Binge Alcohol Exposure Transiently Changes the Endocannabinoid System: A Potential Target to Prevent Alcohol-Induced Neurodegeneration


Abstract
:1. Introduction
2. Materials and Methods
2.1. Binge Ethanol Treatment
2.2. CB1 Receptor Autoradiography
2.3. N-Acylethanolamine Extraction and Quantification
2.4. URB597 Regimen
2.5. FluoroJade B Staining and Quantification
2.6. Statistical Analysis
3. Results
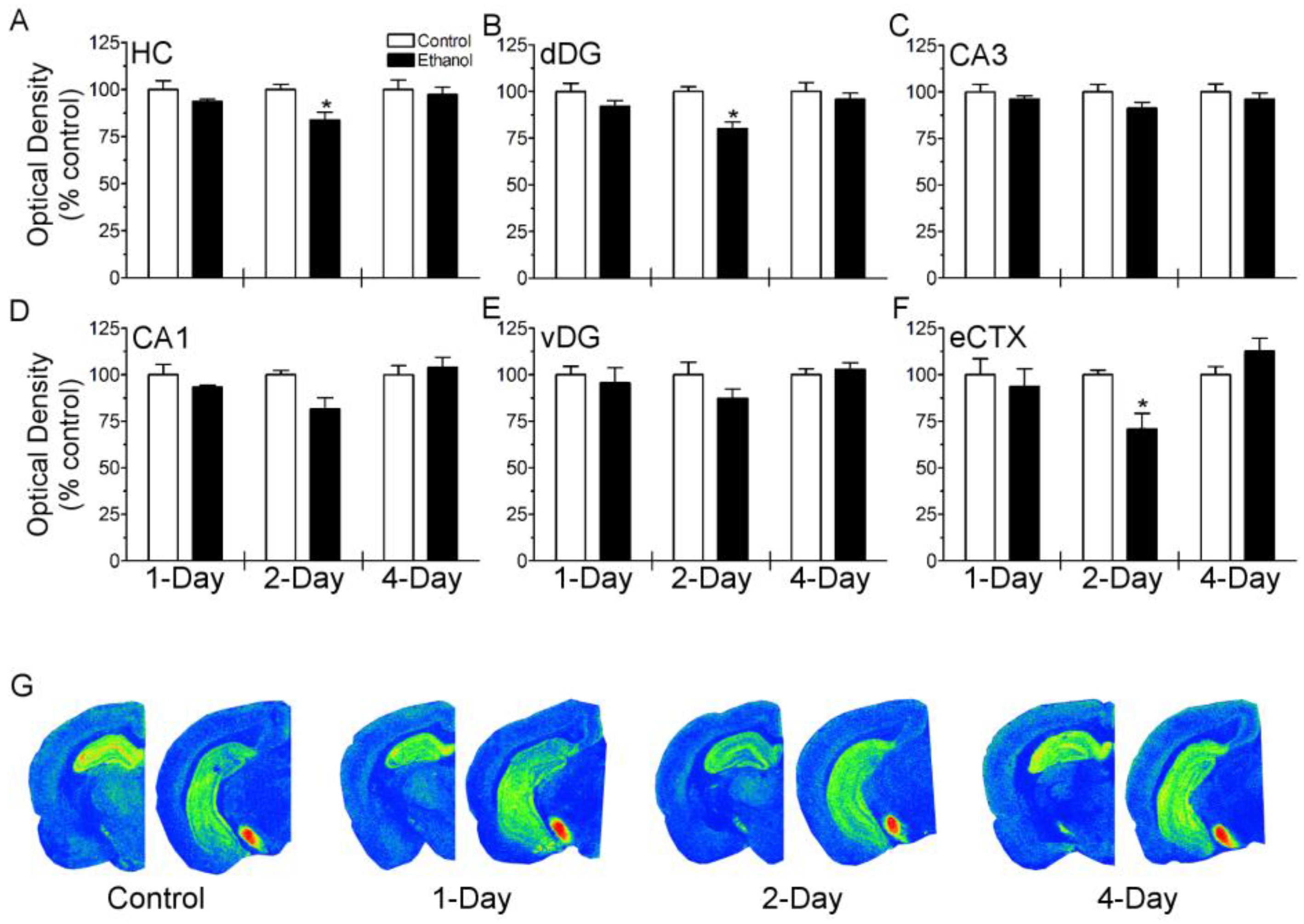
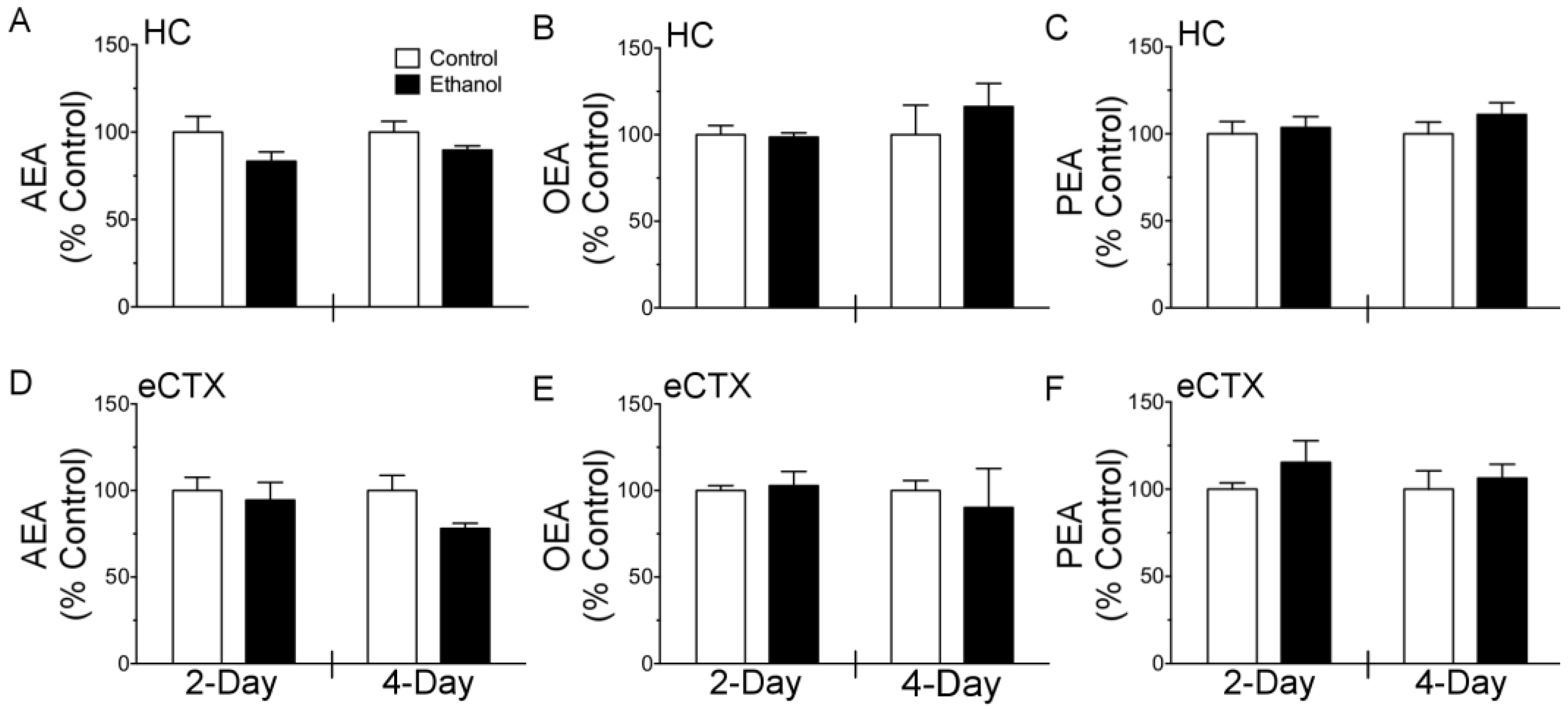
3.1. Binge Ethanol Exposure Transiently Decreases [3H]-CP-55,940 Binding but Does Not Alter NAE Tissue Content
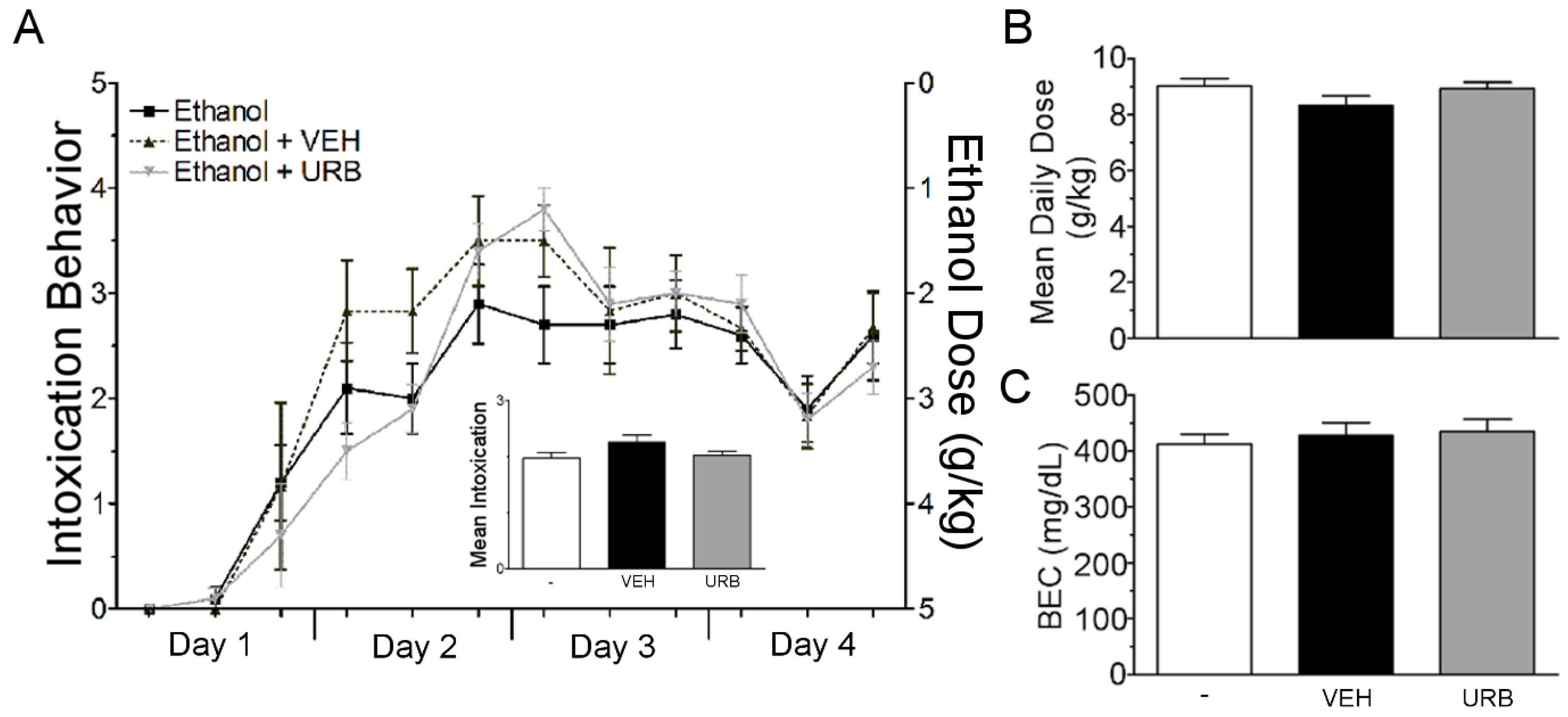
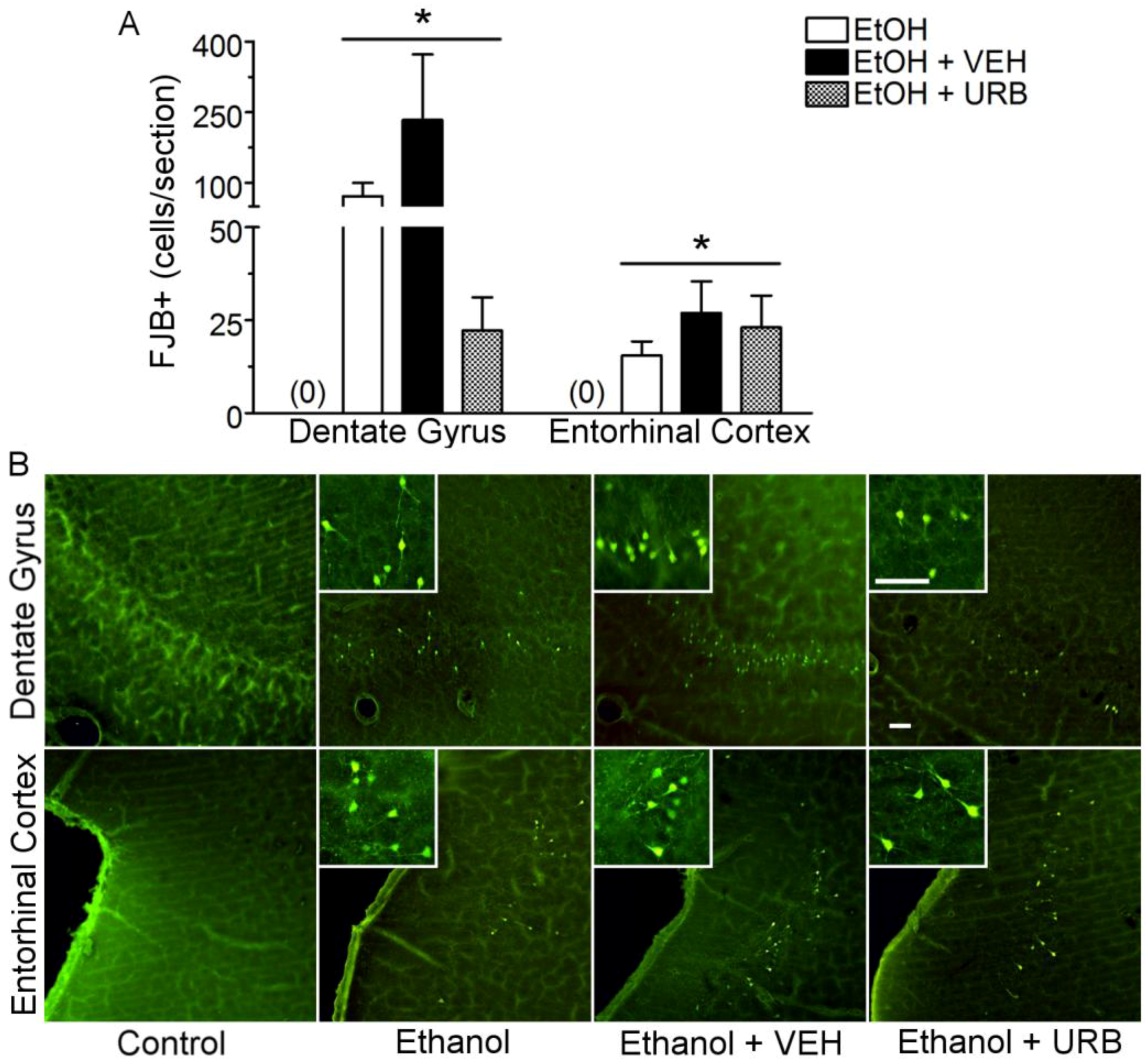
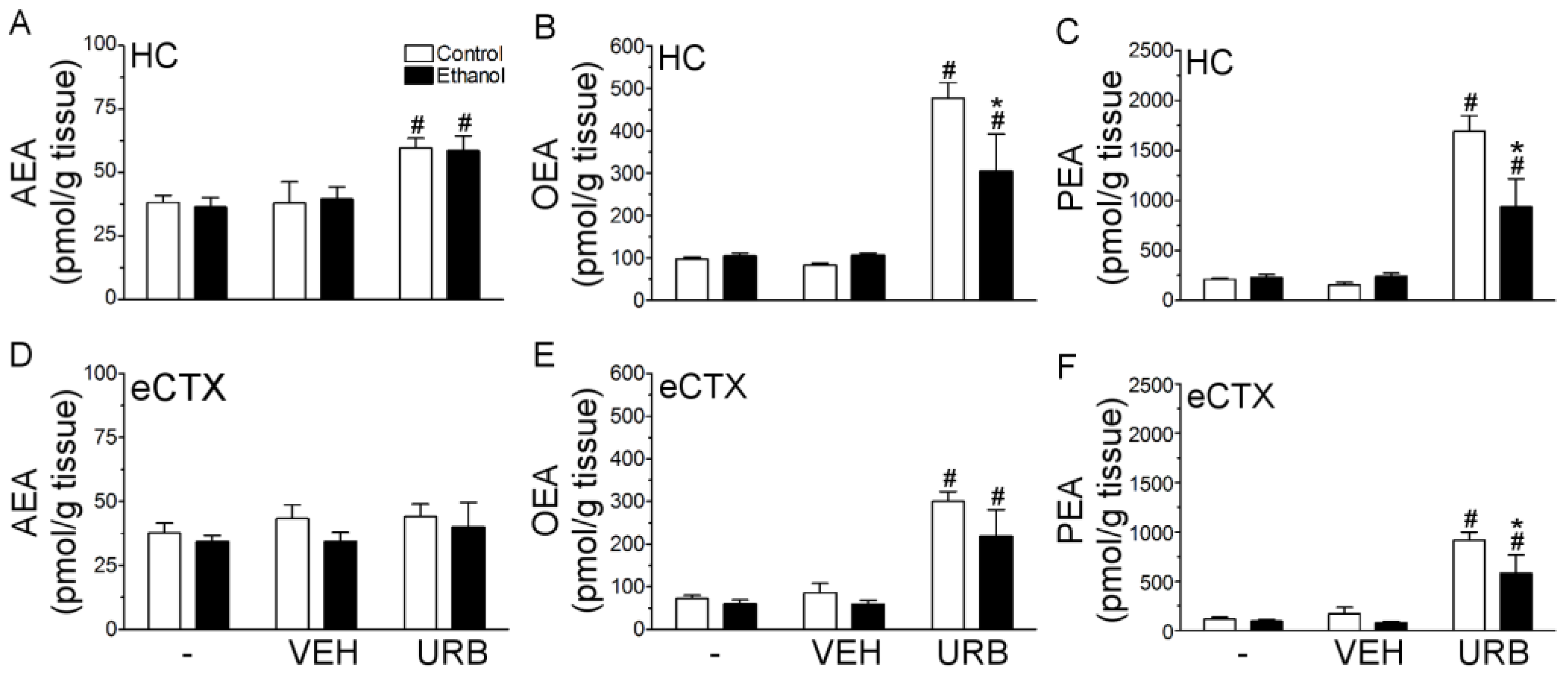
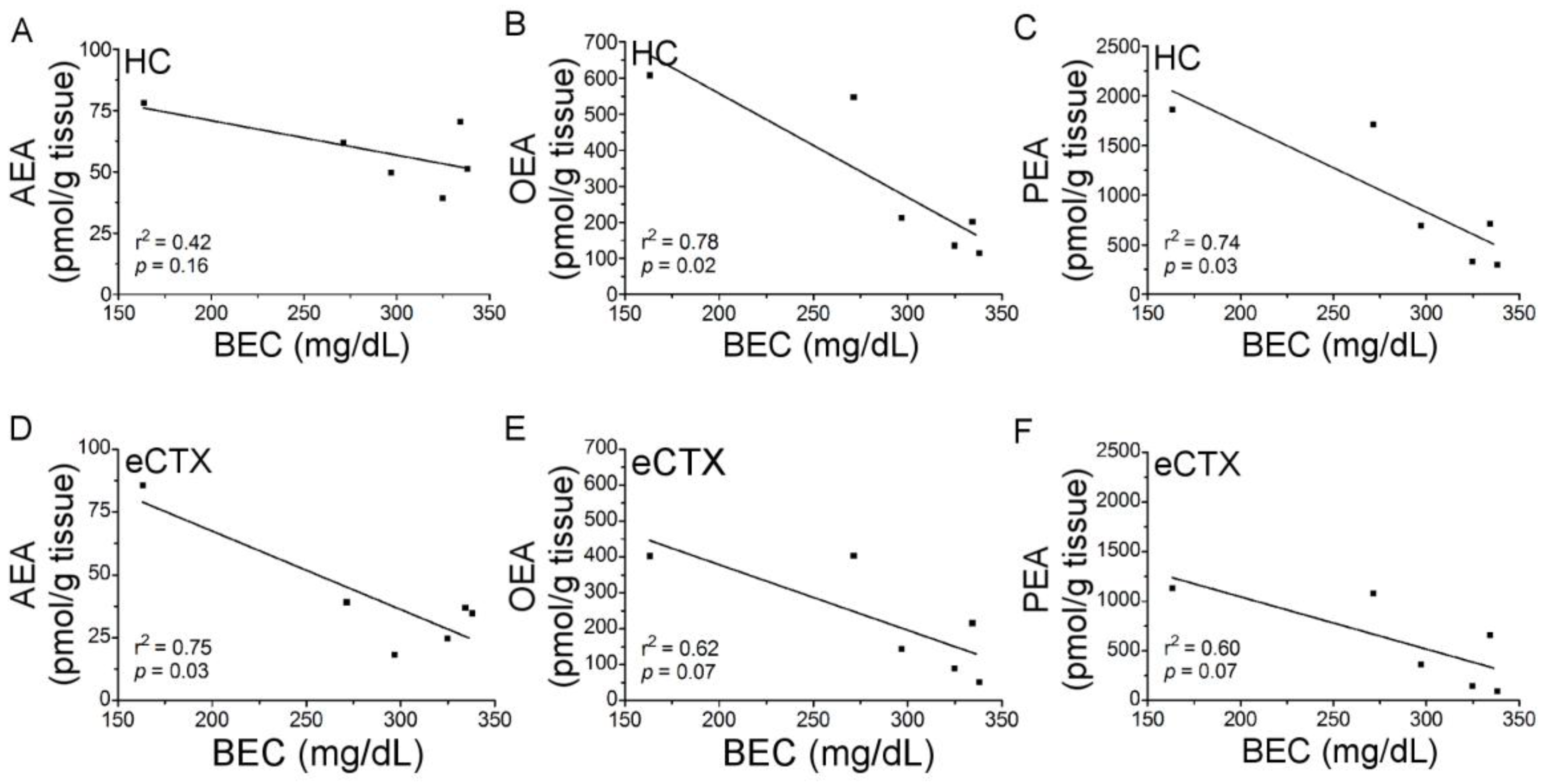
3.2. URB597 Administration Failed to Attenuate Binge Ethanol-Induced Degeneration of the Corticolimbic Pathway
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Grant, B.F.; Goldstein, R.B.; Saha, T.D.; Chou, S.P.; Jung, J.; Zhang, H.; Pickering, R.P.; Ruan, W.J.; Smith, S.M.; Huang, B.; et al. Epidemiology of DSM-5 alcohol use disorder: Results from the national epidemiologic survey on alcohol and related conditions III. JAMA Psychiatry 2015, 72, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Naimi, T.S.; Brewer, R.D.; Mokdad, A.; Denny, C.; Serdula, M.K.; Marks, J.S. Binge drinking among US adults. JAMA 2003, 289, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Robin, R.W.; Long, J.C.; Rasmussen, J.K.; Albaugh, B.; Goldman, D. Relationship of binge drinking to alcohol dependence, other psychiatric disorders, and behavioral problems in an American Indian tribe. Alcohol. Clin. Exp. Res. 1998, 22, 518–523. [Google Scholar] [CrossRef] [PubMed]
- Hunt, W.A. Are binge drinkers more at risk of developing brain damage? Alcohol 1993, 10, 559–561. [Google Scholar] [CrossRef]
- Hasin, D.; Paykin, A.; Endicott, J. Course of DSM-IV alcohol dependence in a community sample: Effects of parental history and binge drinking. Alcohol. Clin. Exp. Res. 2001, 25, 411–414. [Google Scholar] [CrossRef] [PubMed]
- Volicer, L.; Volicer, B.J.; D’Angelo, N. Relationship of family history of alcoholism to patterns of drinking and physical dependence in male alcoholics. Drug Alcohol Depend. 1984, 13, 215–223. [Google Scholar] [CrossRef]
- Garbutt, J.C. The state of pharmacotherapy for the treatment of alcohol dependence. J. Subst. Abuse Treat. 2009, 36, S15–S23. [Google Scholar] [CrossRef] [PubMed]
- Litten, R.Z.; Egli, M.; Heilig, M.; Cui, C.; Fertig, J.B.; Ryan, M.L.; Falk, D.E.; Moss, H.; Huebner, R.; Noronha, A. Medications development to treat alcohol dependence: A vision for the next decade. Addict. Biol. 2012, 17, 513–527. [Google Scholar] [CrossRef] [PubMed]
