Personalized Medicine: Recent Progress in Cancer Therapy


 ,
, 
Abstract
:1. The Revolution of Tumor Treatment: From Tumor Site to Molecular Alterations
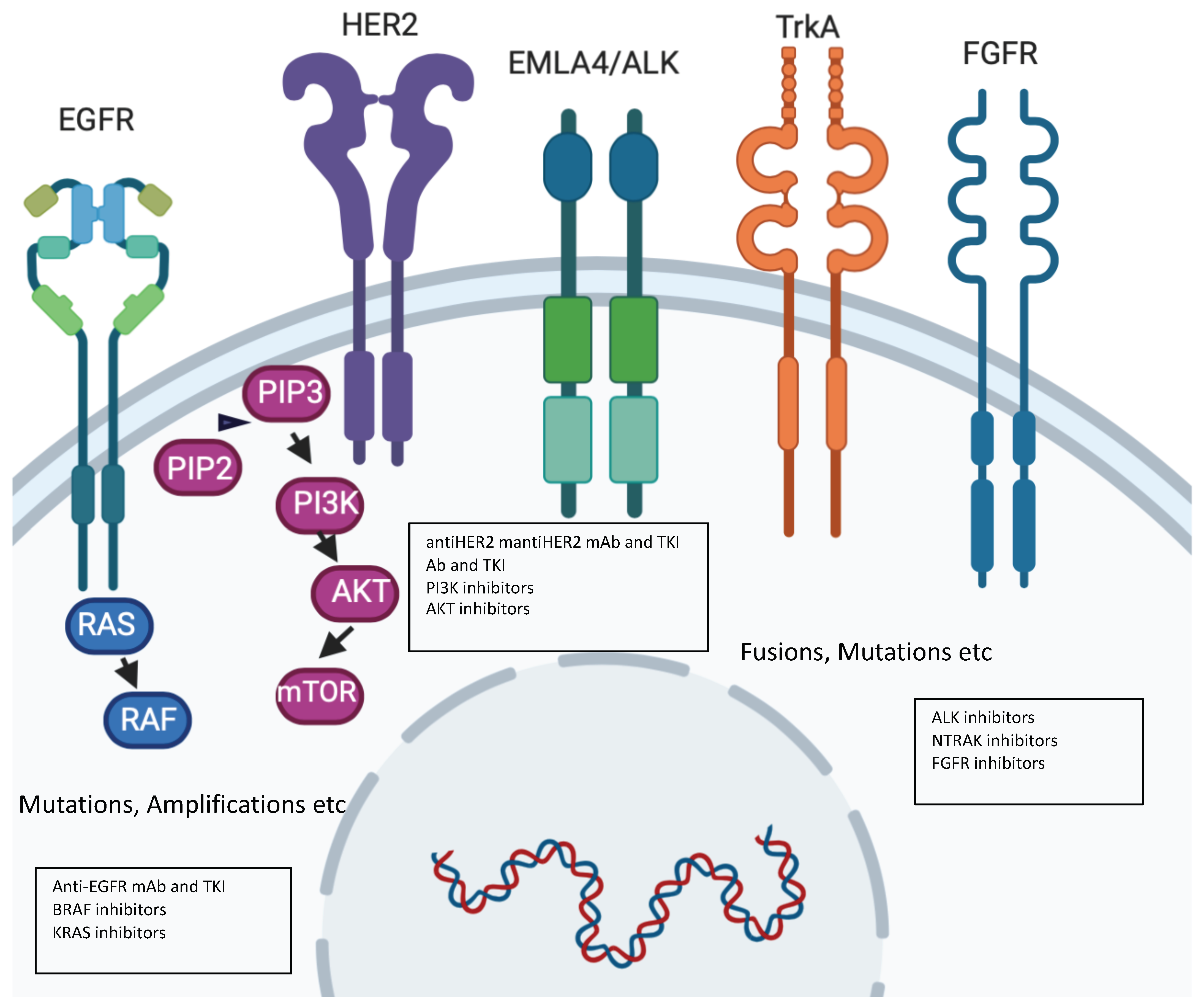
2. Precision Molecular Oncology: Understanding the Role of New Drivers with Novel Drugs
3. Limitations of Molecular Driven Treatments in the Clinic
