Double-Edged Lipid Nanoparticles Combining Liposome-Bound TRAIL and Encapsulated Doxorubicin Showing an Extraordinary Synergistic Pro-Apoptotic Potential

 , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
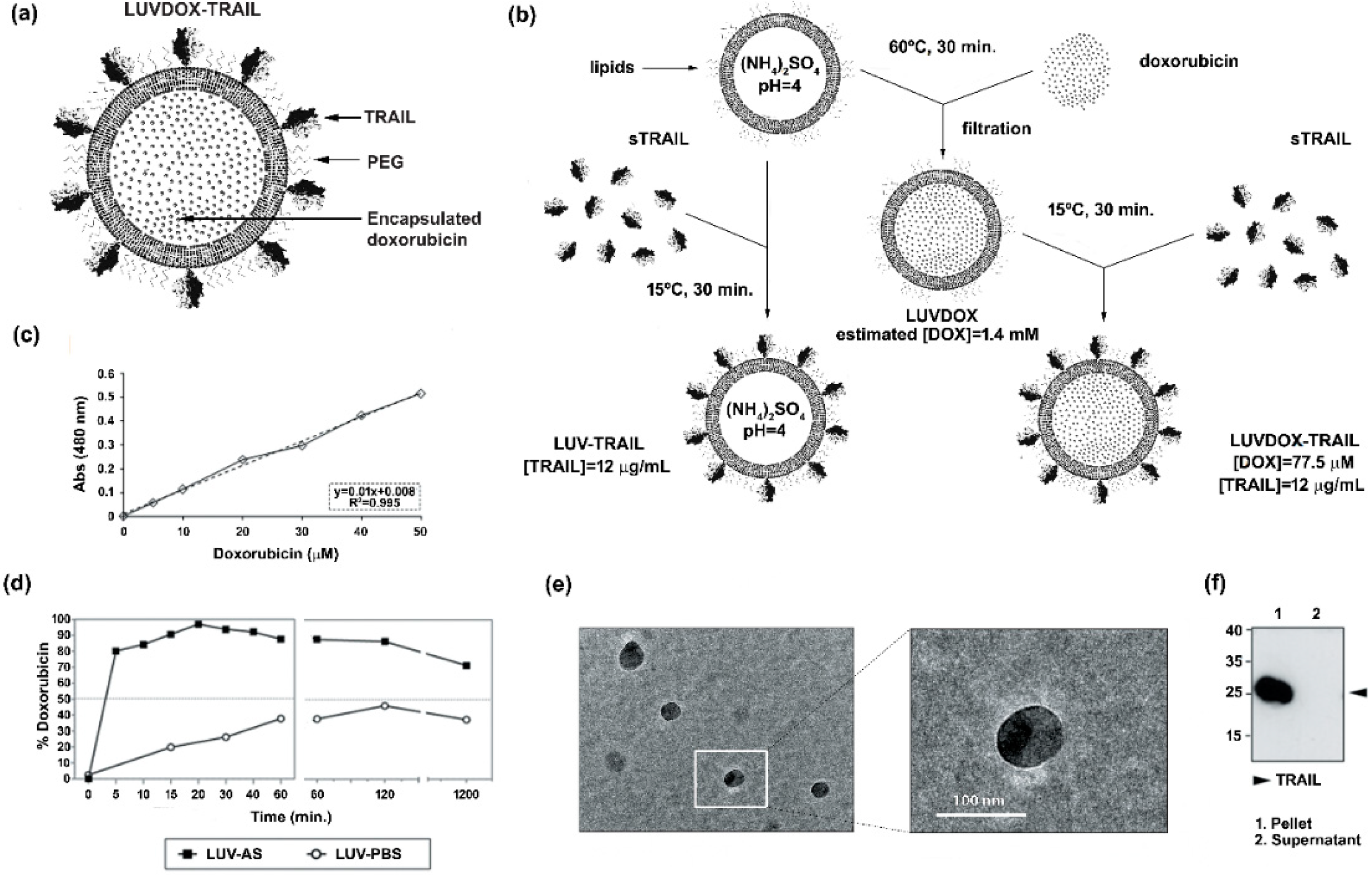
2.1. Synthesis and Characterization of Large Unilamellar Vesicle Doxorubicin-TNF-Related Apoptosis-Inducing Ligand (LUVDOX-TRAIL)
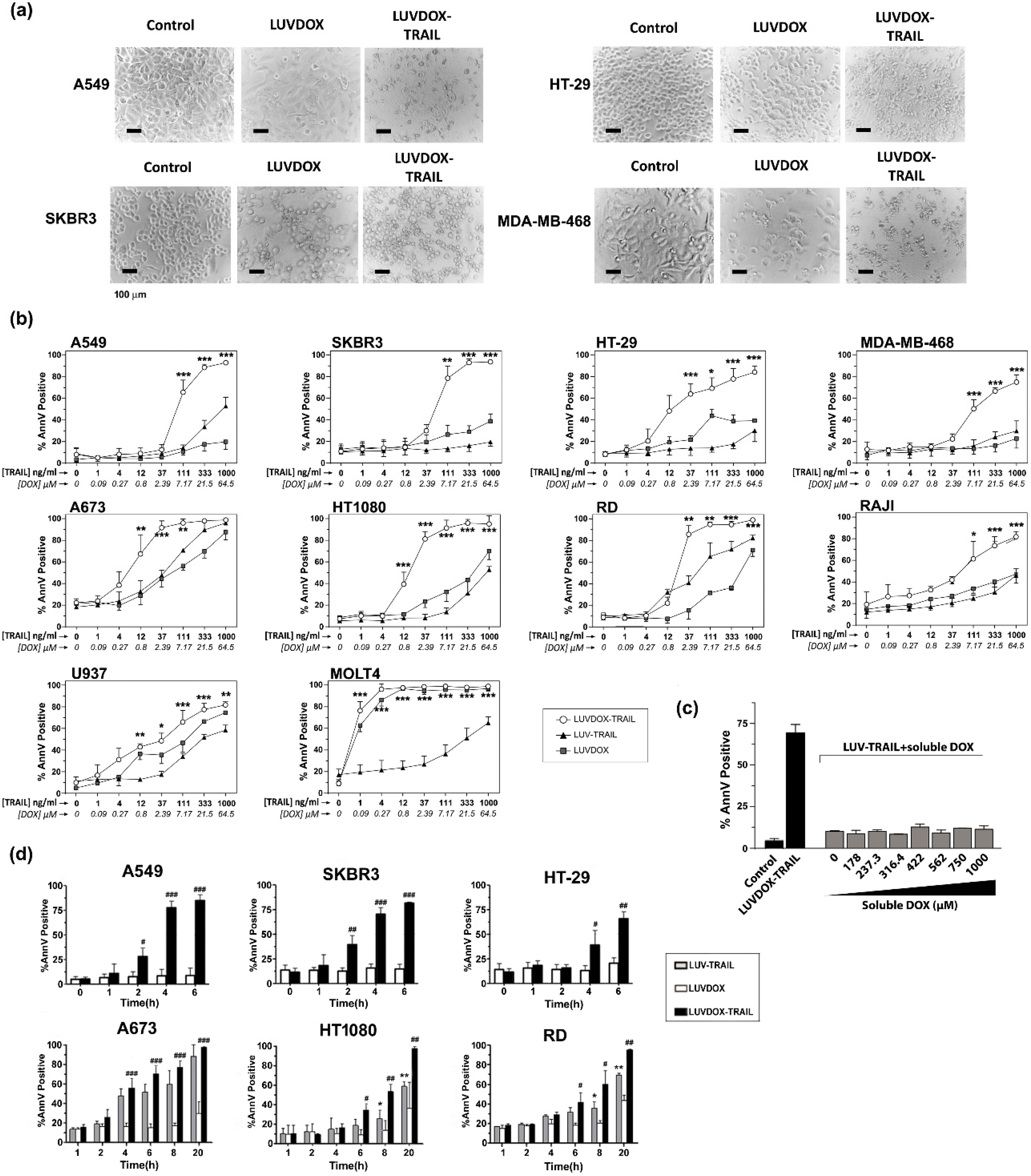
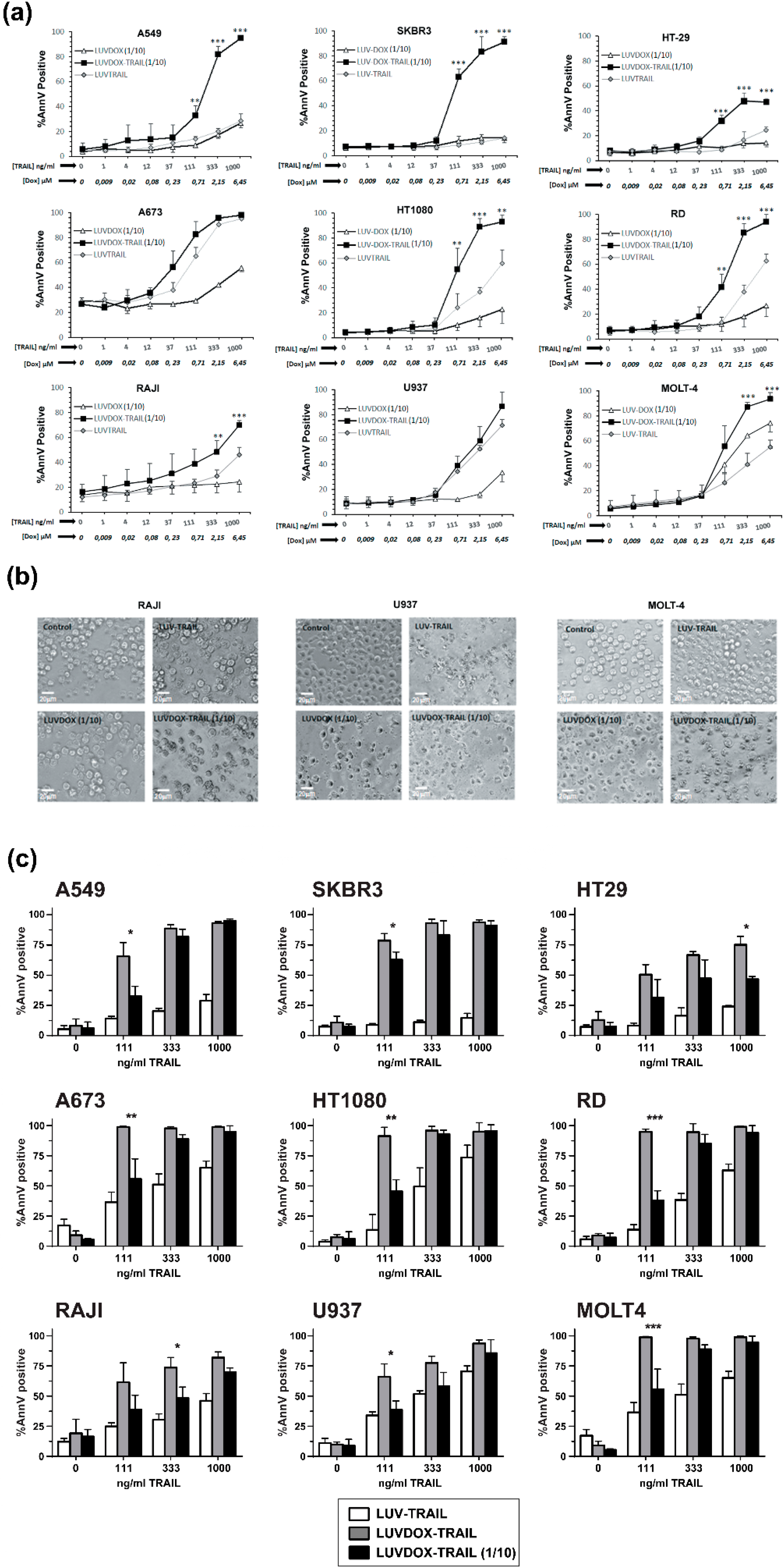
2.2. In Vitro Cytotoxic Potential of LUVDOX-TRAIL
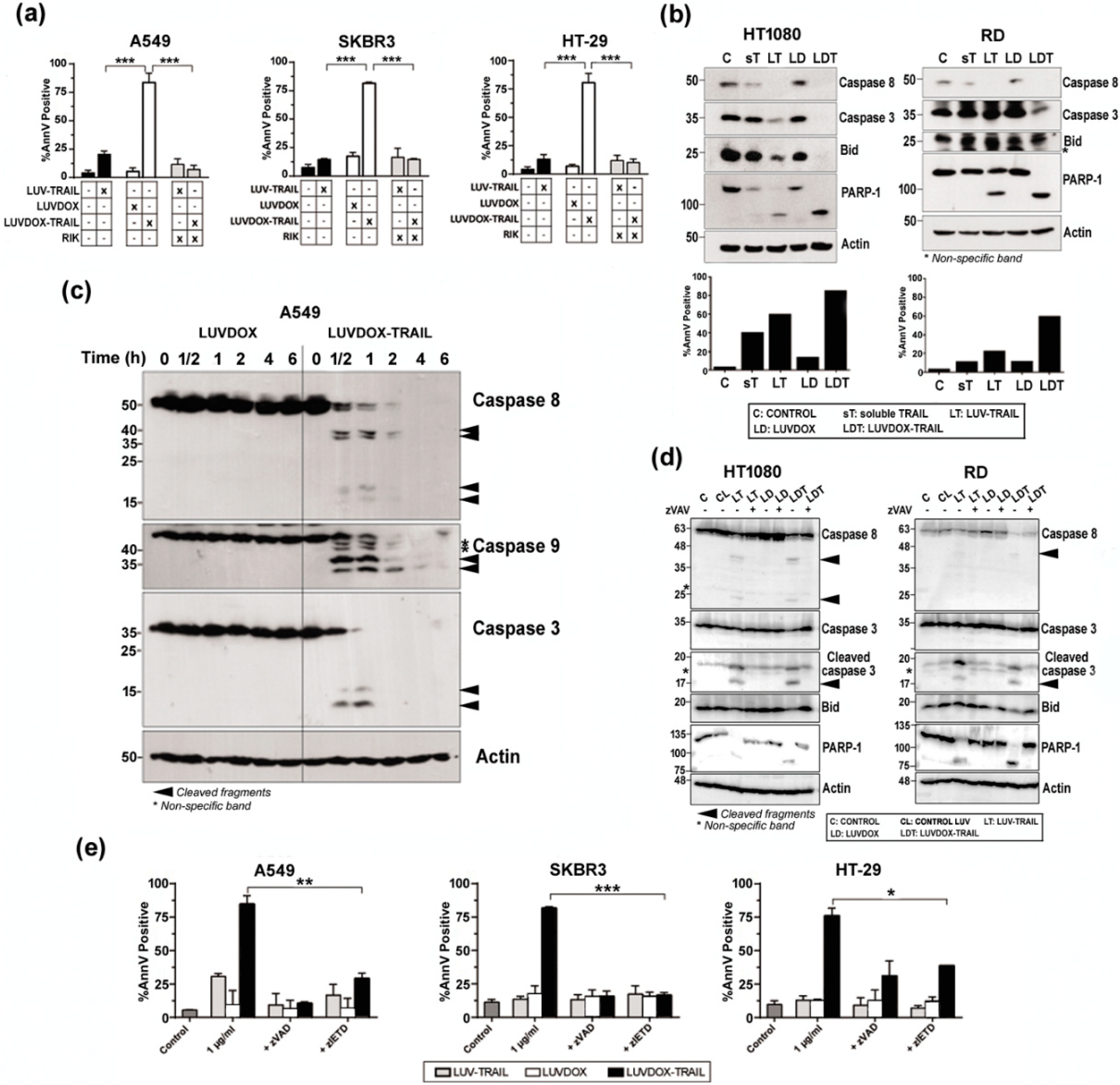
2.3. LUVDOX-TRAIL are able to Induce a Stronger Activation of the Extrinsic Apoptotic Pathway than LUV-TRAIL in Cancer Cells
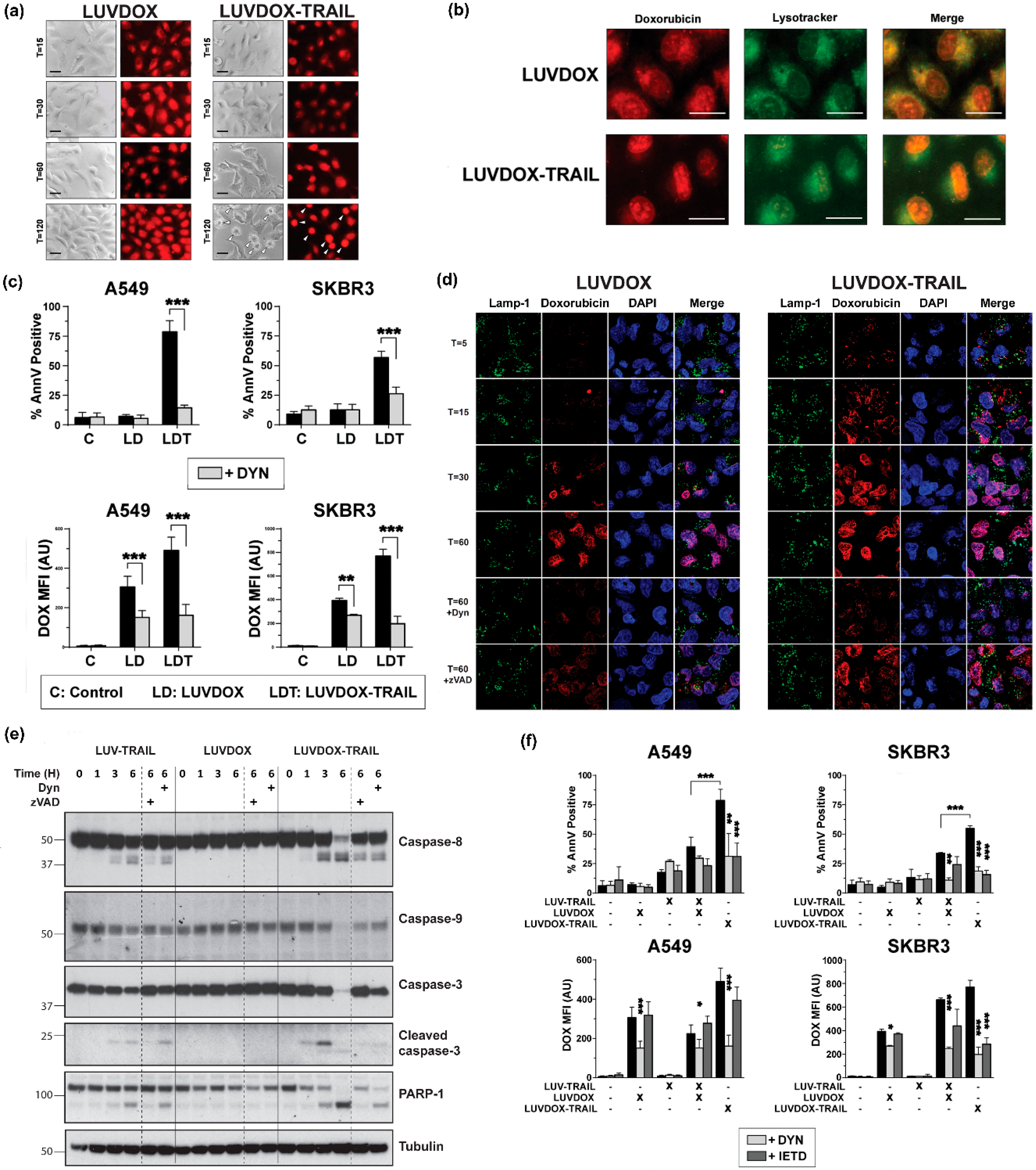
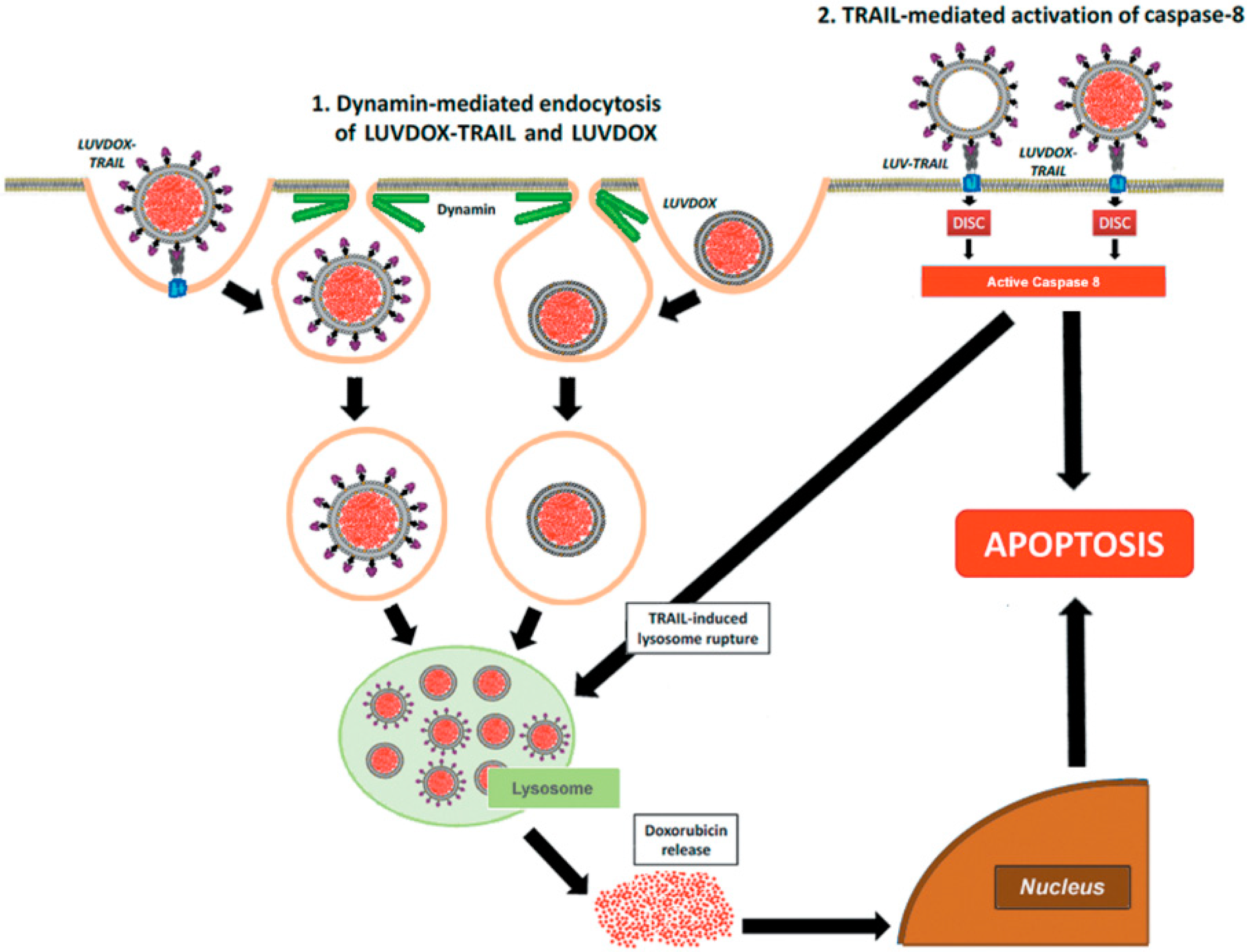
2.4. LUVDOX-TRAIL-Induced Cytotoxicity is a Dynamin-Dependent Mechanism Involving Caspase-8 Activity
2.5. Reduction of DOX Concentration in LUVDOX-TRAIL Reduces Their Toxicity on Normal Peripheral Blood Mononuclear Cells (PBMC) but Maintain Cytotoxicity on Cancer Cells
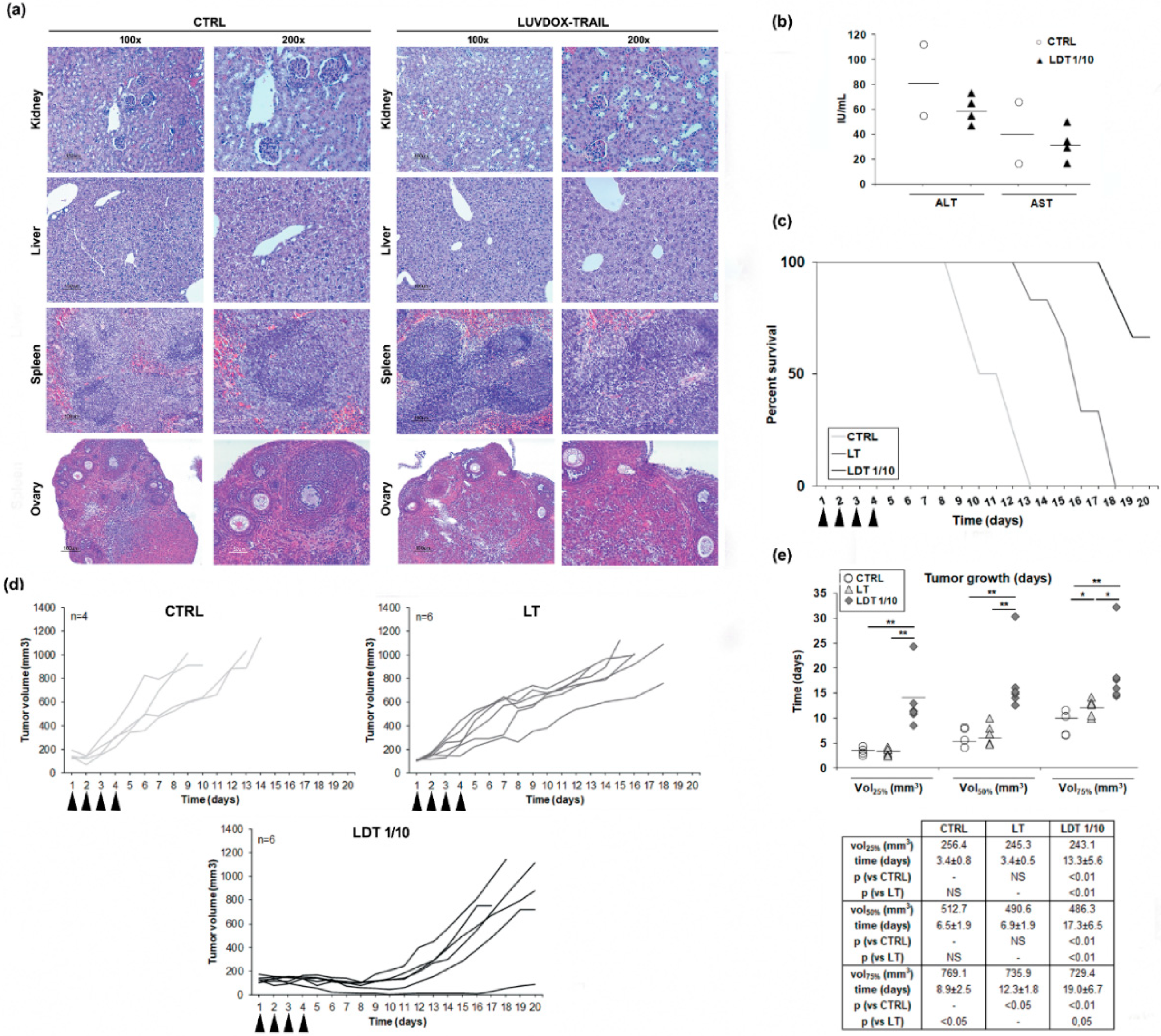
2.6. In Vivo Cytotoxic Potential of LUVDOX-TRAIL
3. Discussion
4. Materials and Methods
4.1. Generation of Different Versions of Lipid Nanoparticles Decorated with TRAIL
4.2. Cell Culture
4.3. Cytotoxicity Assays and Apoptosis Quantification
4.4. Scanning Electron Microscopy
4.5. Western Blot Analysis
4.6. Fluorescence Microscopy Analysis
4.7. Confocal Fluorescence Microscopy Analysis
4.8. Study of the Internalization Mechanism
4.9. In Vivo Study of Toxicity
4.10. In Vivo Anti-Tumor Activity
4.11. Statistical Analysis
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pitti, R.M.; Marsters, S.A.; Ruppert, S.; Donahue, C.J.; Moore, A.; Ashkenazi, A. Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J. Biol. Chem. 1996, 271, 12687–12690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiley, S.R.; Schooley, K.; Smolak, P.J.; Din, W.S.; Huang, C.P.; Nicholl, J.K.; Sutherland, G.R.; Smith, T.D.; Rauch, C.; Smith, C.A.; et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 1995, 3, 673–682. [Google Scholar] [CrossRef] [Green Version]
- Walczak, H.; Miller, R.E.; Ariail, K.; Gliniak, B.; Griffith, T.S.; Kubin, M.; Chin, W.; Jones, J.; Woodward, A.; Le, T.; et al. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat. Med. 1999, 5, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A. Directing cancer cells to self-destruct with pro-apoptotic receptor agonists. Nat. Rev. Drug Discov. 2008, 7, 1001–1012. [Google Scholar] [CrossRef]
- Johnstone, R.W.; Frew, A.J.; Smyth, M.J. The TRAIL apoptotic pathway in cancer onset, progression and therapy. Nat. Rev. Cancer 2008, 8, 782–798. [Google Scholar] [CrossRef]
- von Karstedt, S.; Montinaro, A.; Walczak, H. Exploring the TRAILs less travelled: TRAIL in cancer biology and therapy. Nat. Rev. Cancer 2017, 17, 352–366. [Google Scholar] [CrossRef]
