Identifying Therapies to Combat Epithelial Mesenchymal Plasticity-Associated Chemoresistance to Conventional Breast Cancer Therapies Using An shRNA Library Screen

 , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Cell Line and Growth Conditions
2.2. ‘Polarity Pool’ shRNA Library
2.3. ‘Polarity Pool’ shRNA Library Infection of BL2T-MDAMB468 Cells
2.4. Dose Determination for Growth Inhibition
2.5. RNA Extraction, cDNA Synthesis, and Reverse Transcriptase-Quantitative PCR (RT-qPCR)
2.6. Western Blotting
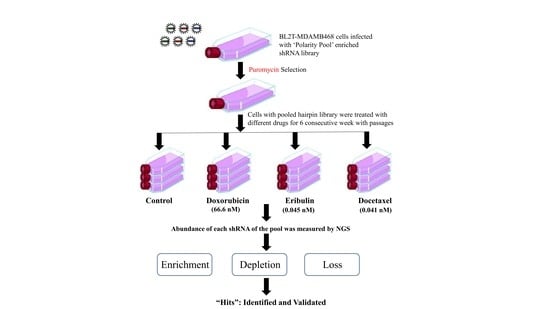
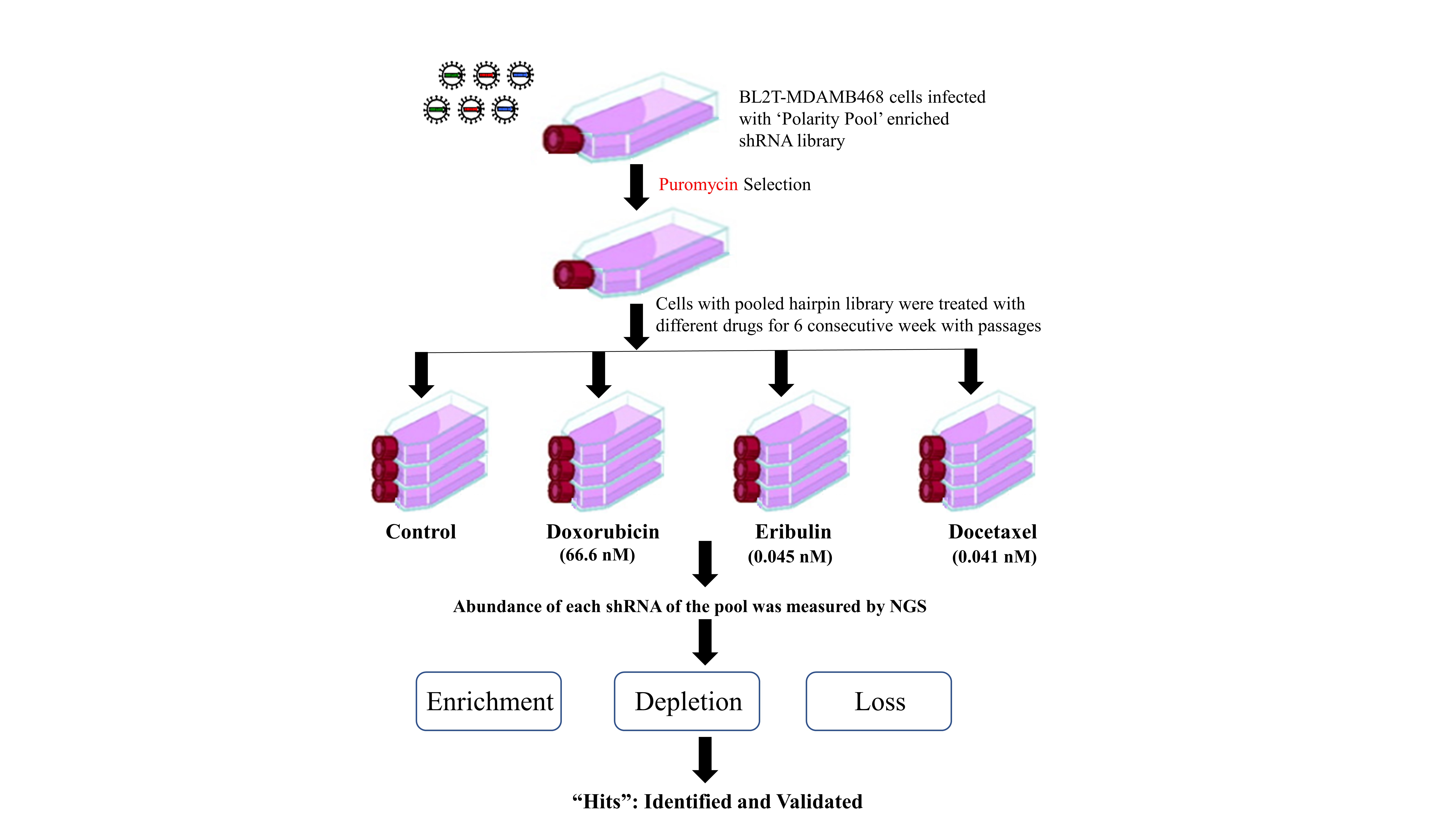
2.7. Drug Treatment for Selection of ‘Polarity Pool’-Enriched Hairpin Library of BLT-MDAMB468 Cells
2.8. Genomic DNA Extraction and shRNA Amplification
2.9. Data Analysis
2.10. Drug Combination Experiments
3. Results
3.1. Mathematical Modelling to Simulate the Distribution of Sampling Error in Regard to Hairpin Abundance in PMC42-LA Cells
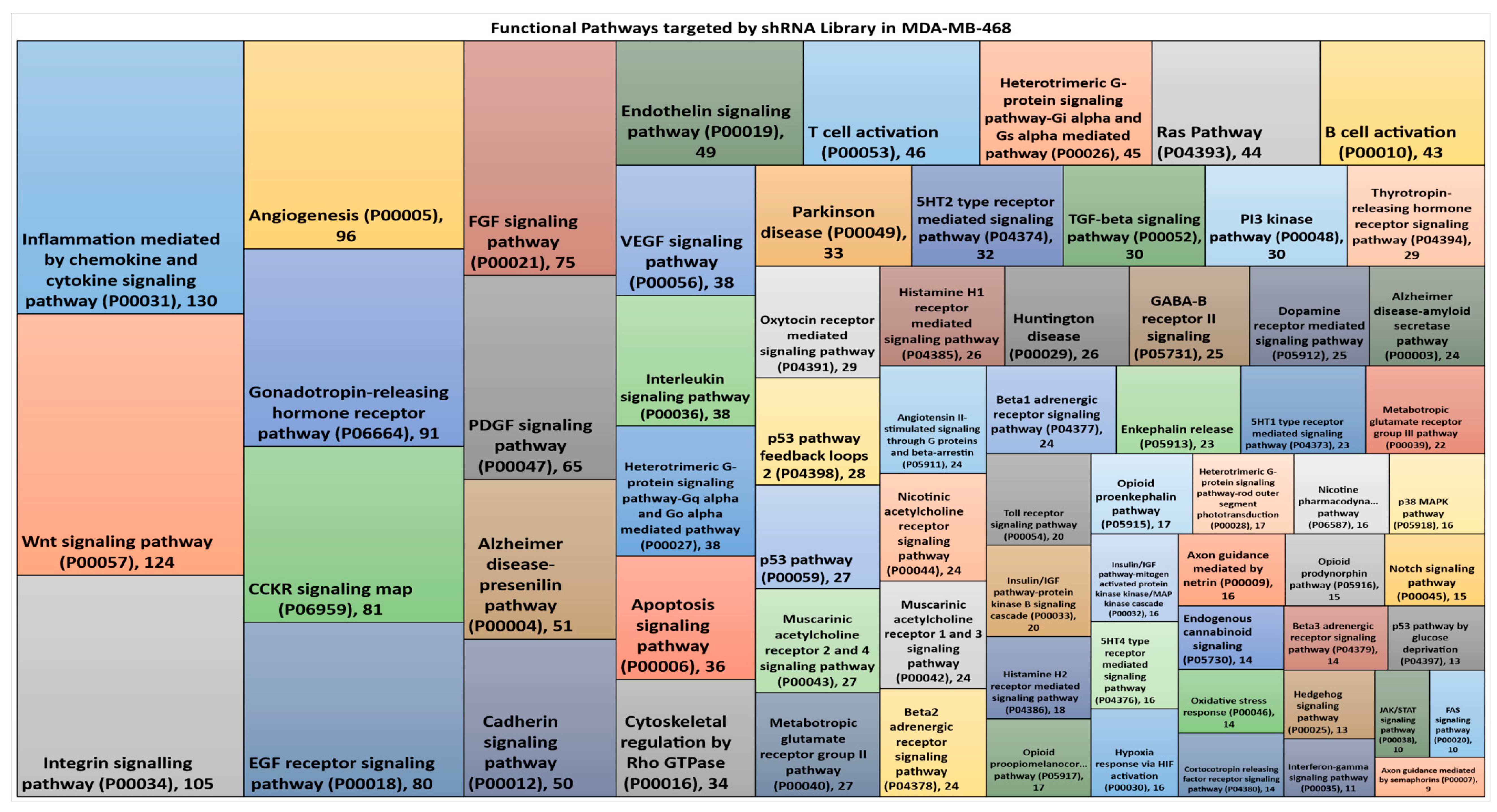
3.2. Hairpin Representation in Functional Pathways
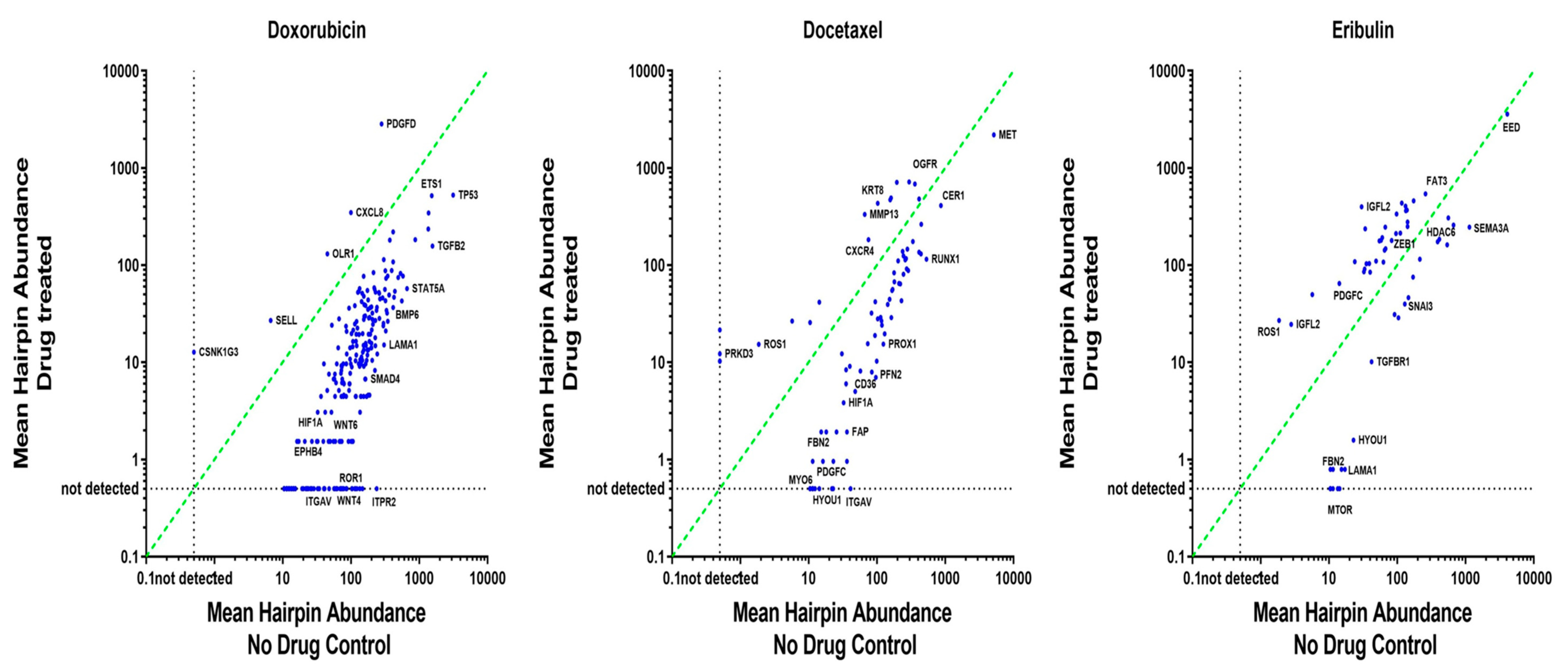
3.3. ‘Polarity Pool’-Associated shRNA Hairpin Lethality Screen Performance
3.4. CMap Analysis Revealed Potential Inhibitors to Be Synergistic with Doxorubicin
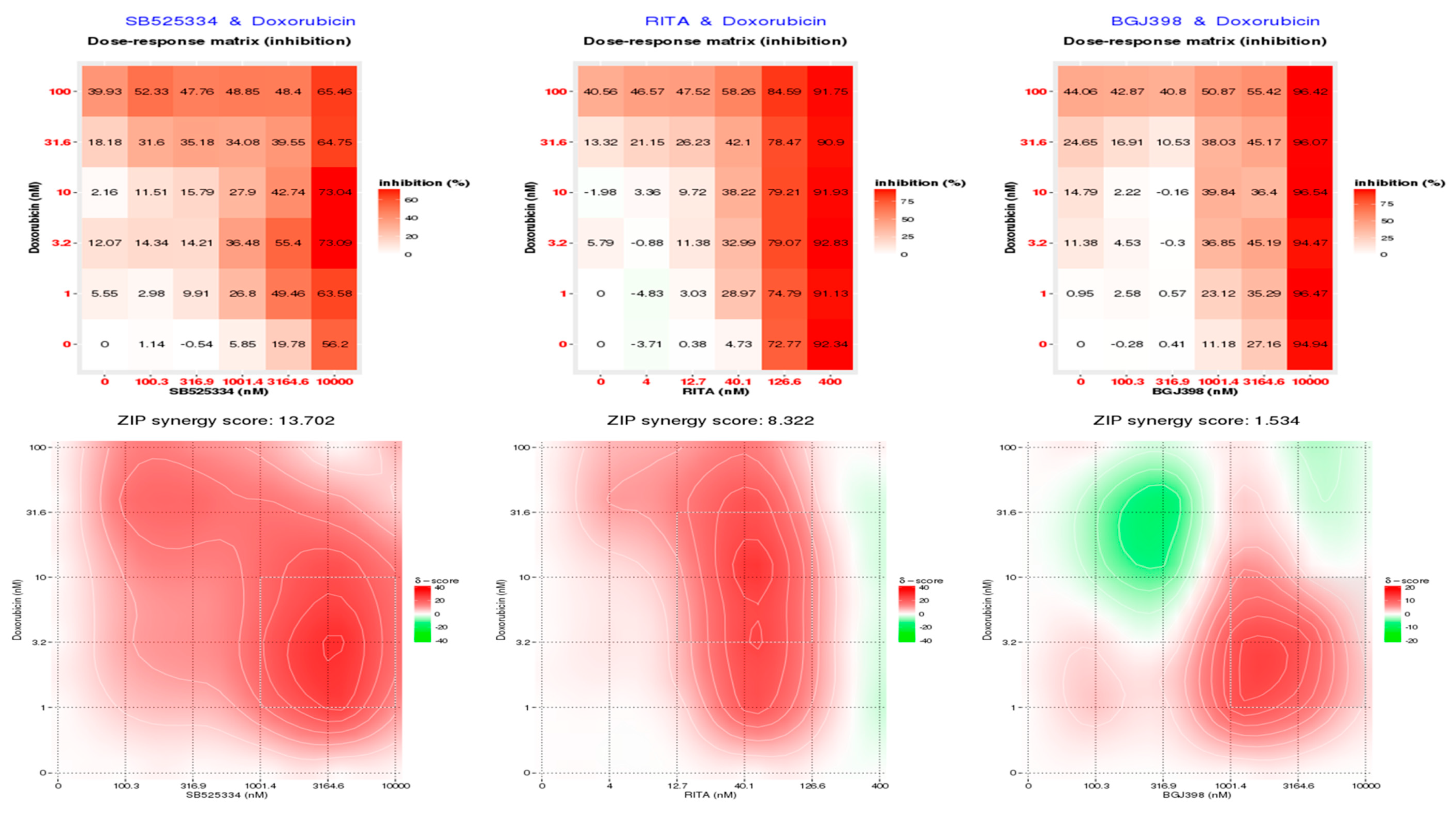
3.5. SB525334 and RITA Inhibitors Synergistically Inhibited MDA-MB-468 Cell Viability in Combination with Doxorubicin
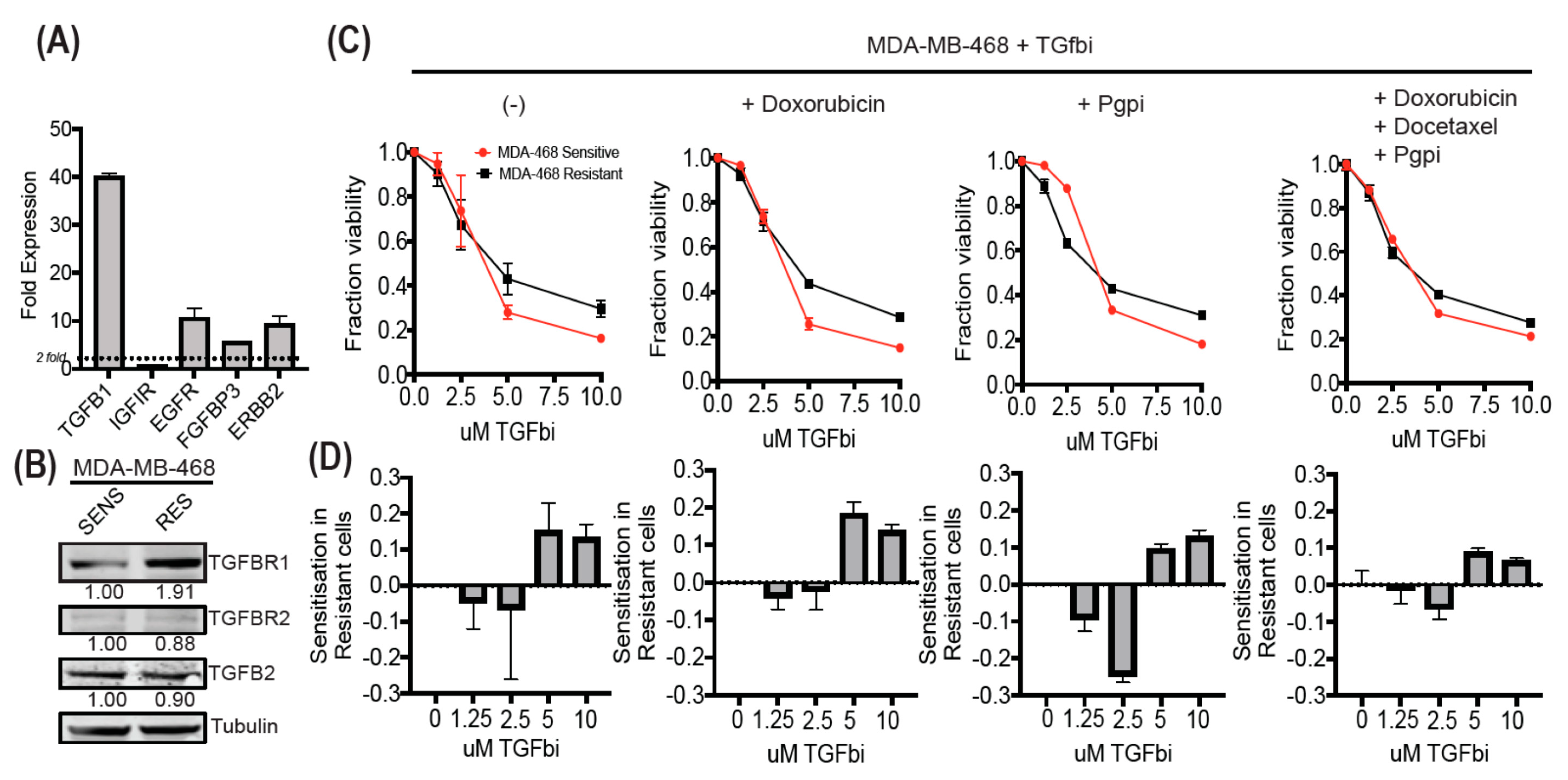
3.6. MDA-MB-468-Resistant Cells Display Enhanced TGF-β Expression and Can Be Sensitized Using SB525334
4. Discussion
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| BC | Breast cancer |
| EMP | Epithelial mesenchymal plasticity |
| CMap | Connectivity Map |
| shRNAs | short hairpin RNAs |
| EMT | Epithelial mesenchymal transition |
| CSC | cancer stem cells |
| RNAi | RNA interference |
| siRNA | small interfering RNA |
| NGS | Next generation sequencing |
| TGFBR | Transforming growth factor β receptor |
| FGFR | Fibroblast growth factor receptor |
| EGF | Epidermal growth factor |
| MDM | Murine double minute |
| WNT | Wingless-related integration site |
References
- Gerashchenko, T.S.; Zavyalova, M.V.; Denisov, E.V.; Krakhmal, N.V.; Pautova, D.N.; Litviakov, N.V.; Vtorushin, S.V.; Cherdyntseva, N.V.; Perelmuter, V.M. Intratumoral Morphological Heterogeneity of Breast Cancer As an Indicator of the Metastatic Potential and Tumor Chemosensitivity. Acta Nat. 2017, 9, 56–67. [Google Scholar] [CrossRef]