Limitations of the Molecular Approach
4. How to Overcome Limitations: Functional Precision Medicine, Liquid Biopsy, and Molecular Tumor Board
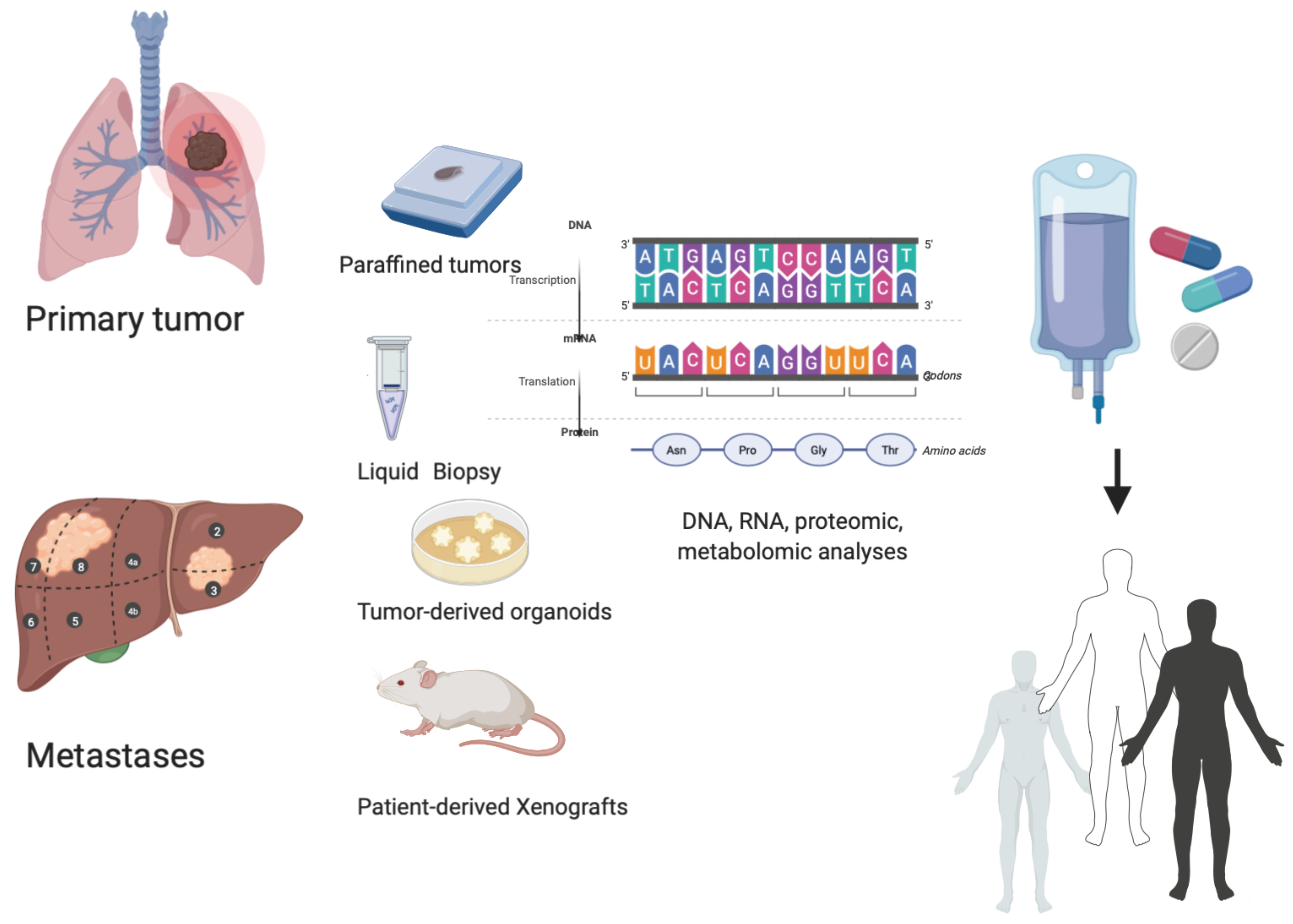
4.1. Functional Precision Medicine: The Role of Patient-Derived Organoids (PDOs) and Patient-Derived Xenografts in a Personalized Approach