- Lemke, J.; von Karstedt, S.; Zinngrebe, J.; Walczak, H. Getting TRAIL back on track for cancer therapy. Cell Death Differ. 2014, 21, 1350–1364. [Google Scholar] [CrossRef] [Green Version]
- Micheau, O.; Shirley, S.; Dufour, F. Death receptors as targets in cancer. Br. J. Pharmacol. 2013, 169, 1723–1744. [Google Scholar] [CrossRef]
- Palacios, C.; Yerbes, R.; Sanchez-Perez, T.; Martin-Perez, R.; Cano-Gonzalez, A.; Lopez-Rivas, A. The long and winding road to cancer treatment: The TRAIL system. Curr. Pharm. Des. 2014, 20, 2819–2833. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, S.; Secchiero, P.; Zauli, G. State of art and recent developments of anti-cancer strategies based on TRAIL. Recent Pat Anticancer Drug Discov. 2012, 7, 207–217. [Google Scholar] [CrossRef]
- den Hollander, M.W.; Gietema, J.A.; de Jong, S.; Walenkamp, A.M.; Reyners, A.K.; Oldenhuis, C.N.; de Vries, E.G. Translating TRAIL-receptor targeting agents to the clinic. Cancer Lett. 2013, 332, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Dimberg, L.Y.; Anderson, C.K.; Camidge, R.; Behbakht, K.; Thorburn, A.; Ford, H.L. On the TRAIL to successful cancer therapy? Predicting and counteracting resistance against TRAIL-based therapeutics. Oncogene 2013, 32, 1341–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, B.; Allen, J.E.; Prabhu, V.V.; Talekar, M.K.; Finnberg, N.K.; El-Deiry, W.S. Targeting TRAIL in the treatment of cancer: New developments. Expert Opin. Ther. Targets 2015, 19, 1171–1185. [Google Scholar] [CrossRef] [PubMed]
- Balsas, P.; Lopez-Royuela, N.; Galan-Malo, P.; Anel, A.; Marzo, I.; Naval, J. Cooperation between Apo2L/TRAIL and bortezomib in multiple myeloma apoptosis. Biochem. Pharmacol. 2009, 77, 804–812. [Google Scholar] [CrossRef] [Green Version]
- Palacios, C.; Yerbes, R.; Lopez-Rivas, A. Flavopiridol induces cellular FLICE-inhibitory protein degradation by the proteasome and promotes TRAIL-induced early signaling and apoptosis in breast tumor cells. Cancer Res. 2006, 66, 8858–8869. [Google Scholar] [CrossRef] [Green Version]
- Frew, A.J.; Lindemann, R.K.; Martin, B.P.; Clarke, C.J.; Sharkey, J.; Anthony, D.A.; Banks, K.M.; Haynes, N.M.; Gangatirkar, P.; Stanley, K.; et al. Combination therapy of established cancer using a histone deacetylase inhibitor and a TRAIL receptor agonist. Proc. Natl. Acad. Sci. USA 2008, 105, 11317–11322. [Google Scholar] [CrossRef] [Green Version]
- Psahoulia, F.H.; Drosopoulos, K.G.; Doubravska, L.; Andera, L.; Pintzas, A. Quercetin enhances TRAIL-mediated apoptosis in colon cancer cells by inducing the accumulation of death receptors in lipid rafts. Mol. Cancer Ther. 2007, 6, 2591–2599. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Pang, Z.; Ye, H.; Qiu, B.; Guo, L.; Li, J.; Ren, J.; Qian, Y.; Zhang, Q.; Chen, J.; et al. Co-delivery of pEGFP-hTRAIL and paclitaxel to brain glioma mediated by an angiopep-conjugated liposome. Biomaterials 2012, 33, 916–924. [Google Scholar] [CrossRef]