- Tsai, J.H.; Yang, J. Epithelial–mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013, 27, 2192–2206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redfern, A.D.; Spalding, L.J.; Thompson, E.W. The Kraken Wakes: Induced EMT as a driver of tumour aggression and poor outcome. Clin. Exp. Metastasis 2018, 35, 285–308. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Gao, R.; Sei, E.; Brandt, R.; Hartman, J.; Hatschek, T.; Crosetto, N.; Foukakis, T.; Navin, N.E. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 2018, 173, 879–893. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [Green Version]
- Shah, A.N.; Summy, J.M.; Zhang, J.; Park, S.I.; Parikh, N.U.; Gallick, G.E. Development and characterization of gemcitabine-resistant pancreatic tumor cells. Ann. Surg. Oncol. 2007, 14, 3629–3637. [Google Scholar] [CrossRef]
- Larocca, C.; Cohen, J.R.; Fernando, R.I.; Huang, B.; Hamilton, D.H.; Palena, C. An autocrine loop between TGF-beta1 and the transcription factor brachyury controls the transition of human carcinoma cells into a mesenchymal phenotype. Mol. Cancer Ther. 2013, 12, 1805–1815. [Google Scholar] [CrossRef] [Green Version]
- Kurrey, N.K.; Jalgaonkar, S.P.; Joglekar, A.V.; Ghanate, A.D.; Chaskar, P.D.; Doiphode, R.Y.; Bapat, S.A. Snail and slug mediate radioresistance and chemoresistance by antagonizing p53-mediated apoptosis and acquiring a stem-like phenotype in ovarian cancer cells. Stem Cells 2009, 27, 2059–2068. [Google Scholar] [CrossRef]
- Kajiyama, H.; Shibata, K.; Terauchi, M.; Yamashita, M.; Ino, K.; Nawa, A.; Kikkawa, F. Chemoresistance to paclitaxel induces epithelial-mesenchymal transition and enhances metastatic potential for epithelial ovarian carcinoma cells. Int. J. Oncol. 2007, 31, 277–283. [Google Scholar] [CrossRef] [Green Version]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef]
- Huang, B.; Cohen, J.R.; Fernando, R.I.; Hamilton, D.H.; Litzinger, M.T.; Hodge, J.W.; Palena, C. The embryonic transcription factor Brachyury blocks cell cycle progression and mediates tumor resistance to conventional antitumor therapies. Cell Death Dis. 2013, 4, e682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, E.D.; Gao, D.; Redfern, A.; Thompson, E.W. Controversies around epithelial–mesenchymal plasticity in cancer metastasis. Nat. Rev. Cancer 2019, 19, 716–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, P.B.; Pastushenko, I.; Skibinski, A.; Blanpain, C.; Kuperwasser, C. Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell Stem Cell 2019, 24, 65–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Park, S.H.; Cieply, B.; Schupp, J.; Killiam, E.; Zhang, F.; Rimm, D.L.; Frisch, S.M. A Pathway for the Control of Anoikis Sensitivity by E-Cadherin and Epithelial-to-Mesenchymal Transition. Mol. Cell. Biol. 2011, 31, 4036–4051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gal, A.; Sjoblom, T.; Fedorova, L.; Imreh, S.; Beug, H.; Moustakas, A. Sustained TGF beta exposure suppresses Smad and non-Smad signalling in mammary epithelial cells, leading to EMT and inhibition of growth arrest and apoptosis. Oncogene 2008, 27, 1218–1230. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.Y.J.; Wong, M.K.; Tan, T.Z.; Kuay, K.T.; Ng, A.H.C.; Chung, V.Y.; Chu, Y.S.; Matsumura, N.; Lai, H.C.; Lee, Y.F.; et al. An EMT spectrum defines an anoikis-resistant and spheroidogenic intermediate mesenchymal state that is sensitive to e-cadherin restoration by a src-kinase inhibitor, saracatinib (AZD0530). Cell Death Dis. 2013, 4, e915. [Google Scholar] [CrossRef]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576. [Google Scholar] [CrossRef] [Green Version]
- Marcotte, R.; Sayad, A.; Brown Kevin, R.; Sanchez-Garcia, F.; Reimand, J.; Haider, M.; Virtanen, C.; Bradner James, E.; Bader Gary, D.; Mills Gordon, B.; et al. Functional Genomic Landscape of Human Breast Cancer Drivers, Vulnerabilities, and Resistance. Cell 2016, 164, 293–309. [Google Scholar] [CrossRef] [Green Version]
- Marcotte, R.; Brown, K.R.; Suarez, F.; Sayad, A.; Karamboulas, K.; Krzyzanowski, P.M.; Sircoulomb, F.; Medrano, M.; Fedyshyn, Y.; Koh, J.L.Y.; et al. Essential gene profiles in breast, pancreatic, and ovarian cancer cells. Cancer Discov. 2012, 2, 172–189. [Google Scholar] [CrossRef] [Green Version]
- Sims, D.; Mendes-Pereira, A.M.; Frankum, J.; Burgess, D.; Cerone, M.A.; Lombardelli, C.; Mitsopoulos, C.; Hakas, J.; Murugaesu, N.; Isacke, C.M.; et al. High-throughput RNA interference screening using pooled shRNA libraries and next generation sequencing. Genome Biol. 2011, 12, R104. [Google Scholar] [CrossRef] [Green Version]
- Rosenbluh, J.; Mercer, J.; Shrestha, Y.; Oliver, R.; Tamayo, P.; Doench, J.G.; Tirosh, I.; Piccioni, F.; Hartenian, E.; Horn, H.J.C. Genetic and proteomic interrogation of lower confidence candidate genes reveals signaling networks in β-catenin-active cancers. Cell Syst. 2016, 3, 302–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iorns, E.; Lord, C.J.; Turner, N.; Ashworth, A. Utilizing RNA interference to enhance cancer drug discovery. Nat. Rev. Drug Discov. 2007, 6, 556–568. [Google Scholar] [CrossRef] [PubMed]
