Activation of ERBB4 in Glioblastoma Can Contribute to Increased Tumorigenicity and Influence Therapeutic Response

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
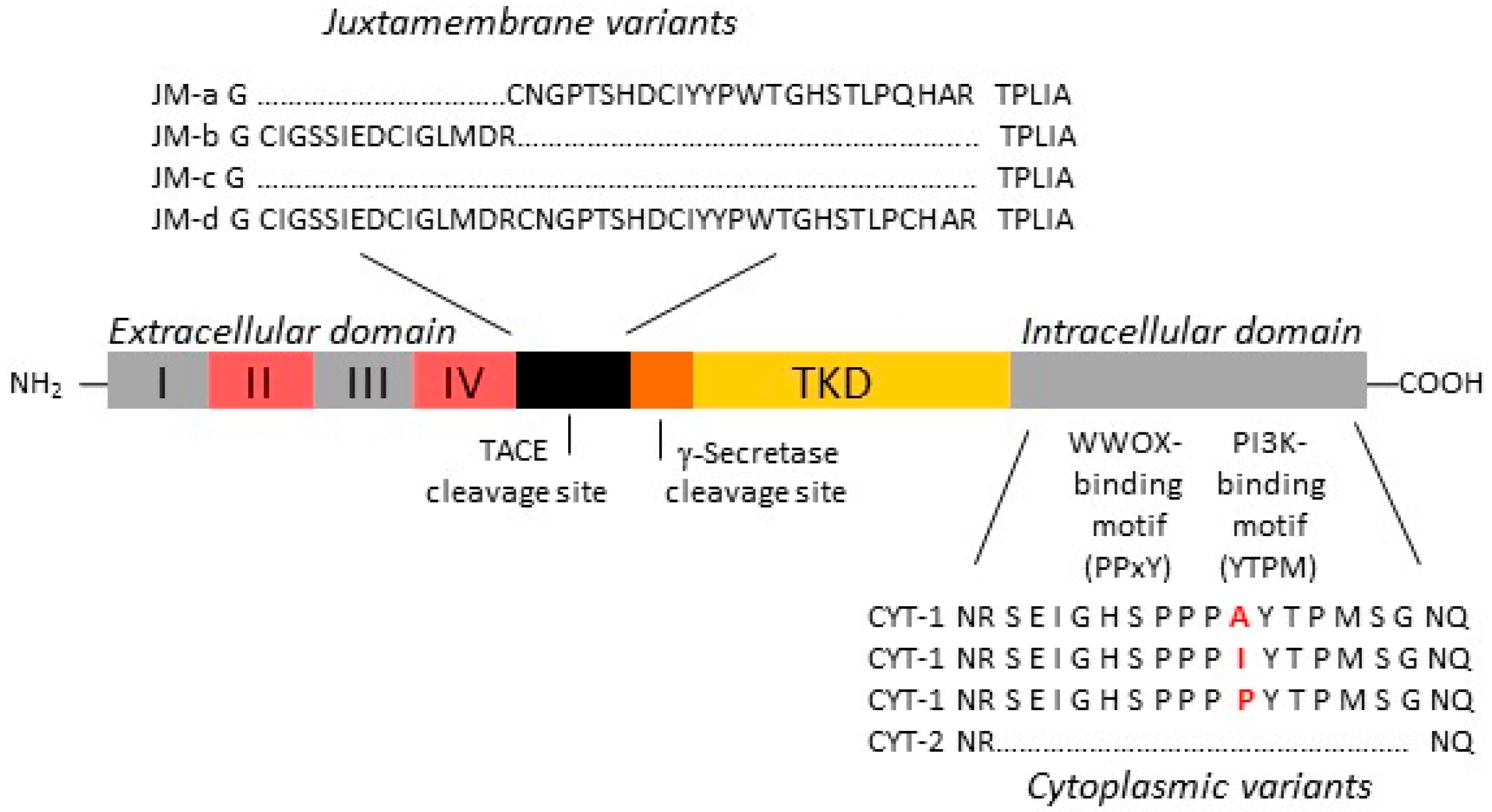
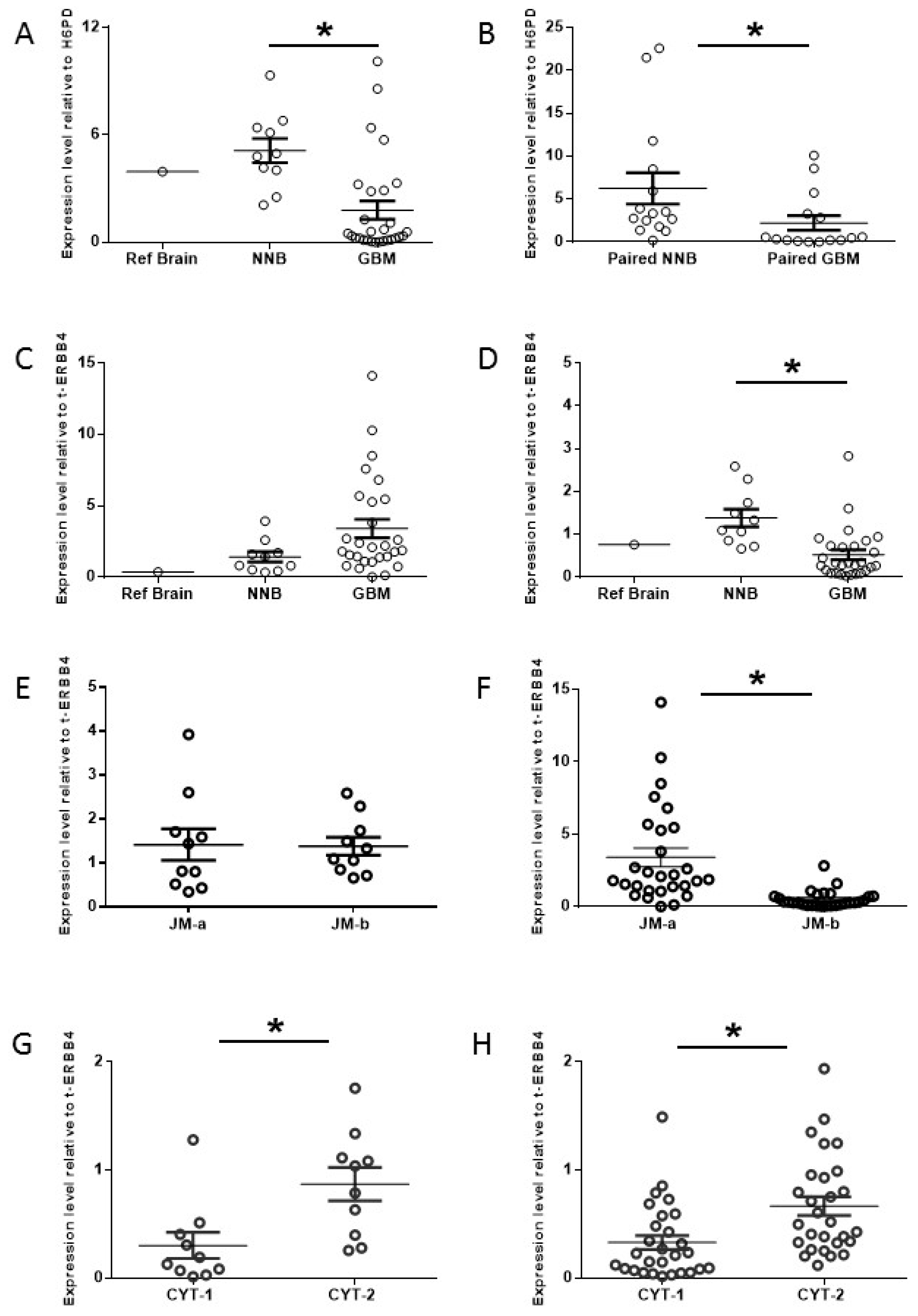
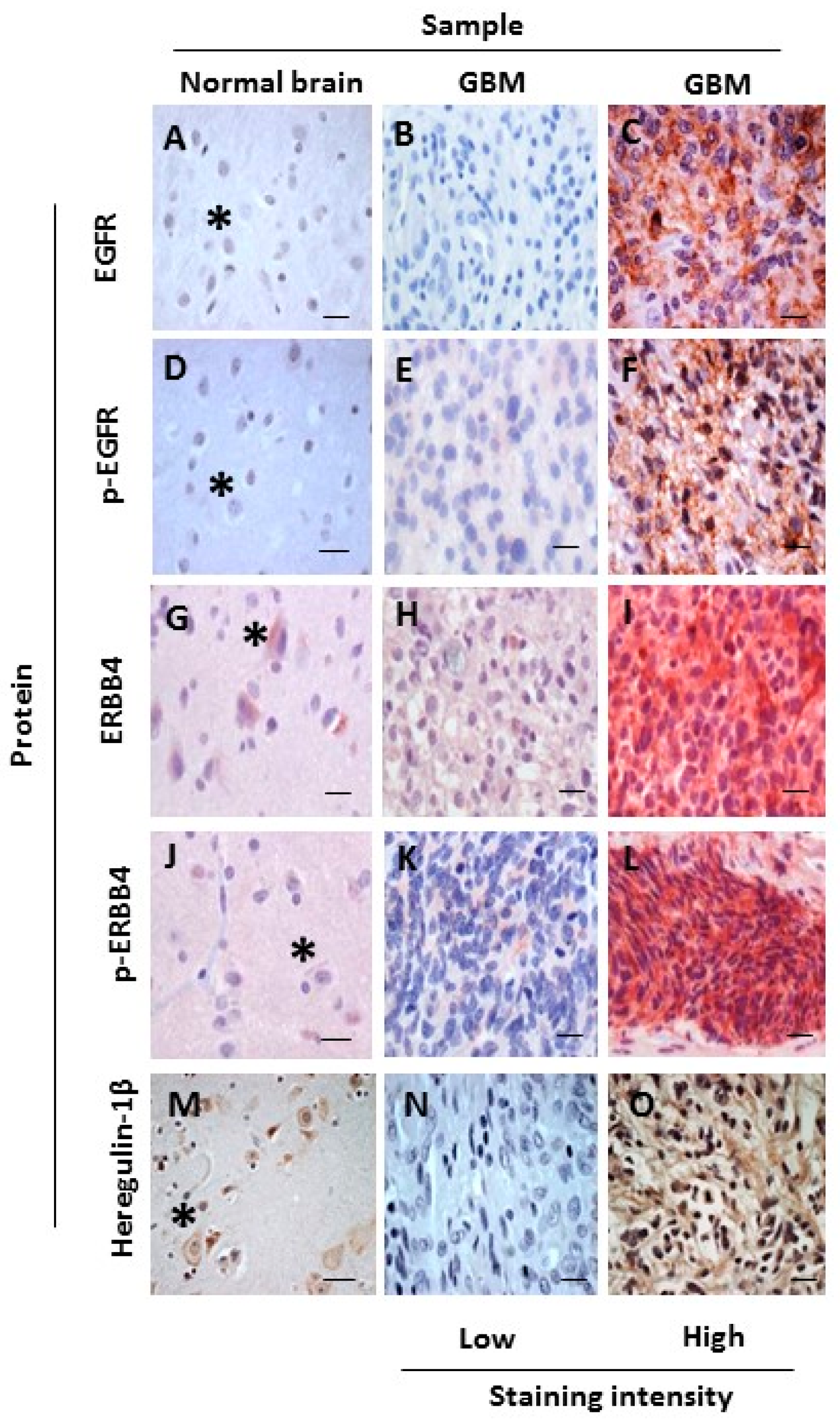
2.1. Increased Oncogenic ERBB4 Variant Expression Is Associated with GBM
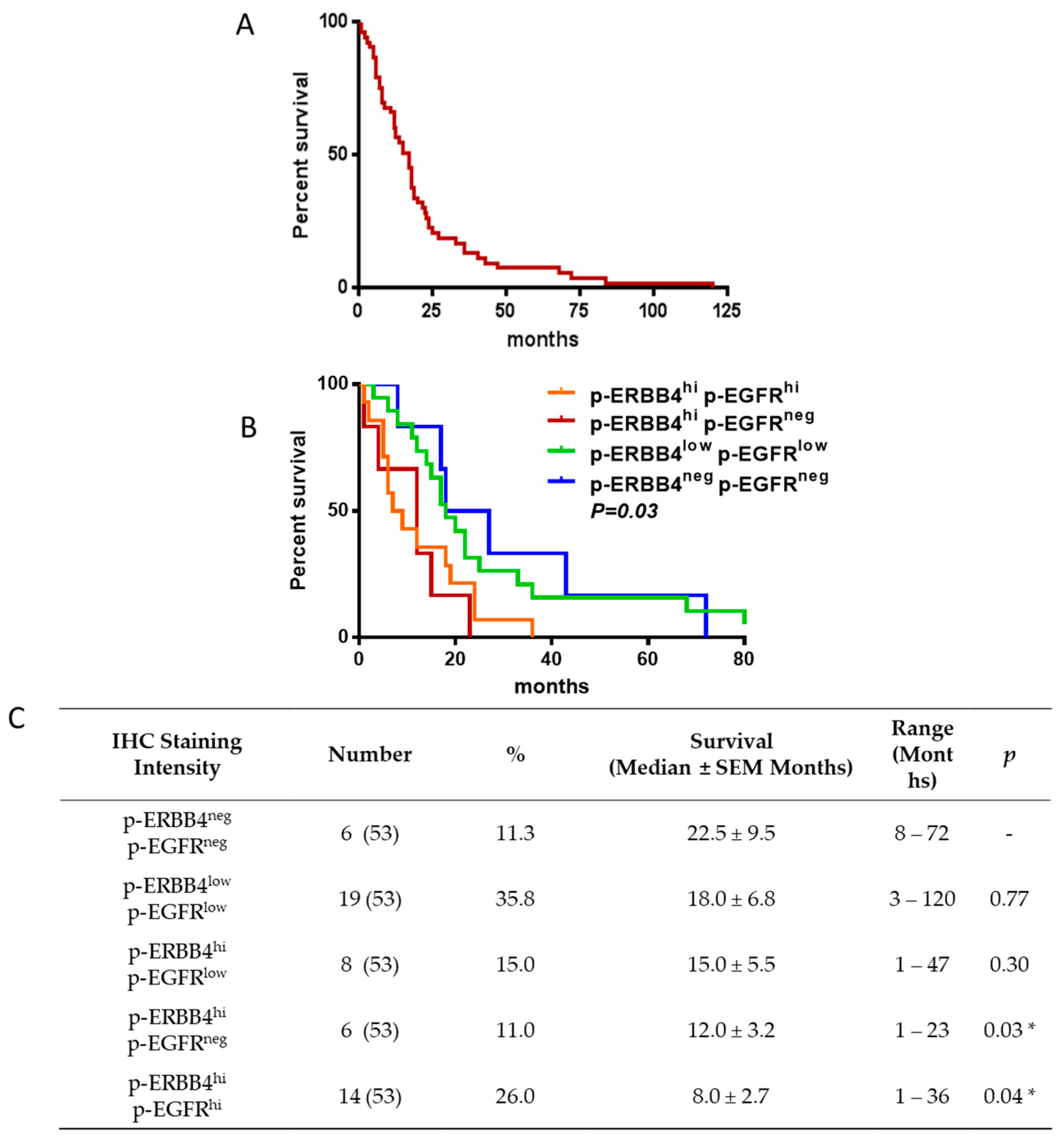
2.2. ERBB4 Activation in GBM Is Associated with Shorter Overall Survival
2.3. ERBB4 Activation Enhances GBM Cell Growth and Tumorigenicity
2.4. Endogenous ERBB4 Activation Permits Tumor Survival During Anti-EGFR Therapy
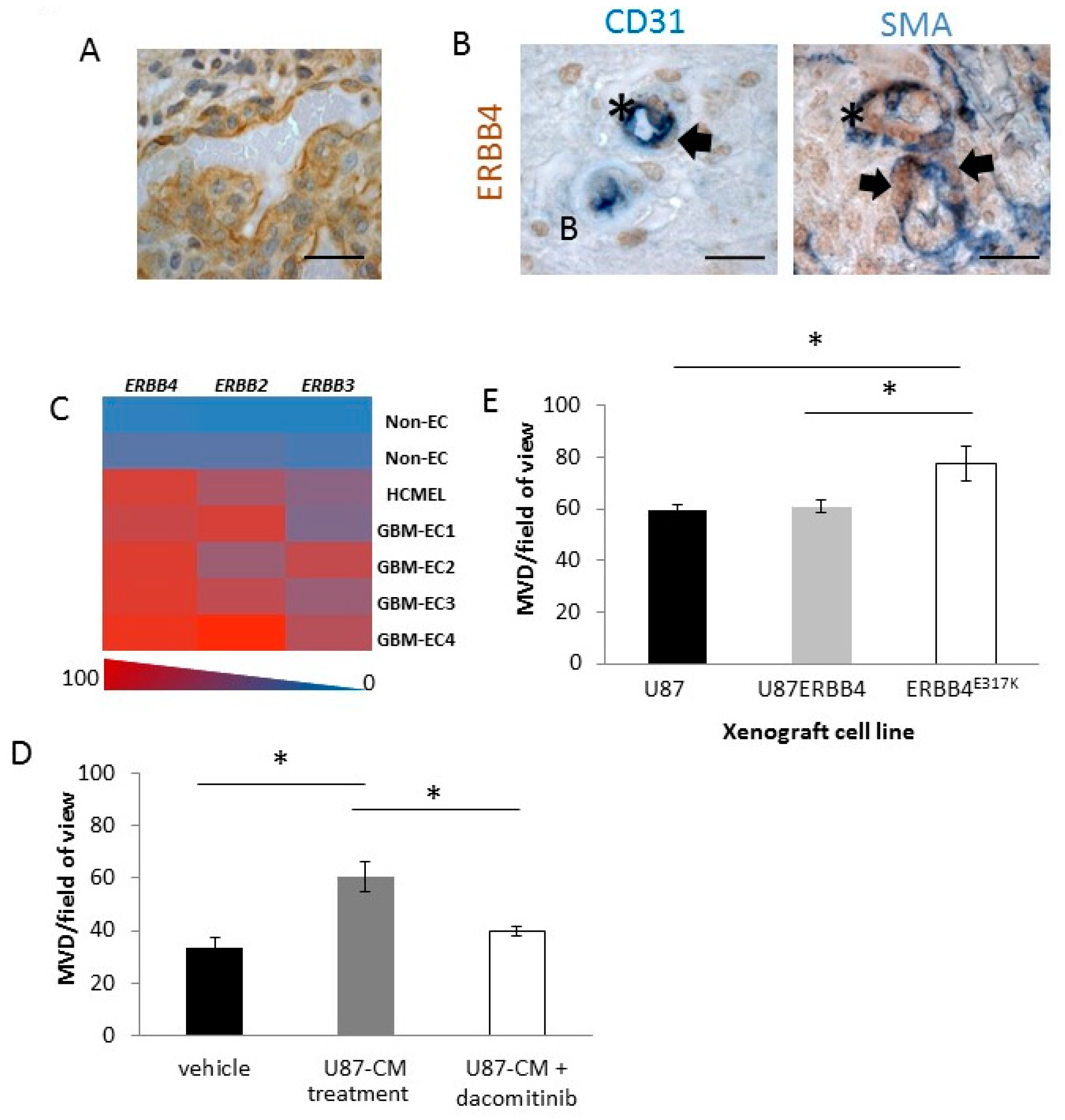
2.5. ERBB4 Has a Vascular Role in GBM
3. Discussion
4. Materials and Methods
4.1. Patient Samples
4.2. Cell Lines, Chemicals and Drugs
4.3. Derivation of U87ERBB4 and U87ERBB4E317K Cell Lines
4.4. RT-qPCR
4.5. Immunohistochemistry (IHC)
4.6. MTS and Bio-Plex Assays
4.7. Gene Expression Profiling of ERBB Proteins in GBM-Derived Endothelial Cells
4.8. Xenograft Studies
4.9. TGCA Analysis
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Maher, E.A.; Furnari, F.B.; Bachoo, R.M.; Rowitch, D.H.; Louis, D.N.; Cavenee, W.K.; DePinho, R.A. Malignant glioma: Genetics and biology of a grave matter. Genes Dev. 2001, 15, 1311–1333. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef] [PubMed]
- Inda, M.M.; Bonavia, R.; Mukasa, A.; Narita, Y.; Sah, D.W.; Vandenberg, S.; Brennan, C.; Johns, T.G.; Bachoo, R.; Hadwiger, P.; et al. Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev. 2010, 24, 1731–1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Bent, M.J.; Brandes, A.A.; Rampling, R.; Kouwenhoven, M.C.; Kros, J.M.; Carpentier, A.F.; Clement, P.M.; Frenay, M.; Campone, M.; Baurain, J.F.; et al. Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. J. Clin. Oncol. 2009, 27, 1268–1274. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.D.; Krishnan, S.; Sarkaria, J.N.; Wu, W.; Jaeckle, K.A.; Uhm, J.H.; Geoffroy, F.J.; Arusell, R.; Kitange, G.; Jenkins, R.B.; et al. Phase I/II trial of erlotinib and temozolomide with radiation therapy in the treatment of newly diagnosed glioblastoma multiforme: North central cancer treatment group study N0177. J. Clin. Oncol. 2008, 26, 5603–5609. [Google Scholar] [CrossRef] [PubMed]
