The Hepatitis B Virus Pre-Core Protein p22 Activates Wnt Signaling

 , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
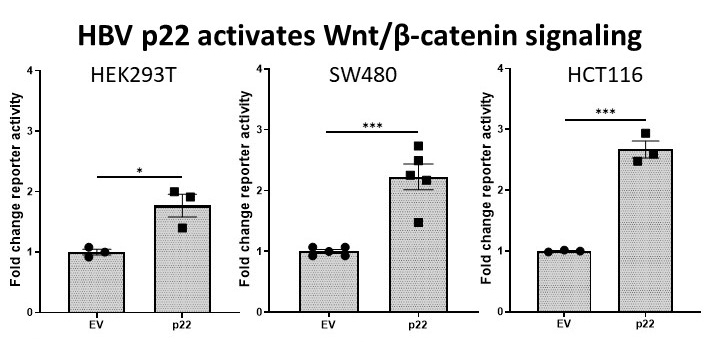
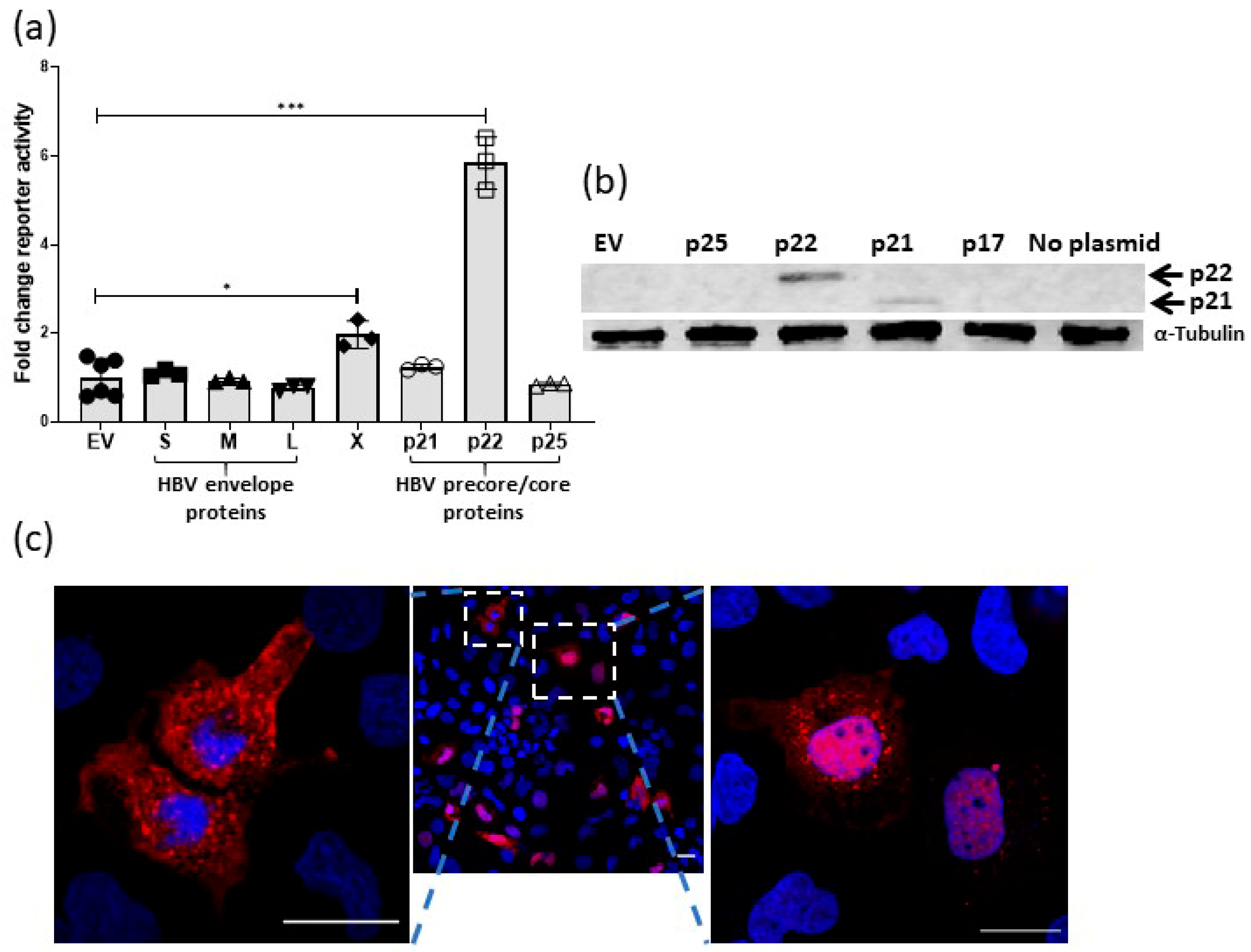
2.1. Effect of HBV Proteins on TCF-β-Catenin Transcription
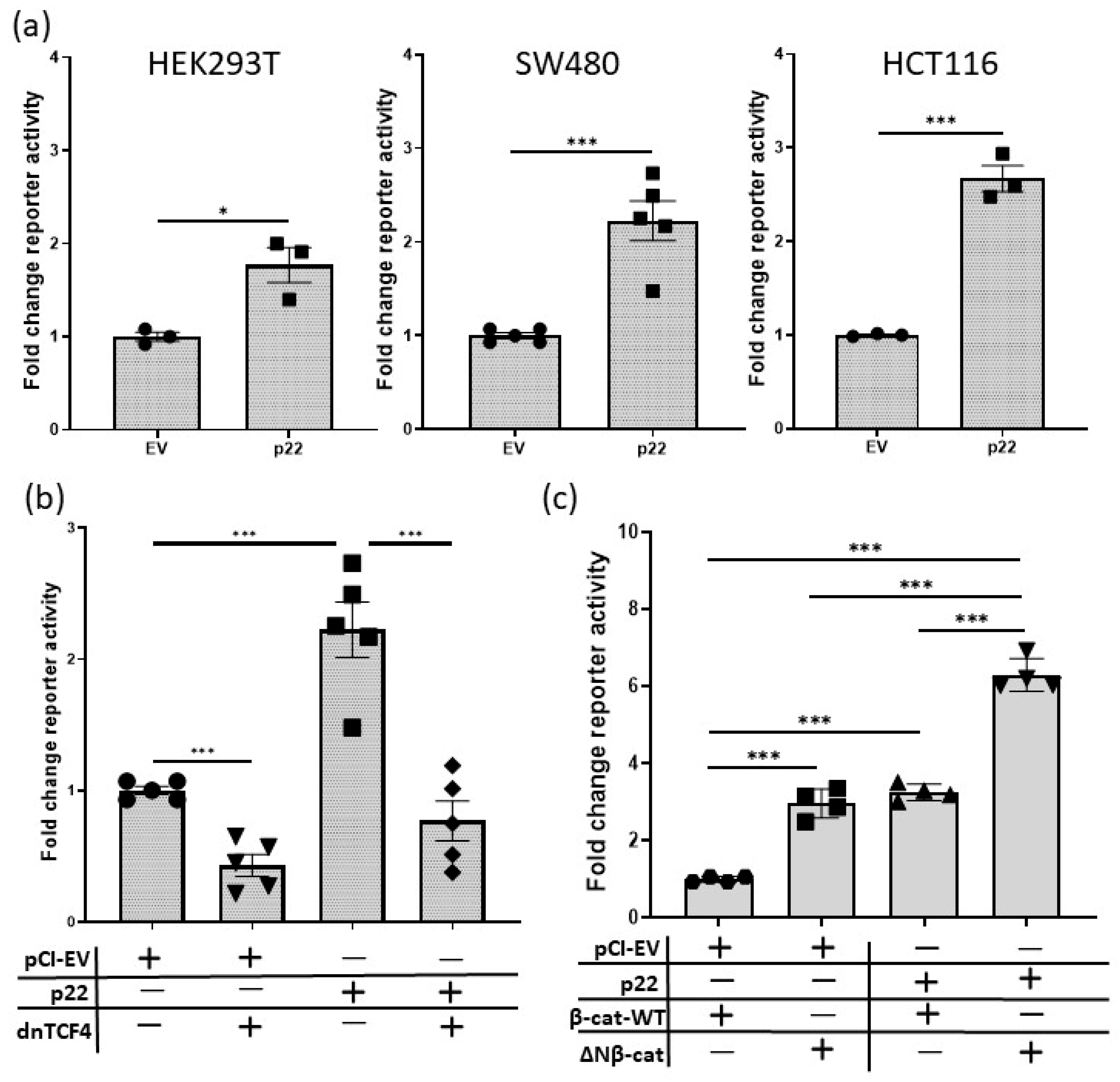