- Mark, T.L.; Kassed, C.A.; Vandivort-Warren, R.; Levit, K.R.; Kranzler, H.R. Alcohol and opioid dependence medications: Prescription trends, overall and by physician specialty. Drug Alcohol Depend. 2009, 99, 345–349. [Google Scholar] [CrossRef] [PubMed]
- McLellan, A.T. Reducing heavy drinking: A public health strategy and a treatment goal? J. Subst. Abuse Treat. 2007, 33, 81–83. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, E.V.; Pfefferbaum, A. Neurocircuitry in alcoholism: A substrate of disruption and repair. Psychopharmacology 2005, 180, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Mechtcheriakov, S.; Brenneis, C.; Egger, K.; Koppelstaetter, F.; Schocke, M.; Marksteiner, J. A widespread distinct pattern of cerebral atrophy in patients with alcohol addiction revealed by voxel-based morphometry. J. Neurol. Neurosurg. Psychiatry 2007, 78, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Pfefferbaum, A.; Sullivan, E.V.; Mathalon, D.H.; Lim, K.O. Frontal lobe volume loss observed with magnetic resonance imaging in older chronic alcoholics. Alcohol. Clin. Exp. Res. 1997, 21, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Harper, C.; Kril, J. Patterns of neuronal loss in the cerebral cortex in chronic alcoholic patients. J. Neurol. Sci. 1989, 92, 81–89. [Google Scholar] [CrossRef]
- Ozsoy, S.; Durak, A.C.; Esel, E. Hippocampal volumes and cognitive functions in adult alcoholic patients with adolescent-onset. Alcohol 2013, 47, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, E.V.; Marsh, L.; Mathalon, D.H.; Lim, K.O.; Pfefferbaum, A. Anterior hippocampal volume deficits in nonamnesic, aging chronic alcoholics. Alcohol. Clin. Exp. Res. 1995, 19, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.; Bair, J.L.; Thomas, K.M.; Iacono, W.G. Problematic alcohol use and reduced hippocampal volume: A meta-analytic review. Psychol. Med. 2017, 47, 2288–2301. [Google Scholar] [CrossRef] [PubMed]
- Stavro, K.; Pelletier, J.; Potvin, S. Widespread and sustained cognitive deficits in alcoholism: A meta-analysis. Addict. Biol. 2012, 18, 203–213. [Google Scholar] [CrossRef] [PubMed]
- O’Daly, O.G.; Trick, L.; Scaife, J.; Marshall, J.; Ball, D.; Phillips, M.L.; Williams, S.S.; Stephens, D.N.; Duka, T. Withdrawal-associated increases and decreases in functional neural connectivity associated with altered emotional regulation in alcoholism. Neuropsychopharmacology 2012, 37, 2267–2276. [Google Scholar] [CrossRef] [PubMed]
- Crews, F.T. Alcohol and Neurodegeneration. CNS Drug Rev. 1999, 5, 379–394. [Google Scholar] [CrossRef]