4.2. Dynamic Evaluation of Tumors: The Role of Liquid Biopsy
4.3. Molecular Tumor Board: Why Do We Need It?
5. Conclusions
Funding
Conflicts of Interest
References
- Berger, M.F.; Mardis, E.R. The emerging clinical relevance of genomics in cancer medicine. Nat. Rev. Clin. Oncol. 2018, 15, 353–365. [Google Scholar] [CrossRef]
- Lambert, A.W.; Pattabiraman, D.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [Green Version]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Primers 2019, 5, 66. [Google Scholar] [CrossRef]
- Soverini, S.; Mancini, M.; Bavaro, L.; Cavo, M.; Martinelli, G. Chronic myeloid leukemia: The paradigm of targeting oncogenic tyrosine kinase signaling and counteracting resistance for successful cancer therapy. Mol. Cancer 2018, 17, 49. [Google Scholar] [CrossRef] [PubMed]
- Hyman, D.M.; Taylor, B.S.; Baselga, J. Implementing Genome-Driven Oncology. Cell 2017, 168, 584–599. [Google Scholar] [CrossRef] [PubMed]
- Ignatiadis, M.; Sotiriou, C. Luminal breast cancer: From biology to treatment. Nat. Rev. Clin. Oncol. 2013, 10, 494–506. [Google Scholar] [CrossRef] [PubMed]
- André, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.; Baselga, J.; Kim, S.-B.; Ro, J.; Semiglazov, V.; Campone, M.; Ciruelos, E.; Ferrero, J.-M.; Schneeweiss, A.; Heeson, S.; et al. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N. Engl. J. Med. 2015, 372, 724–734. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.S.; Crown, J.P.; Lang, I.; Boer, K.; Bondarenko, I.M.; Kulyk, S.O.; Ettl, J.; Patel, R.; Pinter, T.; Schmidt, M.; et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): A randomised phase 2 study. Lancet Oncol. 2015, 16, 25–35. [Google Scholar] [CrossRef]
- Mok, T.S.; Kim, S.W.; Wu, Y.L.; Nakagawa, K.; Yang, J.J.; Ahn, M.J.; Wang, J.; Yang, J.C.H.; Lu, Y.; Atagi, S.; et al. Gefitinib Plus Chemotherapy Versus Chemotherapy in Epidermal Growth Factor Receptor Mutation-Positive Non-Small-Cell Lung Cancer Resistant to First-Line Gefitinib (IMPRESS): Overall Survival and Biomarker Analyses. J. Clin. Oncol. 2017, 35, 4027–4034. [Google Scholar] [CrossRef]
- Solomon, B.; Kim, N.-W.; Mekhail, T.; Paolini, J.; Usari, T.; Reisman, A.; Wilner, K.D.; Tursi, J.; Mok, T.S.; Wu, Y.-L.; et al. First-Line Crizotinib versus Chemotherapy in ALK-Positive Lung Cancer. N. Engl. J. Med. 2014, 371, 2167–2177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus standard chemotherapy as fi rst-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Artic. Lancet Oncol. 2012, 13, 239–285. [Google Scholar] [CrossRef]
- Robert, C.; Grob, J.J.; Stroyakovskiy, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Sileni, V.C.; Schachter, J.; Garbe, C.; Bondarenko, I.; et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N. Engl. J. Med. 2019, 381, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of Mutated, Activated BRAF in Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef] [Green Version]
- Hyman, D.M.; Piha-Paul, S.; Won, H.; Rodon, J.; Saura, C.; Shapiro, G.I.; Juric, D.; Quinn, D.I.; Moreno, V.; Doger, B.; et al. HER kinase inhibition in patients with HER2- and HER3-mutant cancers. Nature 2018, 554, 189–194. [Google Scholar] [CrossRef]
- Smyth, L.M.; Piha-Paul, S.A.; Won, H.H.; Schram, A.M.; Saura, C.; Loi, S.; Lu, J.; Shapiro, G.I.; Juric, D.; Mayer, I.A.; et al. Efficacy and Determinants of Response to HER Kinase Inhibition in HER2-Mutant Metastatic Breast Cancer. Cancer Discov. 2019, 10, 198–213. [Google Scholar] [CrossRef] [Green Version]
- Krop, I.E.; Kim, S.-B.; González-Martín, A.; Lorusso, P.M.; Ferrero, J.-M.; Smitt, M.; Yu, R.; Leung, A.C.F.; Wildiers, H. Trastuzumab emtansine versus treatment of physician’s choice for pretreated HER2-positive advanced breast cancer (TH3RESA): A randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 689–699. [Google Scholar] [CrossRef]