- Kassner, P.D. Discovery of novel targets with high throughput RNA interference screening. Comb. Chem. High Throughput Screen. 2008, 11, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Garnett, M.J.; Edelman, E.J.; Heidorn, S.J.; Greenman, C.D.; Dastur, A.; Lau, K.W.; Greninger, P.; Thompson, I.R.; Luo, X.; Soares, J.; et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 2012, 483, 570–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, J.A.; Ye, F.; Marshall, C.B.; Lehmann, B.D.; Pendleton, C.S.; Shyr, Y.; Arteaga, C.L.; Pietenpol, J.A. RNA interference (RNAi) screening approach identifies agents that enhance paclitaxel activity in breast cancer cells. Breast Cancer Res. BCR 2010, 12, R41. [Google Scholar] [CrossRef] [Green Version]
- Price, C.; Gill, S.; Ho, Z.V.; Davidson, S.M.; Merkel, E.; McFarland, J.M.; Leung, L.; Tang, A.; Kost-Alimova, M.; Tsherniak, A.; et al. Genome-Wide Interrogation of Human Cancers Identifies EGLN1 Dependency in Clear Cell Ovarian Cancers. Cancer Res. 2019, 79, 2564–2579. [Google Scholar] [CrossRef] [Green Version]
- MacKeigan, J.P.; Murphy, L.O.; Blenis, J. Sensitized RNAi screen of human kinases and phosphatases identifies new regulators of apoptosis and chemoresistance. Nat. Cell Biol. 2005, 7, 591–600. [Google Scholar] [CrossRef]
- Diehl, P.; Tedesco, D.; Chenchik, A. Use of RNAi screens to uncover resistance mechanisms in cancer cells and identify synthetic lethal interactions. Drug Discov. Today Technol. 2014, 11, 11–18. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, S.; Monkman, J.; Toh, A.K.L.; Nagaraj, S.H.; Thompson, E.W. Targeting epithelial-mesenchymal plasticity in cancer: Clinical and preclinical advances in therapy and monitoring. Biochem. J. 2017, 474, 3269–3306. [Google Scholar] [CrossRef]
- Yoshida, T.; Ozawa, Y.; Kimura, T.; Sato, Y.; Kuznetsov, G.; Xu, S.; Uesugi, M.; Agoulnik, S.; Taylor, N.; Funahashi, Y.; et al. Eribulin mesilate suppresses experimental metastasis of breast cancer cells by reversing phenotype from epithelial-mesenchymal transition (EMT) to mesenchymal-epithelial transition (MET) states. Br. J. Cancer 2014, 110, 1497–1505. [Google Scholar] [CrossRef]