- Rando, K.; Hong, K.I.; Bhagwagar, Z.; Li, C.S.; Bergquist, K.; Guarnaccia, J.; Sinha, R. Association of frontal and posterior cortical gray matter volume with time to alcohol relapse: A prospective study. Am. J. Psychiatry 2011, 168, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Marcos, M.; Pastor, I.; de la Calle, C.; Barrio-Real, L.; Laso, F.J.; Gonzalez-Sarmiento, R. Cannabinoid receptor 1 gene is associated with alcohol dependence. Alcohol. Clin. Exp. Res. 2012, 36, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Kranzler, H.R.; Luo, X.; Covault, J.; Gelernter, J. CNR1 variation modulates risk for drug and alcohol dependence. Biol. Psychiatry 2007, 62, 616–626. [Google Scholar] [CrossRef] [PubMed]
- Buhler, K.M.; Huertas, E.; Echeverry-Alzate, V.; Gine, E.; Molto, E.; Montoliu, L.; Lopez-Moreno, J.A. Risky alcohol consumption in young people is associated with the fatty acid amide hydrolase gene polymorphism C385A and affective rating of drug pictures. Mol. Genet. Genom. 2014, 289, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Huang, T.; Lee, F.; Kreek, M.J. Involvement of endocannabinoids in alcohol “binge” drinking: Studies of mice with human fatty acid amide hydrolase genetic variation and after CB1 receptor antagonists. Alcohol. Clin. Exp. Res. 2016, 40, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Hirvonen, J.; Zanotti-Fregonara, P.; Umhau, J.C.; George, D.T.; Rallis-Frutos, D.; Lyoo, C.H.; Li, C.T.; Hines, C.S.; Sun, H.; Terry, G.E.; et al. Reduced cannabinoid CB1 receptor binding in alcohol dependence measured with positron emission tomography. Mol. Psychiatry 2013, 18, 916–921. [Google Scholar] [CrossRef] [PubMed]
- Erdozain, A.M.; Callado, L.F. Involvement of the endocannabinoid system in alcohol dependence: The biochemical, behavioral and genetic evidence. Drug Alcohol Depend. 2011, 117, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Pava, M.J.; Woodward, J.J. A review of the interactions between alcohol and the endocannabinoid system: Implications for alcohol dependence and future directions for research. Alcohol 2012, 46, 185–204. [Google Scholar] [CrossRef] [PubMed]
- Kleczkowska, P.; Smaga, I.; Filip, M.; Bujalska-Zadrozny, M. Cannabinoid ligands and alcohol addiction: A promising therapeutic tool or a humbug? Neurotox. Res. 2016, 29, 173–196. [Google Scholar] [CrossRef] [PubMed]
- Gowran, A.; Noonan, J.; Campbell, V.A. The multiplicity of action of cannabinoids: Implications for treating neurodegeneration. CNS Neurosci. Ther. 2011, 17, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Berger, C.; Schmid, P.C.; Schabitz, W.R.; Wolf, M.; Schwab, S.; Schmid, H.H. Massive accumulation of N-acylethanolamines after stroke. Cell signalling in acute cerebral ischemia? J. Neurochem. 2004, 88, 1159–1167. [Google Scholar] [CrossRef] [PubMed]