- Doi, T.; Shitara, K.; Naito, Y.; Shimomura, A.; Fujiwara, Y.; Yonemori, K.; Shimizu, C.; Shimoi, T.; Kuboki, Y.; Matsubara, N.; et al. Safety, pharmacokinetics, and antitumour activity of trastuzumab deruxtecan (DS-8201), a HER2-targeting antibody–drug conjugate, in patients with advanced breast and gastric or gastro-oesophageal tumours: A phase 1 dose-escalation study. Lancet Oncol. 2017, 18, 1512–1522. [Google Scholar] [CrossRef]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, Y.; Diéras, V.; Guardino, E.; et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N. Engl. J. Med. 2012, 367, 1783–1791. [Google Scholar] [CrossRef] [Green Version]
- Paez, J.G. EGFR Mutations in Lung Cancer: Correlation with Clinical Response to Gefitinib Therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [Green Version]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.-I.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4–ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Borger, D.R.; Tanabe, K.K.; Fan, K.C.; Lopez, H.U.; Fantin, V.R.; Straley, K.S.; Schenkein, D.P.; Hezel, A.F.; Ancukiewicz, M.; Liebman, H.M.; et al. Frequent Mutation of Isocitrate Dehydrogenase (IDH)1 and IDH2 in Cholangiocarcinoma Identified Through Broad-Based Tumor Genotyping. Oncology 2011, 17, 72–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Adashek, J.J.; Lorusso, P.M.; Hong, D.S.; Kurzrock, R. Phase I trials as valid therapeutic options for patients with cancer. Nat. Rev. Clin. Oncol. 2019, 16, 773–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Angulo, A.M.; Timms, K.M.; Liu, S.; Chen, H.; Litton, J.K.; Potter, J.; Lanchbury, J.S.; Stemke-Hale, K.; Hennessy, B.T.; Arun, B.K.; et al. Incidence and outcome of BRCA mutations in unselected patients with triple receptor-negative breast cancer. Clin. Cancer Res. 2011, 17, 1082–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alsop, K.; Fereday, S.; Meldrum, C.; DeFazio, A.; Emmanuel, C.; George, J.; Dobrovic, A.; Birrer, M.J.; Webb, P.; Stewart, C.; et al. BRCA Mutation Frequency and Patterns of Treatment Response in BRCA Mutation–Positive Women with Ovarian Cancer: A Report from the Australian Ovarian Cancer Study Group. J. Clin. Oncol. 2012, 30, 2654–2663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dienstmann, R.; Rodon, J.; Prat, A.; Perez-Garcia, J.; Adamo, B.; Felip, E.; Cortés, J.; Iafrate, A.J.; Nuciforo, P.; Tabernero, J. Genomic aberrations in the FGFR pathway: Opportunities for targeted therapies in solid tumors. Ann. Oncol. 2014, 25, 552–563. [Google Scholar] [CrossRef]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; Dubois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.; et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef]
- Kimmelman, J.; Tannock, I. The paradox of precision medicine. Nat. Rev. Clin. Oncol. 2018, 15, 341–342. [Google Scholar] [CrossRef]
- Rodriguez, H.; Pennington, S.R. Revolutionizing Precision Oncology through Collaborative Proteogenomics and Data Sharing. Cell 2018, 173, 535–539. [Google Scholar] [CrossRef] [Green Version]
- Mertins, P.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; Petralia, F.; et al. Proteogenomics connects somatic mutations to signaling in breast cancer. Nature 2016, 534, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puchades-Carrasco, L.; Lucena, A.P. Metabolomics Applications in Precision Medicine: An Oncological Perspective. Curr. Top. Med. Chem. 2017, 17, 2740–2751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clayton, T.A.; Lindon, J.C.; Cloarec, O.; Antti, H.; Charuel, C.; Hanton, G.; Provost, J.P.; Le Net, J.L.; Baker, D.; Walley, R.J.; et al. Pharmaco-metabonomic phenotyping and personalized drug treatment. Nature 2006, 440, 1073–1077. [Google Scholar] [CrossRef] [PubMed]
- Gambardella, V.; Gimeno-Valiente, F.; Tarazona, N.; Martínez-Ciarpaglini, C.; Roda, D.; Fleitas, T.; Tolosa, P.; Cejalvo, J.M.; Huerta, M.; Roselló, S.; et al. NRF2 through RPS6 Activation Is Related to Anti-HER2 Drug Resistance in HER2-Amplified Gastric Cancer. Clin. Cancer Res. 2018, 25, 1639–1649. [Google Scholar] [CrossRef]
- Rojo de Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the hallmarks of cáncer. Cancer Cell 2019, 34, 21–43. [Google Scholar] [CrossRef]
- Garralda, E.; Dienstmann, R.; Piris-Giménez, A.; Braña, I.; Rodon, J.; Tabernero, J. New clinical trial designs in the era of precision medicine. Mol. Oncol. 2019, 13, 549–557. [Google Scholar] [CrossRef]