- Shinojima, N.; Tada, K.; Shiraishi, S.; Kamiryo, T.; Kochi, M.; Nakamura, H.; Makino, K.; Saya, H.; Hirano, H.; Kuratsu, J.; et al. Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer Res. 2003, 63, 6962–6970. [Google Scholar] [PubMed]
- Hatanpaa, K.J.; Burma, S.; Zhao, D.; Habib, A.A. Epidermal growth factor receptor in glioma: Signal transduction, neuropathology, imaging, and radioresistance. Neoplasia 2010, 12, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Heimberger, A.B.; Hlatky, R.; Suki, D.; Yang, D.; Weinberg, J.; Gilbert, M.; Sawaya, R.; Aldape, K. Prognostic effect of epidermal growth factor receptor and EGFRVIII in glioblastoma multiforme patients. Clin. Cancer Res. 2005, 11, 1462–1466. [Google Scholar] [CrossRef] [PubMed]
- Benito, R.; Gil-Benso, R.; Quilis, V.; Perez, M.; Gregori-Romero, M.; Roldan, P.; Gonzalez-Darder, J.; Cerda-Nicolas, M.; Lopez-Gines, C. Primary glioblastomas with and without EGFR amplification: Relationship to genetic alterations and clinicopathological features. Neuropathology 2010, 30, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Andersson, U.; Guo, D.; Malmer, B.; Bergenheim, A.T.; Brannstrom, T.; Hedman, H.; Henriksson, R. Epidermal growth factor receptor family (EGFR, ERBB2–4) in gliomas and meningiomas. Acta Neuropathol. 2004, 108, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Duhem-Tonnelle, V.; Bieche, I.; Vacher, S.; Loyens, A.; Maurage, C.A.; Collier, F.; Baroncini, M.; Blond, S.; Prevot, V.; Sharif, A. Differential distribution of ERBB receptors in human glioblastoma multiforme: Expression of ERBB3 in CD133-positive putative cancer stem cells. J. Neuropathol. Exp. Neurol. 2010, 69, 606–622. [Google Scholar] [CrossRef] [PubMed]