- Twelves, C.; Awada, A.; Cortes, J.; Yelle, L.; Velikova, G.; Olivo, M.S.; Song, J.; Dutcus, C.E.; Kaufman, P.A. Subgroup Analyses from a Phase 3, Open-Label, Randomized Study of Eribulin Mesylate Versus Capecitabine in Pretreated Patients with Advanced or Metastatic Breast Cancer. Breast Cancer Basic Clin. Res. 2016, 10, 77–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, S.; Friedrichs, K.; Noel, D.; Pintér, T.; Van Belle, S.; Vorobiof, D.; Duarte, R.; Gil, M.; Bodrogi, I.; Murray, E.J.J. Prospective randomized trial of docetaxel versus doxorubicin in patients with metastatic breast cancer. J. Clin. Oncol. 1999, 17, 2341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neve, R.M.; Chin, K.; Fridlyand, J.; Yeh, J.; Baehner, F.L.; Fevr, T.; Clark, L.; Bayani, N.; Coppe, J.P.; Tong, F.; et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 2006, 10, 515–527. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Patel, M.R.; Prescher, J.A.; Patsialou, A.; Qian, D.; Lin, J.; Wen, S.; Chang, Y.F.; Bachmann, M.H.; Shimono, Y.; et al. Cancer stem cells from human breast tumors are involved in spontaneous metastases in orthotopic mouse models. Proc. Natl. Acad. Sci. USA 2010, 107, 18115–18120. [Google Scholar] [CrossRef] [Green Version]
- Blick, T.; Hugo, H.; Widodo, E.; Waltham, M.; Pinto, C.; Mani, S.A.; Weinberg, R.A.; Neve, R.M.; Lenburg, M.E.; Thompson, E.W. Epithelial mesenchymal transition traits in human breast cancer cell lines parallel the CD44(hi/)CD24 (lo/-) stem cell phenotype in human breast cancer. J. Mammary Gland Biol. Neoplasia 2010, 15, 235–252. [Google Scholar] [CrossRef]
- Vermeulen, A.; van Brabant Smith, A.; Anderson, S.B.; Beijersbergen, R.L.; Simpson, K.J. Pooled shRNA Screening. In Frontiers in RNAi. Volume 1; Ralph, A., Tripp, J.M.K., Eds.; Bentham Science: Driving Sharjah, UAE, 2014; Volume 21, pp. 58–78. [Google Scholar]
- Cluse, L.A.; Nikolic, I.; Knight, D.; Madhamshettiwar, P.B.; Luu, J.; Cowley, K.J.; Semple, T.; Arnau, G.M.; Shortt, J.; Johnstone, R.W.; et al. A Comprehensive Protocol Resource for Performing Pooled shRNA and CRISPR Screens. Methods Mol. Biol. 2018, 1725, 201–227. [Google Scholar]
- Thomas, P.D.; Campbell, M.J.; Kejariwal, A.; Mi, H.; Karlak, B.; Daverman, R.; Diemer, K.; Muruganujan, A.; Narechania, A. PANTHER: A library of protein families and subfamilies indexed by function. Genome Res. 2003, 13, 2129–2141. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171, 1437–1452. [Google Scholar] [CrossRef]