- Schwaederle, M.; Zhao, M.; Lee, J.J.; Eggermont, A.M.; Schilsky, R.L.; Mendelsohn, J.; Lazar, V.; Kurzrock, R. Impact of Precision Medicine in Diverse Cancers: A Meta-Analysis of Phase II Clinical Trials. J. Clin. Oncol. 2015, 33, 3817–3825. [Google Scholar] [CrossRef]
- Jardim, D.L.; Schwaederle, M.; Wei, C.; Lee, J.J.; Hong, D.S.; Eggermont, A.M.; Schilsky, R.L.; Mendelsohn, J.; Lazar, V.; Kurzrock, R. Impact of a Biomarker-Based Strategy on Oncology Drug Development: A Meta-Analysis of Clinical Trials Leading to FDA Approval. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [Green Version]
- Massard, C.; Michiels, S.; Ferté, C.; Le Deley, M.-C.; Lacroix, L.; Hollebecque, A.; Verlingue, L.; Ileana, E.; Rosellini, S.; Ammari, S.; et al. High-Throughput Genomics and Clinical Outcome in Hard-to-Treat Advanced Cancers: Results of the MOSCATO 01 Trial. Cancer Discov. 2017, 7, 586–595. [Google Scholar] [CrossRef] [Green Version]
- Goncalves, M.D.; Hopkins, B.D.; Cantley, L.C. Phosphatidylinositol 3-Kinase, Growth Disorders, and Cancer. N. Engl. J. Med. 2018, 379, 2052–2062. [Google Scholar] [CrossRef]
- Hong, R.; Edgar, K.; Song, K.; Steven, S.; Young, A.; Hamilton, P.; Arrazate, A.; De La Cruz, C.; Chan, C.; Pang, J.; et al. Abstract PD4-14: GDC-0077 is a selective PI3Kalpha inhibitor that demonstrates robust efficacy in PIK3CA mutant breast cancer models as a single agent and in combination with standard of care therapies. Cancer Res. 2018, 78, PD4-14. [Google Scholar]
- Merlino, G.; Fiascarelli, A.; Bigioni, M.; Bressan, A.; Irrissuto, C.; Pellacani, A.; Scaltriti, M.; Binaschi, M. Abstract 2160: MEN1611, a novel α-selective PI3K inhibitor in solid tumors. Tumor Biol. 2018, 78, 2160. [Google Scholar] [CrossRef]
- Ottensmeier, C.H.; Jones, T.; Sacco, J.J.; McCaul, J.; Brennan, P.; Paterson, C.; Schache, A.; Shaw, R.J.; Singh, R.P.; Davies, J.H.; et al. A randomised, double-blind, placebo-controlled phase IIa trial of AMG319 given orally as neoadjuvant therapy in patients with human papillomavirus (HPV) positive and negative head and neck squamous cell carcinoma (HNSCC). J. Clin. Oncol. 2018, 36, 6068. [Google Scholar] [CrossRef]
- Blagden, S.; Olmin, A.; Josephs, D.; Stavraka, C.; Zivi, A.; Pinato, D.J.; Anthoney, A.; Decordova, S.; Swales, K.; Riisnaes, R.; et al. Correction: First-in-human study of CH5132799, an oral class I PI3K inhibitor, studying toxicity, pharmacokinetics, and pharmacodynamics, in patients with metastatic cancer. Clin. Cancer Res. 2015, 21, 660. [Google Scholar]
- Paik, P.K.; Shen, R.; Berger, M.F.; Ferry, D.; Soria, J.-C.; Mathewson, A.; Rooney, C.; Smith, N.R.; Cullberg, M.; Kilgour, E.; et al. A Phase Ib Open-Label Multicenter Study of AZD4547 in Patients with Advanced Squamous Cell Lung Cancers. Clin. Cancer Res. 2017, 23, 5366–5373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal, S.K.; Rosenberg, J.E.; Hoffman-Censits, J.H.; Berger, R.; Quinn, D.I.; Galsky, M.D.; Wolf, J.; Dittrich, C.; Keam, B.; Delord, J.-P.; et al. Efficacy of BGJ398, a Fibroblast Growth Factor Receptor 1-3 Inhibitor, in Patients with Previously Treated Advanced Urothelial Carcinoma with FGFR3 Alterations. Cancer Discov. 2018, 8, 812–821. [Google Scholar] [CrossRef] [Green Version]
- Koyama, T.; Shimizu, T.; Iwasa, S.; Fujiwara, Y.; Kondo, S.; Kitano, S.; Yonemori, K.; Shimomura, A.; Iizumi, S.; Sasaki, T.; et al. Abstract B160: First-in-human phase 1 study of E7090, a novel selective inhibitor of FGFRs, in patients with advanced solid tumors. Mol. Cancer Ther. 2018, 17, B160. [Google Scholar] [CrossRef]
- Michael, M.; Bang, Y.-J.; Park, Y.S.; Kang, Y.-K.; Kim, T.M.; Hamid, O.; Thornton, N.; Tate, S.C.; Raddad, E.; Tie, J. A Phase 1 Study of LY2874455, an Oral Selective pan-FGFR Inhibitor, in Patients with Advanced Cancer. Target. Oncol. 2017, 12, 463–474. [Google Scholar] [CrossRef]