- Stommel, J.M.; Kimmelman, A.C.; Ying, H.; Nabioullin, R.; Ponugoti, A.H.; Wiedemeyer, R.; Stegh, A.H.; Bradner, J.E.; Ligon, K.L.; Brennan, C.; et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science 2007, 318, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Gassmann, M.; Casagranda, F.; Orioli, D.; Simon, H.; Lai, C.; Klein, R.; Lemke, G. Aberrant neural and cardiac development in mice lacking the ERBB4 neuregulin receptor. Nature 1995, 378, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, R.; Poulsom, R.; Hurst, H.C.; Gullick, W.J. Expression of the c-ERBB-4/HER4 protein and mRNA in normal human fetal and adult tissues and in a survey of nine solid tumour types. J. Pathol. 1998, 185, 236–245. [Google Scholar] [CrossRef]
- Law, A. Schizophrenia, IV: Neuregulin-1 in the human brain. Am. J. Psychiatry 2003, 160, 1392. [Google Scholar] [CrossRef] [PubMed]
- Gilbertson, R.; Hernan, R.; Pietsch, T.; Pinto, L.; Scotting, P.; Allibone, R.; Ellison, D.; Perry, R.; Pearson, A.; Lunec, J. Novel ERBB4 juxtamembrane splice variants are frequently expressed in childhood medulloblastoma. Genes Chromosomes Cancer 2001, 31, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Gilbertson, R.J.; Bentley, L.; Hernan, R.; Junttila, T.T.; Frank, A.J.; Haapasalo, H.; Connelly, M.; Wetmore, C.; Curran, T.; Elenius, K.; et al. ERBB receptor signaling promotes ependymoma cell proliferation and represents a potential novel therapeutic target for this disease. Clin. Cancer Res. 2002, 8, 3054–3064. [Google Scholar] [PubMed]
- Plowman, G.D.; Culouscou, J.M.; Whitney, G.S.; Green, J.M.; Carlton, G.W.; Foy, L.; Neubauer, M.G.; Shoyab, M. Ligand-specific activation of HER4/p180ERBB4, a fourth member of the epidermal growth factor receptor family. Proc. Natl. Acad. Sci. USA 1993, 90, 1746–1750. [Google Scholar] [CrossRef] [PubMed]