- Guo, L.; Fan, L.; Ren, J.; Pang, Z.; Ren, Y.; Li, J.; Wen, Z.; Qian, Y.; Zhang, L.; Ma, H.; et al. Combination of TRAIL and actinomycin D liposomes enhances antitumor effect in non-small cell lung cancer. Int. J. Nanomed. 2012, 7, 1449–1460. [Google Scholar]
- Cuello, M.; Ettenberg, S.A.; Nau, M.M.; Lipkowitz, S. Synergistic induction of apoptosis by the combination of trail and chemotherapy in chemoresistant ovarian cancer cells. Gynecol. Oncol. 2001, 81, 380–390. [Google Scholar] [CrossRef]
- De Miguel, D.; Lemke, J.; Anel, A.; Walczak, H.; Martinez-Lostao, L. Onto better TRAILs for cancer treatment. Cell Death Differ. 2016, 23, 733–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guimaraes, P.P.G.; Gaglione, S.; Sewastianik, T.; Carrasco, R.D.; Langer, R.; Mitchell, M.J. Nanoparticles for Immune Cytokine TRAIL-Based Cancer Therapy. ACS Nano 2018, 12, 912–931. [Google Scholar] [CrossRef] [PubMed]
- Stuckey, D.W.; Shah, K. TRAIL on trial: Preclinical advances in cancer therapy. Trends Mol. Med. 2013, 19, 685–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wajant, H.; Gerspach, J.; Pfizenmaier, K. Engineering death receptor ligands for cancer therapy. Cancer Lett. 2013, 332, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Zakaria, A.B.; Picaud, F.; Rattier, T.; Pudlo, M.; Dufour, F.; Saviot, L.; Chassagnon, R.; Lherminier, J.; Gharbi, T.; Micheau, O.; et al. Nanovectorization of TRAIL with single wall carbon nanotubes enhances tumor cell killing. Nano Lett. 2015, 15, 891–895. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wang, S.; Li, M.; Wang, A.; Zhou, Y.; Li, P.; Wang, Y. Nanocarriers for TRAIL delivery: Driving TRAIL back on track for cancer therapy. Nanoscale 2017, 9, 13879–13904. [Google Scholar] [CrossRef]
- Berg, D.; Lehne, M.; Muller, N.; Siegmund, D.; Munkel, S.; Sebald, W.; Pfizenmaier, K.; Wajant, H. Enforced covalent trimerization increases the activity of the TNF ligand family members TRAIL and CD95L. Cell Death Differ. 2007, 14, 2021–2034. [Google Scholar] [CrossRef] [Green Version]
- de Bruyn, M.; Rybczynska, A.A.; Wei, Y.; Schwenkert, M.; Fey, G.H.; Dierckx, R.A.; van Waarde, A.; Helfrich, W.; Bremer, E. Melanoma-associated Chondroitin Sulfate Proteoglycan (MCSP)-targeted delivery of soluble TRAIL potently inhibits melanoma outgrowth in vitro and in vivo. Mol. Cancer 2010, 9, 301. [Google Scholar] [CrossRef] [Green Version]
- Siegemund, M.; Pollak, N.; Seifert, O.; Wahl, K.; Hanak, K.; Vogel, A.; Nussler, A.K.; Gottsch, D.; Munkel, S.; Bantel, H.; et al. Superior antitumoral activity of dimerized targeted single-chain TRAIL fusion proteins under retention of tumor selectivity. Cell Death Dis. 2012, 3, e295. [Google Scholar] [CrossRef] [Green Version]