- Goyal, L.; Shi, L.; Liu, L.Y.; De La Cruz, F.F.; Lennerz, J.K.; Raghavan, S.; Leschiner, I.; Elagina, L.; Siravegna, G.; Ng, R.; et al. TAS-120 Overcomes Resistance to ATP-Competitive FGFR Inhibitors in Patients with FGFR2 Fusion-Positive Intrahepatic Cholangiocarcinoma. Cancer Discov. 2019, 9, 1064–1079. [Google Scholar] [CrossRef] [Green Version]
- Kim, R.D.; Sarker, D.; Meyer, T.; Yau, T.; Macarulla, T.; Park, J.-W.; Choo, S.P.; Hollebecque, A.; Sung, M.W.; Lim, H.-Y.; et al. First-in-Human Phase I Study of Fisogatinib (BLU-554) Validates Aberrant FGF19 Signaling as a Driver Event in Hepatocellular Carcinoma. Cancer Discov. 2019, 9, 1696–1707. [Google Scholar] [CrossRef] [Green Version]
- Mercade, T.M.; Moreno, V.; John, B.; Morris, J.C.; Sawyer, M.B.; Yong, W.P.; Gutierrez, M.; Karasic, T.B.; Sangro, B.; Sheng-Shun, Y.; et al. A phase I study of H3B-6527 in hepatocellular carcinoma (HCC) or intrahepatic cholangiocarcinoma (ICC) patients (pts). J. Clin. Oncol. 2019, 37, 4095. [Google Scholar] [CrossRef]
- Weiss, A.; Adler, F.; Buhles, A.; Stamm, C.; Fairhurst, R.A.; Kiffe, M.; Sterker, D.; Centeleghe, M.; Wartmann, M.; Kinyamu-Akunda, J.; et al. FGF401, A First-In-Class Highly Selective and Potent FGFR4 Inhibitor for the Treatment of FGF19-Driven Hepatocellular Cancer. Mol. Cancer Ther. 2019, 18, 2194–2206. [Google Scholar] [CrossRef] [PubMed]
- Hyman, D.; Kummar, S.; Farago, A.; Geoerger, B.; Mau-Sorensen, M.; Taylor, M.; Garralda, E.; Nagasubramanian, R.; Natheson, M. Abstract CT127: Phase I and expanded access experience of LOXO-195 (BAY 2731954), a selective next-generation TRK inhibitor (TRKi). Cancer Res. 2019, 79, CT127. [Google Scholar]
- Lin, C.-C.; Arkenau, H.-T.; Lu, S.; Sachdev, J.; Carpeño, J.D.C.; Mita, M.; Dziadziuszko, R.; Su, W.-C.; Bobilev, D.; Hughes, L.; et al. A phase 1, open-label, dose-escalation trial of oral TSR-011 in patients with advanced solid tumours and lymphomas. Br. J. Cancer 2019, 121, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, K.P.; Gandhi, L.; Jänne, P.A.; Ou, S.-H.I.; Shaw, A.; Goldberg, T.R.; Greenberg, J.; Gu, X.; Tachibana, M.; Senaldi, G.; et al. First-in-human study of DS-6051b in patients (pts) with advanced solid tumors (AST) conducted in the US. J. Clin. Oncol. 2018, 36, 2514. [Google Scholar] [CrossRef]
- Dieci, M.; Arnedos, M.; André, F.; Soria, J.-C. Fibroblast Growth Factor Receptor Inhibitors as a Cancer Treatment: From a Biologic Rationale to Medical Perspectives. Cancer Discov. 2013, 3, 264–279. [Google Scholar] [CrossRef] [Green Version]
- Babina, I.; Turner, N.C. Advances and challenges in targeting FGFR signalling in cancer. Nat. Rev. Cancer 2017, 17, 318–332. [Google Scholar] [CrossRef]
- Siefker-Radtke, A.; Curti, B. Immunotherapy in metastatic urothelial carcinoma: Focus on immune checkpoint inhibition. Nat. Rev. Urol. 2017, 15, 112–124. [Google Scholar] [CrossRef]
- Loriot, Y.; Necchi, A.; Park, S.H.; Garcia-Donas, J.; Huddart, R.; Burgess, E.F.; Fleming, M.; Rezazadeh, A.; Mellado, B.; Varlamov, S.; et al. Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med. 2019, 381, 338–348. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinicla sequencing defines the genomic landscape of metastatic colorectal cancer. Cancer Cell 2019, 33, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E-Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Märkl, B.; Hirschbühl, K.; Dhillon, C. NTRK-Fusions—A new kid on the block. Pathol. Res. Pract. 2019, 215, 152572. [Google Scholar] [CrossRef] [PubMed]
- Le Tourneau, C.; Delord, J.-P.; Gonçalves, A.; Gavoille, C.; Dubot, C.; Isambert, N.; Campone, M.; Trédan, O.; Massiani, M.-A.; Mauborgne, C.; et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): A multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol. 2015, 16, 1324–1334. [Google Scholar] [CrossRef]
- Barroilhet, L.; Matulonis, U. The NCI-MATCH trial and precision medicine in gynecologic cancers. Gynecol. Oncol. 2018, 148, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2017, 15, 81–94. [Google Scholar] [CrossRef]
- Eckhardt, S.G.; Lieu, C. Is Precision Medicine an Oxymoron? JAMA Oncol. 2019, 5, 142. [Google Scholar] [CrossRef]