- Junttila, T.T.; Sundvall, M.; Lundin, M.; Lundin, J.; Tanner, M.; Harkonen, P.; Joensuu, H.; Isola, J.; Elenius, K. Cleavable ERBB4 isoform in estrogen receptor-regulated growth of breast cancer cells. Cancer Res. 2005, 65, 1384–1393. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, E.; Di Marcotullio, L.; Gessi, M.; Mattei, T.; Greco, A.; Po, A.; De Smaele, E.; Giangaspero, F.; Riccardi, R.; Di Rocco, C.; et al. Alternative splicing of the ERBB-4 cytoplasmic domain and its regulation by hedgehog signaling identify distinct medulloblastoma subsets. Oncogene 2006, 25, 7267–7273. [Google Scholar] [CrossRef] [PubMed]
- Sartor, C.I.; Zhou, H.; Kozlowska, E.; Guttridge, K.; Kawata, E.; Caskey, L.; Harrelson, J.; Hynes, N.; Ethier, S.; Calvo, B.; et al. Her4 mediates ligand-dependent antiproliferative and differentiation responses in human breast cancer cells. Mol. Cell. Biol. 2001, 21, 4265–4275. [Google Scholar] [CrossRef] [PubMed]
- Naresh, A.; Long, W.; Vidal, G.A.; Wimley, W.C.; Marrero, L.; Sartor, C.I.; Tovey, S.; Cooke, T.G.; Bartlett, J.M.; Jones, F.E. The ERBB4/HER4 intracellular domain 4ICD is a BH3-only protein promoting apoptosis of breast cancer cells. Cancer Res. 2006, 66, 6412–6420. [Google Scholar] [CrossRef] [PubMed]
- Sundvall, M.; Veikkolainen, V.; Kurppa, K.; Salah, Z.; Tvorogov, D.; van Zoelen, E.J.; Aqeilan, R.; Elenius, K. Cell death or survival promoted by alternative isoforms of ERBB4. Mol. Biol. Cell 2010, 21, 4275–4286. [Google Scholar] [CrossRef] [PubMed]
- Maatta, J.A.; Sundvall, M.; Junttila, T.T.; Peri, L.; Laine, V.J.; Isola, J.; Egeblad, M.; Elenius, K. Proteolytic cleavage and phosphorylation of a tumor-associated ERBB4 isoform promote ligand-independent survival and cancer cell growth. Mol. Biol. Cell 2006, 17, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Tal-Or, P.; Erlich, S.; Porat-Shliom, N.; Goldshmit, Y.; Ben-Baruch, G.; Shaharabani, E.; Kloog, Y.; Pinkas-Kramarski, R. Ligand-independent regulation of ERBB4 receptor phosphorylation by activated ras. J. Cell. Biochem. 2006, 98, 1482–1494. [Google Scholar] [CrossRef] [PubMed]