- Degn, M.; Lambertsen, K.L.; Petersen, G.; Meldgaard, M.; Artmann, A.; Clausen, B.H.; Hansen, S.H.; Finsen, B.; Hansen, H.S.; Lund, T.M. Changes in brain levels of N-acylethanolamines and 2-arachidonoylglycerol in focal cerebral ischemia in mice. J. Neurochem. 2007, 103, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Eljaschewitsch, E.; Witting, A.; Mawrin, C.; Lee, T.; Schmidt, P.M.; Wolf, S.; Hoertnagl, H.; Raine, C.S.; Schneider-Stock, R.; Nitsch, R.; et al. The endocannabinoid anandamide protects neurons during CNS inflammation by induction of MKP-1 in microglial cells. Neuron 2006, 49, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Hansen, H.H.; Ikonomidou, C.; Bittigau, P.; Hansen, S.H.; Hansen, H.S. Accumulation of the anandamide precursor and other N-acylethanolamine phospholipids in infant rat models of in vivo necrotic and apoptotic neuronal death. J. Neurochem. 2001, 76, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Marsicano, G.; Goodenough, S.; Monory, K.; Hermann, H.; Eder, M.; Cannich, A.; Azad, S.C.; Cascio, M.G.; Gutiérrez, S.O.; van der Stelt, M.; et al. CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science 2003, 302, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Panikashvili, D.; Simeonidou, C.; Ben-Shabat, S.; Hanus, L.; Breuer, A.; Mechoulam, R.; Shohami, E. An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature 2001, 413, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Loria, F.; Petrosino, S.; Hernangomez, M.; Mestre, L.; Spagnolo, A.; Correa, F.; Di Marzo, V.; Docagne, F.; Guaza, C. An endocannabinoid tone limits excitotoxicity in vitro and in a model of multiple sclerosis. Neurobiol. Dis. 2010, 37, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Hansen, H.S.; Lauritzen, L.; Strand, A.M.; Vinggaard, A.M.; Frandsen, A.; Schousboe, A. Characterization of glutamate-induced formation of N-acylphosphatidylethanolamine and N-acylethanolamine in cultured neocortical neurons. J. Neurochem. 1997, 69, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Parmentier-Batteur, S.; Jin, K.; Mao, X.O.; Xie, L.; Greenberg, D.A. Increased severity of stroke in CB1 cannabinoid receptor knock-out mice. J. Neurosci. 2002, 22, 9771–9775. [Google Scholar] [PubMed]
- Van der Stelt, M.; Di Marzo, V. Cannabinoid receptors and their role in neuroprotection. Neuromol. Med. 2005, 7, 37–50. [Google Scholar] [CrossRef]
- Di Marzo, V.; Stella, N.; Zimmer, A. Endocannabinoid signalling and the deteriorating brain. Nat. Rev. Neurosci. 2015, 16, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Paloczi, J.; Varga, Z.V.; Hasko, G.; Pacher, P. Neuroprotection in oxidative stress-related neurodegenerative dieseases: Role of endocannabinoid system modulation. Antioxid. Redox Signal. 2016. [Google Scholar] [CrossRef]