- Gambardella, V.; Fleitas, T.; Tarazona, N.; Cejalvo, J.; Gimeno-Valiente, F.; Martínez-Ciarpaglini, C.; Huerta, M.; Roselló, S.; Castillo, J.; Roda, D.; et al. Towards precision oncology for HER2 blockade in gastroesophageal adenocarcinoma. Ann. Oncol. 2019, 30, 1254–1264. [Google Scholar] [CrossRef] [Green Version]
- Polasek, T.M.; Shakib, S.; Rostami-hodjegan, A. Expert Review of Clinical Pharmacology Precision dosing in clinical medicine: Present and future. Expert Rev. Clin. Pharmacol. 2018, 11, 743–746. [Google Scholar] [CrossRef] [Green Version]
- Darwich, A.S.; Ogungbenro, K.; Vinks, A.A.; Powell, J.R.; Reny, J.L.; Marsousi, N.; Daali, Y.; Fairman, D.; Cook, J.; Lesko, L.J.; et al. Why has model-informed precision dosing not yet become common clinical reality? Lessons from the past and a roadmap for the future. Clin. Pharmacol. Ther. 2017, 101, 646–656. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell 2015, 27, 15–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz, L.A.; Bardelli, A. Liquid Biopsies: Genotyping Circulating Tumor DNA. J. Clin. Oncol. 2014, 32, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Birkbak, N.J.; Mcgranahan, N. Review Cancer Genome Evolutionary Trajectories in Metastasis. Cancer Cell 2019, 37, 8–19. [Google Scholar] [CrossRef] [PubMed]
- El-Deiry, W.S.; Goldberg, R.M.; Lenz, H.; Shields, A.F.; Gibney, G.T.; Tan, A.R.; Brown, J.; Eisenberg, B.; Heath, E.I.; Phuphanich, S.; et al. The current state of molecular testing in the treatment of patients with solid tumors, 2019. CA A Cancer J. Clin. 2019, 69, 305–343. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Stephenson, J.J.; Rosen, P.; Loesch, D.M.; Borad, M.J.; Anthony, S.; Jameson, G.S.; Brown, S.; Cantafio, N.; Richards, D.; et al. Pilot Study Using Molecular Profiling of Patients’ Tumors to Find Potential Targets and Select Treatments for Their Refractory Cancers. J. Clin. Oncol. 2010, 28, 4877–4883. [Google Scholar] [CrossRef] [PubMed]
- Tsimberidou, A.M.; Iskander, N.G.; Hong, D.S.; Wheler, J.J.; Falchook, G.S.; Fu, S.; Piha-Paul, S.; Naing, A.; Janku, F.; Luthra, R.; et al. Personalized medicine in a phase I clinical trials program: The MD Anderson Cancer Center initiative. Clin. Cancer Res. 2012, 18, 6373–6383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hainsworth, J.D.; Meric-Bernstam, F.; Swanton, C.; Hurwitz, H.; Spigel, D.R.; Sweeney, C.; Burris, H.A.; Bose, R.; Yoo, B.; Stein, A.; et al. Targeted Therapy for Advanced Solid Tumors on the Basis of Molecular Profiles: Results from MyPathway, an Open-Label, Phase IIa Multiple Basket Study. J. Clin. Oncol. 2018, 36, 536–542. [Google Scholar] [CrossRef]
- Trédan, O.; Wang, Q.; Pissaloux, D.; Cassier, P.; De La Fouchardière, A.; Fayette, J.; Desseigne, F.; Ray-Coquard, I.; Frappaz, D.; Heudel, P.-E.; et al. Molecular screening program to select molecular-based recommended therapies for metastatic cancer patients: Analysis from the ProfiLER trial. Ann. Oncol. 2019, 30, 757–765. [Google Scholar] [CrossRef]
- Sicklick, J.K.; Kato, S.; Okamura, R.; Schwaederle, M.; Hahn, M.E.; Williams, C.B.; De, P.; Krie, A.; Piccioni, D.E.; Miller, V.A.; et al. Molecular profiling of cancer patients enables personalized combination therapy: The I-PREDICT study. Nat. Med. 2019, 25, 744–750. [Google Scholar] [CrossRef]
- Rodon, J.; Soria, J.-C.; Berger, R.; Miller, W.H.; Rubin, E.; Kugel, A.; Tsimberidou, A.; Saintigny, P.; Ackerstein, A.; Braña, I.; et al. Genomic and transcriptomic profiling expands precision cancer medicine: The WINTHER trial. Nat. Med. 2019, 25, 751–758. [Google Scholar] [CrossRef]
- Clevers, H. Modeling Development and Disease with Organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letai, A. Functional precision cancer medicine—Moving beyond pure genomics. Nat. Med. 2017, 23, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Vaeteewoottacharn, K.; Kariya, R. Establishment of a Patient-Derived Tumor Xenograft Model and Application for Precision Cancer Medicine. Chem. Pharm. Bull. 2018, 66, 225–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhimani, J.; Ball, K.; Stebbing, J. Patient-derived xenograft models—The future of personalised cancer treatment. Br. J. Cancer 2020, 122, 601–602. [Google Scholar] [CrossRef]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernández-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef] [Green Version]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of Circulating Tumor DNA in Early- and Late-Stage Human Malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef] [Green Version]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. N. Engl. J. 2012, 366, 883–892. [Google Scholar] [CrossRef] [Green Version]