- Zeng, F.; Zhang, M.Z.; Singh, A.B.; Zent, R.; Harris, R.C. ERBB4 isoforms selectively regulate growth factor induced madin-darby canine kidney cell tubulogenesis. Mol. Biol. Cell 2007, 18, 4446–4456. [Google Scholar] [CrossRef] [PubMed]
- Sundvall, M.; Peri, L.; Maatta, J.A.; Tvorogov, D.; Paatero, I.; Savisalo, M.; Silvennoinen, O.; Yarden, Y.; Elenius, K. Differential nuclear localization and kinase activity of alternative ERBB4 intracellular domains. Oncogene 2007, 26, 6905–6914. [Google Scholar] [CrossRef] [PubMed]
- Prickett, T.D.; Agrawal, N.S.; Wei, X.; Yates, K.E.; Lin, J.C.; Wunderlich, J.R.; Cronin, J.C.; Cruz, P.; Rosenberg, S.A.; Samuels, Y. Analysis of the tyrosine kinome in melanoma reveals recurrent mutations in ERBB4. Nat. Genet. 2009, 41, 1127–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimoto, K.; Ma, X.; Guan, Y.; Mizoguchi, M.; Nakamizo, A.; Amano, T.; Hata, N.; Kuga, D.; Sasaki, T. Expression of stem cell marker and receptor kinase genes in glioblastoma tissue quantified by real-time RT-PCR. Brain Tumor Pathol. 2011, 28, 291–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veikkolainen, V.; Vaparanta, K.; Halkilahti, K.; Iljin, K.; Sundvall, M.; Elenius, K. Function of ERBB4 is determined by alternative splicing. Cell Cycle 2011, 10, 2647–2657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elenius, K.; Choi, C.J.; Paul, S.; Santiestevan, E.; Nishi, E.; Klagsbrun, M. Characterization of a naturally occurring ERBB4 isoform that does not bind or activate phosphatidyl inositol 3-kinase. Oncogene 1999, 18, 2607–2615. [Google Scholar] [CrossRef] [PubMed]
- Haas-Kogan, D.A.; Prados, M.D.; Tihan, T.; Eberhard, D.A.; Jelluma, N.; Arvold, N.D.; Baumber, R.; Lamborn, K.R.; Kapadia, A.; Malec, M.; et al. Epidermal growth factor receptor, protein kinase B/Akt, and glioma response to erlotinib. J. Natl. Cancer Inst. 2005, 97, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.F.; Tatter, S.B.; Willingham, M.C.; Yang, M.; Hu, J.J.; Frankel, A.E. Growth factor receptor expression varies among high-grade gliomas and normal brain: Epidermal growth factor receptor has excellent properties for interstitial fusion protein therapy. Mol. Cancer Ther. 2003, 2, 783–787. [Google Scholar] [PubMed]