- Bremer, E.; van Dam, G.M.; de Bruyn, M.; van Riezen, M.; Dijkstra, M.; Kamps, G.; Helfrich, W.; Haisma, H. Potent systemic anticancer activity of adenovirally expressed EGFR-selective TRAIL fusion protein. Mol. Ther. 2008, 16, 1919–1926. [Google Scholar] [CrossRef] [Green Version]
- Ten Cate, B.; Bremer, E.; de Bruyn, M.; Bijma, T.; Samplonius, D.; Schwemmlein, M.; Huls, G.; Fey, G.; Helfrich, W. A novel AML-selective TRAIL fusion protein that is superior to Gemtuzumab Ozogamicin in terms of in vitro selectivity, activity and stability. Leukemia 2009, 23, 1389–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Bruyn, M.; Wei, Y.; Wiersma, V.R.; Samplonius, D.F.; Klip, H.G.; van der Zee, A.G.; Yang, B.; Helfrich, W.; Bremer, E. Cell surface delivery of TRAIL strongly augments the tumoricidal activity of T cells. Clin. Cancer Res. 2011, 17, 5626–5637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Mesery, M.; Trebing, J.; Schafer, V.; Weisenberger, D.; Siegmund, D.; Wajant, H. CD40-directed scFv-TRAIL fusion proteins induce CD40-restricted tumor cell death and activate dendritic cells. Cell Death Dis. 2013, 4, e916. [Google Scholar] [CrossRef] [PubMed]
- Gasparian, M.E.; Bychkov, M.L.; Yagolovich, A.V.; Dolgikh, D.A.; Kirpichnikov, M.P. Mutations Enhancing Selectivity of Antitumor Cytokine TRAIL to DR5 Receptor Increase Its Cytotoxicity against Tumor Cells. Biochemistry 2015, 80, 1080–1091. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Zhu, H.; Yi, C.; Yan, J.; Wei, L.; Yang, X.; Chen, S.; Huang, Y. A novel TRAIL mutant-TRAIL-Mu3 enhances the antitumor effects by the increased affinity and the up-expression of DR5 in pancreatic cancer. Cancer Chemother. Pharmacol. 2018, 82, 829–838. [Google Scholar] [CrossRef]
- Chae, S.Y.; Kim, T.H.; Park, K.; Jin, C.H.; Son, S.; Lee, S.; Youn, Y.S.; Kim, K.; Jo, D.G.; Kwon, I.C.; et al. Improved antitumor activity and tumor targeting of NH(2)-terminal-specific PEGylated tumor necrosis factor-related apoptosis-inducing ligand. Mol. Cancer Ther. 2010, 9, 1719–1729. [Google Scholar] [CrossRef] [Green Version]
- De Miguel, D.; Basanez, G.; Sanchez, D.; Malo, P.G.; Marzo, I.; Larrad, L.; Naval, J.; Pardo, J.; Anel, A.; Martinez-Lostao, L. Liposomes decorated with Apo2L/TRAIL overcome chemoresistance of human hematologic tumor cells. Mol. Pharm. 2013, 10, 893–904. [Google Scholar] [CrossRef]
- Guo, L.; Fan, L.; Pang, Z.; Ren, J.; Ren, Y.; Li, J.; Chen, J.; Wen, Z.; Jiang, X. TRAIL and doxorubicin combination enhances anti-glioblastoma effect based on passive tumor targeting of liposomes. J. Control. Release 2011, 154, 93–102. [Google Scholar] [CrossRef]
- Perlstein, B.; Finniss, S.A.; Miller, C.; Okhrimenko, H.; Kazimirsky, G.; Cazacu, S.; Lee, H.K.; Lemke, N.; Brodie, S.; Umansky, F.; et al. TRAIL conjugated to nanoparticles exhibits increased anti-tumor activities in glioma cells and glioma stem cells in vitro and in vivo. Neuro Oncol. 2013, 15, 29–40. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.H.; Jiang, H.H.; Youn, Y.S.; Park, C.W.; Lim, S.M.; Jin, C.H.; Tak, K.K.; Lee, H.S.; Lee, K.C. Preparation and characterization of Apo2L/TNF-related apoptosis-inducing ligand-loaded human serum albumin nanoparticles with improved stability and tumor distribution. J. Pharm. Sci. 2011, 100, 482–491. [Google Scholar] [CrossRef]