- Ianevski, A.; He, L.; Aittokallio, T.; Tang, J. SynergyFinder: A web application for analyzing drug combination dose–response matrix data. Bioinformatics 2017, 33, 2413–2415. [Google Scholar] [CrossRef]
- Yadav, B.; Wennerberg, K.; Aittokallio, T.; Tang, J. Searching for drug synergy in complex dose–response landscapes using an interaction potency model. Comput. Struct. Biotechnol. J. 2015, 13, 504–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Risom, T.; Langer, E.M.; Chapman, M.P.; Rantala, J.; Fields, A.J.; Boniface, C.; Alvarez, M.J.; Kendsersky, N.D.; Pelz, C.R.; Johnson-Camacho, K.; et al. Differentiation-state plasticity is a targetable resistance mechanism in basal-like breast cancer. Nat. Commun. 2018, 9, 3815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farmer, P.; Bonnefoi, H.; Anderle, P.; Cameron, D.; Wirapati, P.; Becette, V.; Andre, S.; Piccart, M.; Campone, M.; Brain, E.; et al. A stroma-related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer. Nat. Med. 2009, 15, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Byers, L.A.; Diao, L.; Wang, J.; Saintigny, P.; Girard, L.; Peyton, M.; Shen, L.; Fan, Y.; Giri, U.; Tumula, P.K.; et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin. Cancer Res. 2013, 19, 279–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fredebohm, J.; Wolf, J.; Hoheisel, J.D.; Boettcher, M. Depletion of RAD17 sensitizes pancreatic cancer cells to gemcitabine. J. Cell Sci. 2013, 126, 3380–3389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudo, M.; Mori, S.; Madan, V.; Yang, H.; Leong, G.; Koeffler, H.P. Short-hairpin RNA library: Identification of therapeutic partners for gefitinib-resistant non-small cell lung cancer. Oncotarget 2015, 6, 814–824. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Pan, Y.; Gang, Y.; Wang, H.; Jin, H.; Tie, J.; Xia, L.; Zhang, Y.; He, L.; Yao, L.; et al. Identification of GAS1 as an epirubicin resistance-related gene in human gastric cancer cells with a partially randomized small interfering RNA library. J. Biol. Chem. 2009, 284, 26273–26285. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y.; Deng, W.; Ekmekcioglu, S.; Grimm, E.A. Identification of unique sensitizing targets for anti-inflammatory CDDO-Me in metastatic melanoma by a large-scale synthetic lethal RNAi screening. Pigment Cell Melanoma Res. 2013, 26, 97–112. [Google Scholar] [CrossRef]