- Parikh, A.R.; Leshchiner, I.; Elagina, L.; Goyal, L.; Levovitz, C.; Siravegna, G.; Livitz, D.; Rhrissorrakrai, K.; Martin, E.E.; Van Seventer, E.E.; et al. Liquid versus tissue biopsy for detecting acquired resistance and tumor heterogeneity in gastrointestinal cancers. Nat. Med. 2019, 25, 1415–1421. [Google Scholar] [CrossRef]
- Van Der Velden, D.; Van Herpen, C.; Van Laarhoven, H.; Smit, E.; Groen, H.; Willems, S.; Nederlof, P.; Langenberg, M.; Cuppen, E.; Sleijfer, S.; et al. Molecular Tumor Boards: Current practice and future needs. Ann. Oncol. 2017, 28, 3070–3075. [Google Scholar] [CrossRef]
- Van De Haar, J.; Hoes, L.; Voest, E. Advancing molecular tumour boards: Highly needed to maximise the impact of precision medicine. ESMO Open 2019, 4, e000516. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Class of Inhibitors | Novel Targeted Agents under Development for Solid Tumors in Phase I Trials | ||
|---|---|---|---|
| Compound Name | Mechanism of Action | Phase | |
| PI3K inhibitors | GDC0077 [41] | Potent PI3K alpha inhibitor | Ib |
| MEN1611 (PA799) [42] | PI3K alpha inhibitor | Ib | |
| AMG319 [43] | AMG319 is a PI3Kδ inhibitor. Preclinically, target inhibition abrogates Treg-mediated immunosuppression, augmenting CD8+ T-cell antitumor activity | IIa | |
| CH5132799 [44] | Oral pan-PI3 kinase inhibitor | Ia/b | |
| FGFR inhibitors | AZD4547 [45] | Potent and selective inhibitor of FGFR 1, 2, and 3 | I |
| NVP-BGJ398 [46] | Oral, selective, ATP-competitive inhibitor of FGFR1, 2, and 3 | I | |
| E-7090 [47] | Oral and selective inhibitor of FGFR1, 2, and 3 | I | |
| LY2874455 [48] | Inhibitor of FGFR 1, 2, 3, and 4 | I | |
| TAS-120 [49] | Potent and highly specific against wildtype FGFR1–4 as well as against some FGFR2 kinase domain mutations | I | |
| BLU-554 [50] | Potent and selective inhibitor of FGFR4 | I | |
| H3B-6527 [51] | Selective and covalent inhibitor of FGFR4 | I | |
| FGF-401 [52] | Potent and selective, reversible-covalent small-molecule inhibitor of FGFR4 | I | |
| NTRK inhibitor | LOXO-195 [53] | Selective inhibitor of TRK | I |
| TSR-011 [54] | Dual ALK4 and TRK inhibitor | I | |
| DS-6051b [55] | Inhibitor with high affinity for ROS1 5 and TRK | I | |
| Molecular Tools for Selecting Patients in Precision Medicine-Based Basket Trials | |
|---|---|
| Clinical Trial | Molecular Tools |
| Bisgrove [75] | Immunohistochemistry, Fluorescence in situ hybridization microarray |
| IMPACT [76] | PCR-based genomics and NGS |
| SHIVA [64] | Targeted NGS-based |
| MOSCATO [39] | Targeted NGS-based, RNA Seq |
| MyPathway [77] | Genomic testing |
| Profiler [78] | Targeted NGS-based |
| I-PREDICT [79] | Targeted NGS-based, ctDNA |
| WINTHER [80] | Targeted NGS-based, Transcriptomic |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gambardella, V.; Tarazona, N.; Cejalvo, J.M.; Lombardi, P.; Huerta, M.; Roselló, S.; Fleitas, T.; Roda, D.; Cervantes, A. Personalized Medicine: Recent Progress in Cancer Therapy. Cancers 2020, 12, 1009. https://doi.org/10.3390/cancers12041009
Gambardella V, Tarazona N, Cejalvo JM, Lombardi P, Huerta M, Roselló S, Fleitas T, Roda D, Cervantes A. Personalized Medicine: Recent Progress in Cancer Therapy. Cancers. 2020; 12(4):1009. https://doi.org/10.3390/cancers12041009
Chicago/Turabian StyleGambardella, Valentina, Noelia Tarazona, Juan Miguel Cejalvo, Pasquale Lombardi, Marisol Huerta, Susana Roselló, Tania Fleitas, Desamparados Roda, and Andres Cervantes. 2020. "Personalized Medicine: Recent Progress in Cancer Therapy" Cancers 12, no. 4: 1009. https://doi.org/10.3390/cancers12041009