- Crews, F.T.; Nixon, K. Mechanisms of neurodegeneration and regeneration in alcoholism. Alcohol Alcohol. 2009, 44, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Croxford, J.L. Therapeutic potential of cannabinoids in CNS disease. CNS Drugs 2003, 17, 179–202. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.Y.; Ju, C.; Jalin, A.M.A.; Lee, D.I.; Prather, P.L.; Kim, W.K. Activation of cannabinoid CB2 receptor-mediated AMPK/CREB pathway reduces cerebral Ischemic Injury. Am. J. Pathol. 2013, 182, 928–939. [Google Scholar] [CrossRef] [PubMed]
- Karanian, D.A.; Brown, Q.B.; Makriyannis, A.; Kosten, T.A.; Bahr, B.A. Dual modulation of endocannabinoid transport and fatty acid amide hydrolase protects against excitotoxicity. J. Neurosci. 2005, 25, 7813–7820. [Google Scholar] [CrossRef] [PubMed]
- Karanian, D.A.; Karim, S.L.; Wood, J.T.; Williams, J.S.; Lin, S.; Makriyannis, A.; Bahr, B.A. Endocannabinoid enhancement protects against kainic acid-induced seizures and associated brain damage. J. Pharmacol. Exp. Ther. 2007, 322, 1059–1066. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, V.; Nikas, S.P.; Karanian, D.A.; Hwang, J.; Zhao, J.; Wood, J.T.; Alapafuja, S.O.; Vadivel, S.K.; Butler, D.; Makriyannis, A.; et al. A new generation fatty acid amide hydrolase inhibitor protects against kainate-induced excitotoxicity. J. Mol. Neurosci. 2011, 43, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Bortolato, M.; Mangieri, R.A.; Fu, J.; Kim, J.H.; Arguello, O.; Duranti, A.; Tontini, A.; Mor, M.; Tarzia, G.; Piomelli, D. Antidepressant-like activity of the fatty acid amide hydrolase inhibitor URB597 in a rat model of chronic mild stress. Biol. Psychiatry 2007, 62, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Adamson, C.; Butler, D.; Janero, D.R.; Makriyannis, A.; Bahr, B.A. Enhancement of endocannabinoid signaling by fatty acid amide hydrolase inhibition: A neuroprotective therapeutic modality. Life Sci. 2010, 86, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Rubio, M.; Villain, H.; Docagne, F.; Roussel, B.D.; Ramos, J.A.; Vivien, D.; Fernandez-Ruiz, J.; Ali, C. Pharmacological activation/inhibition of the cannabinoid system affects alcohol withdrawal-induced neuronal hypersensitivity to excitotoxic insults. PLoS ONE 2011, 6, e23690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cadete-Leite, A.; Tavares, M.A.; Uylings, H.B.; Paula-Barbosa, M. Granule cell loss and dendritic regrowth in the hippocampal dentate gyrus of the rat after chronic alcohol consumption. Brain Res. 1988, 473, 1–14. [Google Scholar] [CrossRef]