- Nair, P.M.; Flores, H.; Gogineni, A.; Marsters, S.; Lawrence, D.A.; Kelley, R.F.; Ngu, H.; Sagolla, M.; Komuves, L.; Bourgon, R.; et al. Enhancing the antitumor efficacy of a cell-surface death ligand by covalent membrane display. Proc. Natl. Acad. Sci. USA 2015, 112, 5679–5684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seifert, O.; Pollak, N.; Nusser, A.; Steiniger, F.; Ruger, R.; Pfizenmaier, K.; Kontermann, R.E. Immuno-LipoTRAIL: Targeted delivery of TRAIL-functionalized liposomal nanoparticles. Bioconjug. Chem. 2014, 25, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lorenzo, M.J.; Anel, A.; Gamen, S.; Monle n, I.; Lasierra, P.; Larrad, L.; Pineiro, A.; Alava, M.A.; Naval, J. Activated human T cells release bioactive Fas ligand and APO2 ligand in microvesicles. J. Immunol. 1999, 163, 1274–1281. [Google Scholar] [PubMed]
- Monleon, I.; Martinez-Lorenzo, M.J.; Monteagudo, L.; Lasierra, P.; Taules, M.; Iturralde, M.; Pineiro, A.; Larrad, L.; Alava, M.A.; Naval, J.; et al. Differential secretion of Fas ligand- or APO2 ligand/TNF-related apoptosis-inducing ligand-carrying microvesicles during activation-induced death of human T cells. J. Immunol. 2001, 167, 6736–6744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Miguel, D.; Gallego-Lleyda, A.; Anel, A.; Martinez-Lostao, L. Liposome-bound TRAIL induces superior DR5 clustering and enhanced DISC recruitment in histiocytic lymphoma U937 cells. Leuk. Res. 2015, 39, 657–666. [Google Scholar] [CrossRef] [PubMed]
- De Miguel, D.; Gallego-Lleyda, A.; Galan-Malo, P.; Rodriguez-Vigil, C.; Marzo, I.; Anel, A.; Martinez-Lostao, L. Immunotherapy with liposome-bound TRAIL overcome partial protection to soluble TRAIL-induced apoptosis offered by down-regulation of Bim in leukemic cells. Clin. Transl. Oncol. 2015, 17, 657–667. [Google Scholar] [CrossRef]
- De Miguel, D.; Gallego-Lleyda, A.; Ayuso, J.M.; Erviti-Ardanaz, S.; Pazo-Cid, R.; del Agua, C.; Fernandez, L.J.; Ochoa, I.; Anel, A.; Martinez-Lostao, L. TRAIL-coated lipid-nanoparticles overcome resistance to soluble recombinant TRAIL in non-small cell lung cancer cells. Nanotechnology 2016, 27, 185101. [Google Scholar] [CrossRef]
- De Miguel, D.; Gallego-Lleyda, A.; Ayuso, J.M.; Pawlak, A.; Conde, B.; Ochoa, I.; Fernandez, L.J.; Anel, A.; Martinez-Lostao, L. Improved Anti-Tumor Activity of Novel Highly Bioactive Liposome-Bound TRAIL in Breast Cancer Cells. Recent Pat Anticancer Drug Discov. 2016, 11, 197–214. [Google Scholar] [CrossRef]