- Taylor, T.E.; Furnari, F.B.; Cavenee, W.K. Targeting EGFR for treatment of glioblastoma: Molecular basis to overcome resistance. Curr. Cancer Drug Targets 2012, 12, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Koziolek, E.J.; Donoghue, J.F.; Bentley, J.D.; Lovrecz, G.; Dolezal, O.; Ward, C.W.; Rothacker, J.; Nice, E.C.; Burgess, A.W.; Hafner, M.; et al. A high-affinity ERBB4fc fusion protein is a potent antagonist of heregulin-mediated receptor activation. Growth Factors 2012, 30, 310–319. [Google Scholar] [CrossRef] [PubMed]
- Muraoka-Cook, R.S.; Sandahl, M.A.; Strunk, K.E.; Miraglia, L.C.; Husted, C.; Hunter, D.M.; Elenius, K.; Chodosh, L.A.; Earp, H.S., III. ERBB4 splice variants CYT1 and CYT2 differ by 16 amino acids and exert opposing effects on the mammary epithelium in vivo. Mol. Cell. Biol. 2009, 29, 4935–4948. [Google Scholar] [CrossRef] [PubMed]
- Kainulainen, V.; Sundvall, M.; Maatta, J.A.; Santiestevan, E.; Klagsbrun, M.; Elenius, K. A natural ERBB4 isoform that does not activate phosphoinositide 3-kinase mediates proliferation but not survival or chemotaxis. J. Biol. Chem. 2000, 275, 8641–8649. [Google Scholar] [CrossRef] [PubMed]
- Schuchardt, B.J.; Bhat, V.; Mikles, D.C.; McDonald, C.B.; Sudol, M.; Farooq, A. Molecular basis of the binding of yap transcriptional regulator to the ERBB4 receptor tyrosine kinase. Biochimie 2014, 101, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Sundvall, M.; Korhonen, A.; Paatero, I.; Gaudio, E.; Melino, G.; Croce, C.M.; Aqeilan, R.I.; Elenius, K. Isoform-specific monoubiquitination, endocytosis, and degradation of alternatively spliced ERBB4 isoforms. Proc. Natl. Acad. Sci. USA 2008, 105, 4162–4167. [Google Scholar] [CrossRef] [PubMed]