- Corso, T.D.; Mostafa, H.M.; Collins, M.A.; Neafsey, E.J. Brain neuronal degeneration caused by episodic alcohol intoxication in rats: Effects of nimodipine, 6,7-dinitro-quinoxaline-2,3-dione, and MK-801. Alcohol. Clin. Exp. Res. 1998, 22, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Hayes, D.M.; Deeny, M.A.; Shaner, C.A.; Nixon, K. Determining the threshold for alcohol-induced brain damage: New evidence with gliosis markers. Alcohol. Clin. Exp. Res. 2013, 37, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Obernier, J.A.; Bouldin, T.W.; Crews, F.T. Binge ethanol exposure in adult rats causes necrotic cell death. Alcohol. Clin. Exp. Res. 2002, 26, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Pelicao, R.; Santos, M.C.; Freitas-Lima, L.C.; Meyrelles, S.S.; Vasquez, E.C.; Nakamura-Palacios, E.M.; Rodrigues, L.C. URB597 inhibits oxidative stress induced by alcohol binging in the prefrontal cortex of adolescent rats. Neurosci. Lett. 2016, 624, 17–22. [Google Scholar] [CrossRef] [PubMed]
- National Research Council. Guide for the Care and Use of Laboratory Animals; National Academies Press: Washington, DC, USA, 2010. [Google Scholar]
- Morris, S.A.; Kelso, M.L.; Liput, D.J.; Marshall, S.A.; Nixon, K. Similar withdrawal severity in adolescents and adults in a rat model of alcohol dependence. Alcohol 2010, 44, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Urso, T.; Gavaler, J.S.; Van Thiel, D.H. Blood ethanol levels in sober alcohol users seen in an emergency room. Life Sci. 1981, 28, 1053–1056. [Google Scholar] [CrossRef]
- Van Hoof, J.J.; Lely, N.; Pereira, R.R.; van Dalen, W.E. Adolescent alcohol intoxication in the Dutch hospital Departments of Pediatrics. J. Stud. Alcohol Drugs 2010, 71, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.A.; Corso, T.D.; Neafsey, E.J. Neuronal degeneration in rat cerebrocortical and olfactory regions during subchronic “binge” intoxication with ethanol: Possible explanation for olfactory deficits in alcoholics. Alcohol. Clin. Exp. Res. 1996, 20, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Kelso, M.L.; Liput, D.J.; Eaves, D.W.; Nixon, K. Upregulated vimentin suggests new areas of neurodegeneration in a model of an alcohol use disorder. Neuroscience 2011, 197, 381–393. [Google Scholar] [CrossRef] [PubMed]
- Crews, F.T.; Braun, C.J.; Hoplight, B.; Switzer, R.C., 3rd; Knapp, D.J. Binge ethanol consumption causes differential brain damage in young adolescent rats compared with adult rats. Alcohol. Clin. Exp. Res. 2000, 24, 1712–1723. [Google Scholar] [CrossRef] [PubMed]
- Liput, D.J.; Hammell, D.C.; Stinchcomb, A.L.; Nixon, K. Transdermal delivery of cannabidiol attenuates binge alcohol-induced neurodegeneration in a rodent model of an alcohol use disorder. Pharm. Biochem. Behav. 2013, 111, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Majchrowicz, E. Induction of physical dependence upon ethanol and the associated behavioral changes in rats. Psychopharmacology 1975, 43, 245–254. [Google Scholar] [CrossRef]