- De Miguel, D.; Gallego-Lleyda, A.; Ayuso, J.M.; Pejenaute-Ochoa, D.; Jarauta, V.; Marzo, I.; Fernandez, L.J.; Ochoa, I.; Conde, B.; Anel, A.; et al. High-order TRAIL oligomer formation in TRAIL-coated lipid nanoparticles enhances DR5 cross-linking and increases antitumour effect against colon cancer. Cancer Lett. 2016, 383, 250–260. [Google Scholar] [CrossRef] [Green Version]
- Gabizon, A.A.; Patil, Y.; La-Beck, N.M. New insights and evolving role of pegylated liposomal doxorubicin in cancer therapy. Drug Resist Updat. 2016, 29, 90–106. [Google Scholar] [CrossRef]
- Zhao, Z.; Ukidve, A.; Gao, Y.; Kim, J.; Mitragotri, S. Erythrocyte leveraged chemotherapy (ELeCt): Nanoparticle assembly on erythrocyte surface to combat lung metastasis. Sci. Adv. 2019, 5, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Ren, W.; Liu, J.; Lahat, G.; Torres, K.; Lopez, G.; Lazar, A.J.; Hayes-Jordan, A.; Liu, K.; Bankson, J.; et al. TRAIL and doxorubicin combination induces proapoptotic and antiangiogenic effects in soft tissue sarcoma in vivo. Clin. Cancer Res. 2010, 16, 2591–2604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumitru, C.A.; Carpinteiro, A.; Trarbach, T.; Hengge, U.R.; Gulbins, E. Doxorubicin enhances TRAIL-induced cell death via ceramide-enriched membrane platforms. Apoptosis 2007, 12, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Kelly, M.M.; Hoel, B.D.; Voelkel-Johnson, C. Doxorubicin pretreatment sensitizes prostate cancer cell lines to TRAIL induced apoptosis which correlates with the loss of c-FLIP expression. Cancer Biol. Ther. 2002, 1, 520–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.L.; Dhillon, S.H.; Wang, Y.; Pervaiz, S.; Fan, W.; Yang, Y.Y. Synergistic anti-cancer effects via co-delivery of TNF-related apoptosis-inducing ligand (TRAIL/Apo2L) and doxorubicin using micellar nanoparticles. Mol. Biosyst. 2011, 7, 1512–1522. [Google Scholar] [CrossRef] [PubMed]
- Barenholz, Y. Doxil(R)––The first FDA–approved nano–drug: Lessons learned. J. Control Release 2012, 160, 117–134. [Google Scholar] [CrossRef]
- Martinez–Lostao, L.; Garcia–Alvarez, F.; Basanez, G.; Alegre–Aguaron, E.; Desportes, P.; Larrad, L.; Naval, J.; Jose Martinez–Lorenzo, M.; Anel, A. Liposome–bound APO2L/TRAIL is an effective treatment in a rheumatoid arthritis model. Arthritis Rheum 2010, 62, 2272–2282. [Google Scholar] [CrossRef]
- Gallego–Lleyda, A.; De Miguel, D.; Anel, A.; Martinez–Lostao, L. Lipid Nanoparticles Decorated with TNF–Related Aptosis–Inducing Ligand (TRAIL) Are More Cytotoxic than Soluble Recombinant TRAIL in Sarcoma. Int. J. Mol. Sci. 2018, 19, 1449. [Google Scholar] [CrossRef] [Green Version]
- Kang, Z.; Chen, J.J.; Yu, Y.; Li, B.; Sun, S.Y.; Zhang, B.; Cao, L. Drozitumab, a human antibody to death receptor 5, has potent antitumor activity against rhabdomyosarcoma with the expression of caspase–8 predictive of response. Clin. Cancer Res. 2011, 17, 3181–3192. [Google Scholar] [CrossRef] [Green Version]