- Mills, J.R.; Malina, A.; Lee, T.; Di Paola, D.; Larsson, O.; Miething, C.; Grosse, F.; Tang, H.; Zannis-Hadjopoulos, M.; Lowe, S.W.; et al. RNAi screening uncovers Dhx9 as a modifier of ABT-737 resistance in an Emu-myc/Bcl-2 mouse model. Blood 2013, 121, 3402–3412. [Google Scholar] [CrossRef] [Green Version]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.P.; Subramanian, A.; Ross, K.N.; et al. The Connectivity Map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef] [Green Version]
- Chiu, H.-C.; Chou, D.-L.; Huang, C.-T.; Lin, W.-H.; Lien, T.-W.; Yen, K.-J.; Hsu, J.T.A. Suppression of Stat3 activity sensitizes gefitinib-resistant non small cell lung cancer cells. Biochem. Pharmacol. 2011, 81, 1263–1270. [Google Scholar] [CrossRef] [PubMed]
- Kazemi, A.; Safa, M.; Shahbazi, A. RITA enhances chemosensivity of pre-B ALL cells to doxorubicin by inducing p53-dependent apoptosis. Hematology 2011, 16, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Navarro, R.; Busnadiego, I.; Ruiz-Larrea, M.B.; Ruiz-Sanz, J.I. Superoxide Anions Are Involved in Doxorubicin-Induced ERK Activation in Hepatocyte Cultures. Ann. N. Y. Acad. Sci. 2006, 1090, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, A.; Wang, L.; Agyin, J.; Tang, Y.; Lin, S.; Yeh, I.T.; De, K.; Sun, L.Z. Doxorubicin in combination with a small TGFbeta inhibitor: A potential novel therapy for metastatic breast cancer in mouse models. PLoS ONE 2010, 5, e10365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Liao, S.; Diop-Frimpong, B.; Chen, W.; Goel, S.; Naxerova, K.; Ancukiewicz, M.; Boucher, Y.; Jain, R.K.; Xu, L. TGF-beta blockade improves the distribution and efficacy of therapeutics in breast carcinoma by normalizing the tumor stroma. Proc. Natl. Acad. Sci. USA 2012, 109, 16618–16623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, C.D.; Parvani, J.G.; Schiemann, W.P. The relevance of the TGF-beta Paradox to EMT-MET programs. Cancer Lett. 2013, 341, 30–40. [Google Scholar] [CrossRef] [Green Version]
- Goswami, R.; Subramanian, G.; Silayeva, L.; Newkirk, I.; Doctor, D.; Chawla, K.; Chattopadhyay, S.; Chandra, D.; Chilukuri, N.; Betapudi, V. Gene Therapy Leaves a Vicious Cycle. Front. Oncol. 2019, 9, 297. [Google Scholar] [CrossRef]
- Foucquier, J.; Guedj, M. Analysis of drug combinations: Current methodological landscape. Pharmacol. Res. Perspect. 2015, 3, e00149. [Google Scholar] [CrossRef]
- Vakil, V.; Trappe, W. Drug Combinations: Mathematical Modeling and Networking Methods. Pharmaceutics 2019, 11, e208. [Google Scholar] [CrossRef] [Green Version]