- Herkenham, M.; Lynn, A.B.; Johnson, M.R.; Melvin, L.S.; de Costa, B.R.; Rice, K.C. Characterization and localization of cannabinoid receptors in rat brain: A quantitative in vitro autoradiographic study. J. Neurosci. 1991, 11, 563–583. [Google Scholar] [PubMed]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates; Academic Press: London, UK, 2009. [Google Scholar]
- Liput, D.J.; Tsakalozou, E.; Hammell, D.C.; Paudel, K.S.; Nixon, K.; Stinchcomb, A.L. Quantification of anandamide, oleoylethanolamide and palmitoylethanolamide in rodent brain tissue using high performance liquid chromatography-electrospray mass spectroscopy. J. Pharm. Anal. 2014, 4, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Fegley, D.; Gaetani, S.; Duranti, A.; Tontini, A.; Mor, M.; Tarzia, G.; Piomelli, D. Characterization of the fatty acid amide hydrolase inhibitor cyclohexyl carbamic acid 3′-carbamoyl-biphenyl-3-yl ester (URB597): Effects on anandamide and oleoylethanolamide deactivation. J. Pharmacol. Exp. Ther. 2005, 313, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Leasure, J.L.; Nixon, K. Exercise neuroprotection in a rat model of binge alcohol consumption. Alcohol. Clin. Exp. Res. 2010, 34, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Vinod, K.Y.; Yalamanchili, R.; Xie, S.; Cooper, T.B.; Hungund, B.L. Effect of chronic ethanol exposure and its withdrawal on the endocannabinoid system. Neurochem. Int. 2006, 49, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Basavarajappa, B.S.; Cooper, T.B.; Hungund, B.L. Chronic ethanol administration down-regulates cannabinoid receptors in mouse brain synaptic plasma membrane. Brain Res. 1998, 793, 212–218. [Google Scholar] [CrossRef]
- Mitrirattanakul, S.; Lopez-Valdes, H.E.; Liang, J.; Matsuka, Y.; Mackie, K.; Faull, K.F.; Spigelman, I. Bidirectional alterations of hippocampal cannabinoid 1 receptors and their endogenous ligands in a rat model of alcohol withdrawal and dependence. Alcohol. Clin. Exp. Res. 2007, 31, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Pava, M.J.; Blake, E.M.; Green, S.T.; Mizroch, B.J.; Mulholland, P.J.; Woodward, J.J. Tolerance to cannabinoid-induced behaviors in mice treated chronically with ethanol. Psychopharmacology 2012, 219, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Hansen, H.S.; Moesgaard, B.; Petersen, G.; Hansen, H.H. Putative neuroprotective actions of N-acyl-ethanolamines. Pharmacol. Ther. 2002, 95, 119–126. [Google Scholar] [CrossRef]
- Collins, M.A.; Zou, J.Y.; Neafsey, E.J. Brain damage due to episodic alcohol exposure in vivo and in vitro: Furosemide neuroprotection implicates edema-based mechanism. FASEB J. 1998, 12, 221–230. [Google Scholar] [PubMed]