- Seynhaeve, A.L.B.; Dicheva, B.M.; Hoving, S.; Koning, G.A.; Ten Hagen, T.L.M. Intact Doxil is taken up intracellularly and released doxorubicin sequesters in the lysosome: Evaluated by in vitro/in vivo live cell imaging. J. Control Release 2013, 172, 330–340. [Google Scholar] [CrossRef]
- Kirchhausen, T.; Macia, E.; Pelish, H.E. Use of dynasore, the small molecule inhibitor of dynamin, in the regulation of endocytosis. Methods Enzymol. 2008, 438, 77–93. [Google Scholar] [PubMed] [Green Version]
- Lawrence, D.; Shahrokh, Z.; Marsters, S.; Achilles, K.; Shih, D.; Mounho, B.; Hillan, K.; Totpal, K.; DeForge, L.; Schow, P.; et al. Differential hepatocyte toxicity of recombinant Apo2L/TRAIL versions. Nat. Med. 2001, 7, 383–385. [Google Scholar] [CrossRef] [PubMed]
- Keane, M.M.; Ettenberg, S.A.; Nau, M.M.; Russell, E.K.; Lipkowitz, S. Chemotherapy augments TRAIL–induced apoptosis in breast cell lines. Cancer Res. 1999, 59, 734–741. [Google Scholar] [PubMed]
- Vaculova, A.; Kaminskyy, V.; Jalalvand, E.; Surova, O.; Zhivotovsky, B. Doxorubicin and etoposide sensitize small cell lung carcinoma cells expressing caspase–8 to TRAIL. Mol. Cancer 2010, 9, 87. [Google Scholar] [CrossRef] [Green Version]
- Akazawa, Y.; Mott, J.L.; Bronk, S.F.; Werneburg, N.W.; Kahraman, A.; Guicciardi, M.E.; Meng, X.W.; Kohno, S.; Shah, V.H.; Kaufmann, S.H.; et al. Death receptor 5 internalization is required for lysosomal permeabilization by TRAIL in malignant liver cell lines. Gastroenterology 2009, 136, 2365–2376. [Google Scholar] [CrossRef] [Green Version]
- Werneburg, N.W.; Guicciardi, M.E.; Bronk, S.F.; Kaufmann, S.H.; Gores, G.J. Tumor necrosis factor–related apoptosis–inducing ligand activates a lysosomal pathway of apoptosis that is regulated by Bcl–2 proteins. J. Biol. Chem. 2007, 282, 28960–28970. [Google Scholar] [CrossRef] [Green Version]
- Guicciardi, M.E.; Bronk, S.F.; Werneburg, N.W.; Gores, G.J. cFLIPL prevents TRAIL–induced apoptosis of hepatocellular carcinoma cells by inhibiting the lysosomal pathway of apoptosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G1337–G1346. [Google Scholar] [CrossRef] [Green Version]
- Bosque, A.; Pardo, J.; Martinez–Lorenzo, M.J.; Iturralde, M.; Marzo, I.; Pineiro, A.; Alava, M.A.; Naval, J.; Anel, A. Down–regulation of normal human T cell blast activation: Roles of APO2L/TRAIL, FasL, and c– FLIP, Bim, or Bcl–x isoform expression. J. Leukoc. Biol. 2005, 77, 568–578. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazi, A.; Pai, R.C.; Fong, S.; Leung, S.; Lawrence, D.A.; Marsters, S.A.; Blackie, C.; Chang, L.; McMurtrey, A.E.; Hebert, A.; et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J. Clin. Invest. 1999, 104, 155–162. [Google Scholar] [CrossRef]
- Ganten, T.M.; Koschny, R.; Sykora, J.; Schulze–Bergkamen, H.; Buchler, P.; Haas, T.L.; Schader, M.B.; Untergasser, A.; Stremmel, W.; Walczak, H. Preclinical differentiation between apparently safe and potentially hepatotoxic applications of TRAIL either alone or in combination with chemotherapeutic drugs. Clin. Cancer Res. 2006, 12, 2640–2646. [Google Scholar] [CrossRef] [Green Version]