- Zeng, F.; Xu, J.; Harris, R.C. NEDD4 mediates ERBB4 JM-a/CYT-1 ICD ubiquitination and degradation in MDCK II cells. FASEB J. 2009, 23, 1935–1945. [Google Scholar] [CrossRef] [PubMed]
- Kiuchi, T.; Ortiz-Zapater, E.; Monypenny, J.; Matthews, D.R.; Nguyen, L.K.; Barbeau, J.; Coban, O.; Lawler, K.; Burford, B.; Rolfe, D.J.; et al. The ERBB4 CYT2 variant protects EGFR from ligand-induced degradation to enhance cancer cell motility. Sci. Signal. 2014, 7, ra78. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Peereboom, D.M.; Shepard, D.R.; Ahluwalia, M.S.; Brewer, C.J.; Agarwal, N.; Stevens, G.H.; Suh, J.H.; Toms, S.A.; Vogelbaum, M.A.; Weil, R.J.; et al. Phase II trial of erlotinib with temozolomide and radiation in patients with newly diagnosed glioblastoma multiforme. J. Neuro-Oncol. 2010, 98, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Raizer, J.J.; Abrey, L.E.; Lassman, A.B.; Chang, S.M.; Lamborn, K.R.; Kuhn, J.G.; Yung, W.K.; Gilbert, M.R.; Aldape, K.A.; Wen, P.Y.; et al. A phase ii trial of erlotinib in patients with recurrent malignant gliomas and nonprogressive glioblastoma multiforme postradiation therapy. Neuro-Oncology 2010, 12, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Lievre, A.; Bachet, J.B.; Le Corre, D.; Boige, V.; Landi, B.; Emile, J.F.; Cote, J.F.; Tomasic, G.; Penna, C.; Ducreux, M.; et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006, 66, 3992–3995. [Google Scholar] [CrossRef] [PubMed]
- Pillay, V.; Allaf, L.; Wilding, A.L.; Donoghue, J.F.; Court, N.W.; Greenall, S.A.; Scott, A.M.; Johns, T.G. The plasticity of oncogene addiction: Implications for targeted therapies directed to receptor tyrosine kinases. Neoplasia 2009, 11, 448–458. [Google Scholar] [CrossRef] [PubMed]
- Mishima, K.; Higashiyama, S.; Asai, A.; Yamaoka, K.; Nagashima, Y.; Taniguchi, N.; Kitanaka, C.; Kirino, T.; Kuchino, Y. Heparin-binding epidermal growth factor-like growth factor stimulates mitogenic signaling and is highly expressed in human malignant gliomas. Acta Neuropathol. 1998, 96, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Russell, K.S.; Stern, D.F.; Polverini, P.J.; Bender, J.R. Neuregulin activation of ERBB receptors in vascular endothelium leads to angiogenesis. Am. J. Physiol. 1999, 277, H2205–H2211. [Google Scholar] [CrossRef] [PubMed]