- Hamelink, C.; Hampson, A.; Wink, D.A.; Eiden, L.E.; Eskay, R.L. Comparison of cannabidiol, antioxidants, and diuretics in reversing binge ethanol-induced neurotoxicity. J. Pharmacol. Exp. Ther. 2005, 314, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Okine, B.N.; Norris, L.M.; Woodhams, S.; Burston, J.; Patel, A.; Alexander, S.P.; Barrett, D.A.; Kendall, D.A.; Bennett, A.J.; Chapman, V. Lack of effect of chronic pre-treatment with the FAAH inhibitor URB597 on inflammatory pain behaviour: Evidence for plastic changes in the endocannabinoid system. Br. J. Pharmacol. 2012, 167, 627–640. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, B.; Bermudez-Silva, F.J.; Bilbao, A.; Alvarez-Jaimes, L.; Sanchez-Vera, I.; Giuffrida, A.; Serrano, A.; Baixeras, E.; Khaturia, S.; Navarro, M.; et al. Regulation of brain anandamide by acute administration of ethanol. Biochem. J. 2007, 404, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Egertova, M.; Cravatt, B.F.; Elphick, M.R. Comparative analysis of fatty acid amide hydrolase and cb1 cannabinoid receptor expression in the mouse brain: Evidence of a widespread role for fatty acid amide hydrolase in regulation of endocannabinoid signaling. Neuroscience 2003, 119, 481–496. [Google Scholar] [CrossRef]
- Hansen, H.S.; Moesgaard, B.; Hansen, H.H.; Petersen, G. N-Acylethanolamines and precursor phospholipids—Relation to cell injury. Chem. Phys. Lipids 2000, 108, 135–150. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Intoxication Behavior * | Ethanol Dose (g/kg/day) | BEC (mg/dL) |
|---|---|---|---|
| CB1 Autoradiography | |||
| 1-Day (n = 5) | 0.5 ± 0.1 | 13.4 ± 0.4 | 304.1 ± 19.0 |
| 2-Day (n = 6) | 1.6 ± 0.1 | 10.3 ± 0.3 | 403.2 ± 28.0 |
| 4-Day (n = 6) | 2.1 ± 0.2 | 8.8 ± 0.4 | 404.9 ± 24.4 |
| NAE quantification | |||
| 2-Day (n = 6) | 1.4 ± 0.2 | 10.8 ± 0.6 | 443.8 ± 10.5 |
| 4-Day (n = 7) | 2.3 ± 0.1 | 8.1 ± 0.3 | 420.5 ± 24.1 |
| Group | Intoxication Behavior * | Ethanol Dose (g/kg/day) | BEC (mg/dL) |
|---|---|---|---|
| No Injection (n = 5) | 0.3 ± 0.1 | 13.8 ± 0.4 | 331.8 ± 27.8 |
| Vehicle (n = 6) | 0.1 ± 0.1 | 14.7 ± 0.2 | 304.3 ± 18.4 |
| URB597 (n = 6) | 0.2 ± 0.2 | 14.3 ± 0.2 | 291.4 ± 28.4 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liput, D.J.; Pauly, J.R.; Stinchcomb, A.L.; Nixon, K. Binge Alcohol Exposure Transiently Changes the Endocannabinoid System: A Potential Target to Prevent Alcohol-Induced Neurodegeneration. Brain Sci. 2017, 7, 158. https://doi.org/10.3390/brainsci7120158
Liput DJ, Pauly JR, Stinchcomb AL, Nixon K. Binge Alcohol Exposure Transiently Changes the Endocannabinoid System: A Potential Target to Prevent Alcohol-Induced Neurodegeneration. Brain Sciences. 2017; 7(12):158. https://doi.org/10.3390/brainsci7120158
Chicago/Turabian StyleLiput, Daniel J., James R. Pauly, Audra L. Stinchcomb, and Kimberly Nixon. 2017. "Binge Alcohol Exposure Transiently Changes the Endocannabinoid System: A Potential Target to Prevent Alcohol-Induced Neurodegeneration" Brain Sciences 7, no. 12: 158. https://doi.org/10.3390/brainsci7120158