- Griner, L.A.M.; Guha, R.; Shinn, P.; Young, R.M.; Keller, J.M.; Liu, D.; Goldlust, I.S.; Yasgar, A.; McKnight, C.; Boxer, M.B.J. High-throughput combinatorial screening identifies drugs that cooperate with ibrutinib to kill activated B-cell–like diffuse large B-cell lymphoma cells. Proc. Natl. Acad. Sci. USA 2014, 111, 2349–2354. [Google Scholar] [CrossRef] [Green Version]
- Lehár, J.; Krueger, A.S.; Avery, W.; Heilbut, A.M.; Johansen, L.M.; Price, E.R.; Rickles, R.J.; Short Iii, G.F.; Staunton, J.E.; Jin, X.; et al. Synergistic drug combinations tend to improve therapeutically relevant selectivity. Nat. Biotechnol. 2009, 27, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Amzallag, A.; Ramaswamy, S.; Benes, C.H. Statistical assessment and visualization of synergies for large-scale sparse drug combination datasets. BMC Bioinform. 2019, 20, 83. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CMap Classes | Sets of Compound Perturbagens with Enrichment Scores above 90 (Similar) and below -90 (Opposing) | Pharmacologic included Drug Numbers |
|---|---|---|
| Topoisomerase inhibitor | 94.01 | 16 |
| ATPase inhibitor | 92.45 | 16 |
| TGF beta receptor inhibitor | −92.12 | 4 |
| FGFR inhibitor | −94.27 | 4 |
| Bile acid | −94.89 | 4 |
| MDM inhibitor | −99.78 | 4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhatia, S.; Blick, T.; Pinto, C.; Waltham, M.; Monkman, J.; Ivanova, E.; Pollock, P.M.; Nagaraj, S.H.; Wiegmans, A.P.; Haviv, I.; et al. Identifying Therapies to Combat Epithelial Mesenchymal Plasticity-Associated Chemoresistance to Conventional Breast Cancer Therapies Using An shRNA Library Screen. Cancers 2020, 12, 1123. https://doi.org/10.3390/cancers12051123
Bhatia S, Blick T, Pinto C, Waltham M, Monkman J, Ivanova E, Pollock PM, Nagaraj SH, Wiegmans AP, Haviv I, et al. Identifying Therapies to Combat Epithelial Mesenchymal Plasticity-Associated Chemoresistance to Conventional Breast Cancer Therapies Using An shRNA Library Screen. Cancers. 2020; 12(5):1123. https://doi.org/10.3390/cancers12051123
Chicago/Turabian StyleBhatia, Sugandha, Tony Blick, Cletus Pinto, Mark Waltham, James Monkman, Ekaterina Ivanova, Pamela M. Pollock, Shivashankar H. Nagaraj, Adrian P. Wiegmans, Izhak Haviv, and et al. 2020. "Identifying Therapies to Combat Epithelial Mesenchymal Plasticity-Associated Chemoresistance to Conventional Breast Cancer Therapies Using An shRNA Library Screen" Cancers 12, no. 5: 1123. https://doi.org/10.3390/cancers12051123