in human fatty liver. JCI Insight 2019, 4, e127737. [Google Scholar] [CrossRef] [PubMed]
- Mariappan, M.M.; Prasad, S.; D’Silva, K.; Cedillo, E.; Sataranatarajan, K.; Barnes, J.L.; Choudhury, G.G.; Kasinath, B.S. Activation of glycogen synthase kinase 3beta ameliorates diabetes-induced kidney injury. J. Biol. Chem. 2014, 289, 35363–35375. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hidalgo-Gutiérrez, A.; Barriocanal-Casado, E.; Díaz-Casado, M.E.; González-García, P.; Zenezini Chiozzi, R.; Acuña-Castroviejo, D.; López, L.C. β-RA Targets Mitochondrial Metabolism and Adipogenesis, Leading to Therapeutic Benefits against CoQ Deficiency and Age-Related Overweight. Biomedicines 2021, 9, 1457. https://doi.org/10.3390/biomedicines9101457
Hidalgo-Gutiérrez A, Barriocanal-Casado E, Díaz-Casado ME, González-García P, Zenezini Chiozzi R, Acuña-Castroviejo D, López LC. β-RA Targets Mitochondrial Metabolism and Adipogenesis, Leading to Therapeutic Benefits against CoQ Deficiency and Age-Related Overweight. Biomedicines. 2021; 9(10):1457. https://doi.org/10.3390/biomedicines9101457
Chicago/Turabian StyleHidalgo-Gutiérrez, Agustín, Eliana Barriocanal-Casado, María Elena Díaz-Casado, Pilar González-García, Riccardo Zenezini Chiozzi, Darío Acuña-Castroviejo, and Luis Carlos López. 2021. "β-RA Targets Mitochondrial Metabolism and Adipogenesis, Leading to Therapeutic Benefits against CoQ Deficiency and Age-Related Overweight" Biomedicines 9, no. 10: 1457. https://doi.org/10.3390/biomedicines9101457