- MacFarlane, M.; Ahmad, M.; Srinivasula, S.M.; Fernandes–Alnemri, T.; Cohen, G.M.; Alnemri, E.S. Identification and molecular cloning of two novel receptors for the cytotoxic ligand TRAIL. J. Biol. Chem. 1997, 272, 25417–25420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, S.A.; Waterhouse, D.N.; Mayer, L.D.; Cullis, P.R.; Madden, T.D.; Bally, M.B. The liposomal formulation of doxorubicin. Methods Enzymol. 2005, 391, 71–97. [Google Scholar] [PubMed]
- Haran, G.; Cohen, R.; Bar, L.K.; Barenholz, Y. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochim. Biophys. Acta 1993, 1151, 201–215. [Google Scholar] [CrossRef]
- Bae, S.; Ma, K.; Kim, T.H.; Lee, E.S.; Oh, K.T.; Park, E.S.; Lee, K.C.; Youn, Y.S. Doxorubicin–loaded human serum albumin nanoparticles surface–modified with TNF–related apoptosis–inducing ligand and transferrin for targeting multiple tumor types. Biomaterials 2012, 33, 1536–1546. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Fan, L.; Ren, J.; Pang, Z.; Ren, Y.; Li, J.; Wen, Z.; Jiang, X. A novel combination of TRAIL and doxorubicin enhances antitumor effect based on passive tumor–targeting of liposomes. Nanotechnology 2011, 22, 265105. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.H.; Kim, T.H.; Lee, S.; Chen, X.; Youn, Y.S.; Lee, K.C. PEGylated TNF–related apoptosis–inducing ligand (TRAIL) for effective tumor combination therapy. Biomaterials 2011, 32, 8529–8537. [Google Scholar] [CrossRef] [PubMed]
- Thao le, Q.; Byeon, H.J.; Lee, C.; Lee, S.; Lee, E.S.; Choi, Y.W.; Choi, H.G.; Park, E.S.; Lee, K.C.; Youn, Y.S. Doxorubicin–Bound Albumin Nanoparticles Containing a TRAIL Protein for Targeted Treatment of Colon Cancer. Pharm. Res. 2016, 33, 615–626. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Miguel, D.; Gallego-Lleyda, A.; Martinez-Ara, M.; Plou, J.; Anel, A.; Martinez-Lostao, L. Double-Edged Lipid Nanoparticles Combining Liposome-Bound TRAIL and Encapsulated Doxorubicin Showing an Extraordinary Synergistic Pro-Apoptotic Potential. Cancers 2019, 11, 1948. https://doi.org/10.3390/cancers11121948
De Miguel D, Gallego-Lleyda A, Martinez-Ara M, Plou J, Anel A, Martinez-Lostao L. Double-Edged Lipid Nanoparticles Combining Liposome-Bound TRAIL and Encapsulated Doxorubicin Showing an Extraordinary Synergistic Pro-Apoptotic Potential. Cancers. 2019; 11(12):1948. https://doi.org/10.3390/cancers11121948
Chicago/Turabian StyleDe Miguel, Diego, Ana Gallego-Lleyda, Miguel Martinez-Ara, Javier Plou, Alberto Anel, and Luis Martinez-Lostao. 2019. "Double-Edged Lipid Nanoparticles Combining Liposome-Bound TRAIL and Encapsulated Doxorubicin Showing an Extraordinary Synergistic Pro-Apoptotic Potential" Cancers 11, no. 12: 1948. https://doi.org/10.3390/cancers11121948