- Morgenstern, J.P.; Land, H. Advanced mammalian gene transfer: High titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 1990, 18, 3587–3596. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Chadalavada, K.; Wilshire, J.; Kowalik, U.; Hovinga, K.E.; Geber, A.; Fligelman, B.; Leversha, M.; Brennan, C.; Tabar, V. Glioblastoma stem-like cells give rise to tumour endothelium. Nature 2010, 468, 829–833. [Google Scholar] [CrossRef] [PubMed]
- Poller, B.; Gutmann, H.; Krahenbuhl, S.; Weksler, B.; Romero, I.; Couraud, P.O.; Tuffin, G.; Drewe, J.; Huwyler, J. The human brain endothelial cell line hCMEC/D3 as a human blood-brain barrier model for drug transport studies. J. Neurochem. 2008, 107, 1358–1368. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donoghue, J.F.; Kerr, L.T.; Alexander, N.W.; Greenall, S.A.; Longano, A.B.; Gottardo, N.G.; Wang, R.; Tabar, V.; Adams, T.E.; Mischel, P.S.; et al. Activation of ERBB4 in Glioblastoma Can Contribute to Increased Tumorigenicity and Influence Therapeutic Response. Cancers 2018, 10, 243. https://doi.org/10.3390/cancers10080243
Donoghue JF, Kerr LT, Alexander NW, Greenall SA, Longano AB, Gottardo NG, Wang R, Tabar V, Adams TE, Mischel PS, et al. Activation of ERBB4 in Glioblastoma Can Contribute to Increased Tumorigenicity and Influence Therapeutic Response. Cancers. 2018; 10(8):243. https://doi.org/10.3390/cancers10080243
Chicago/Turabian StyleDonoghue, Jacqueline F., Lauren T. Kerr, Naomi W. Alexander, Sameer A. Greenall, Anthony B. Longano, Nicholas G. Gottardo, Rong Wang, Viviane Tabar, Timothy E. Adams, Paul S. Mischel, and et al. 2018. "Activation of ERBB4 in Glioblastoma Can Contribute to Increased Tumorigenicity and Influence Therapeutic Response" Cancers 10, no. 8: 243. https://doi.org/10.3390/cancers10080243