2.2. HBV p22 Activates TCF-β-Catenin Transcription
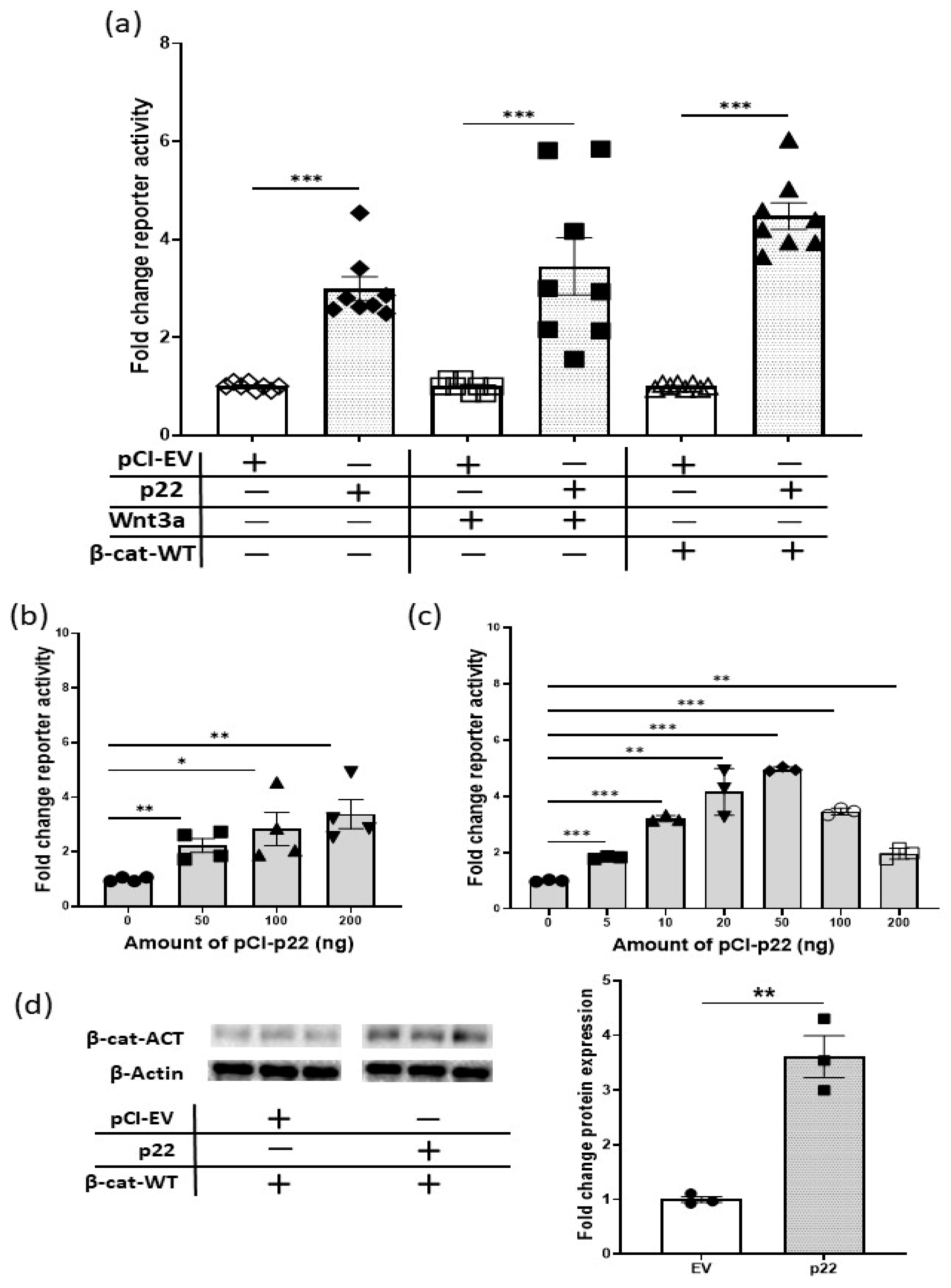
2.3. HBV p22 Activates Native TCF/β-Catenin Promoters
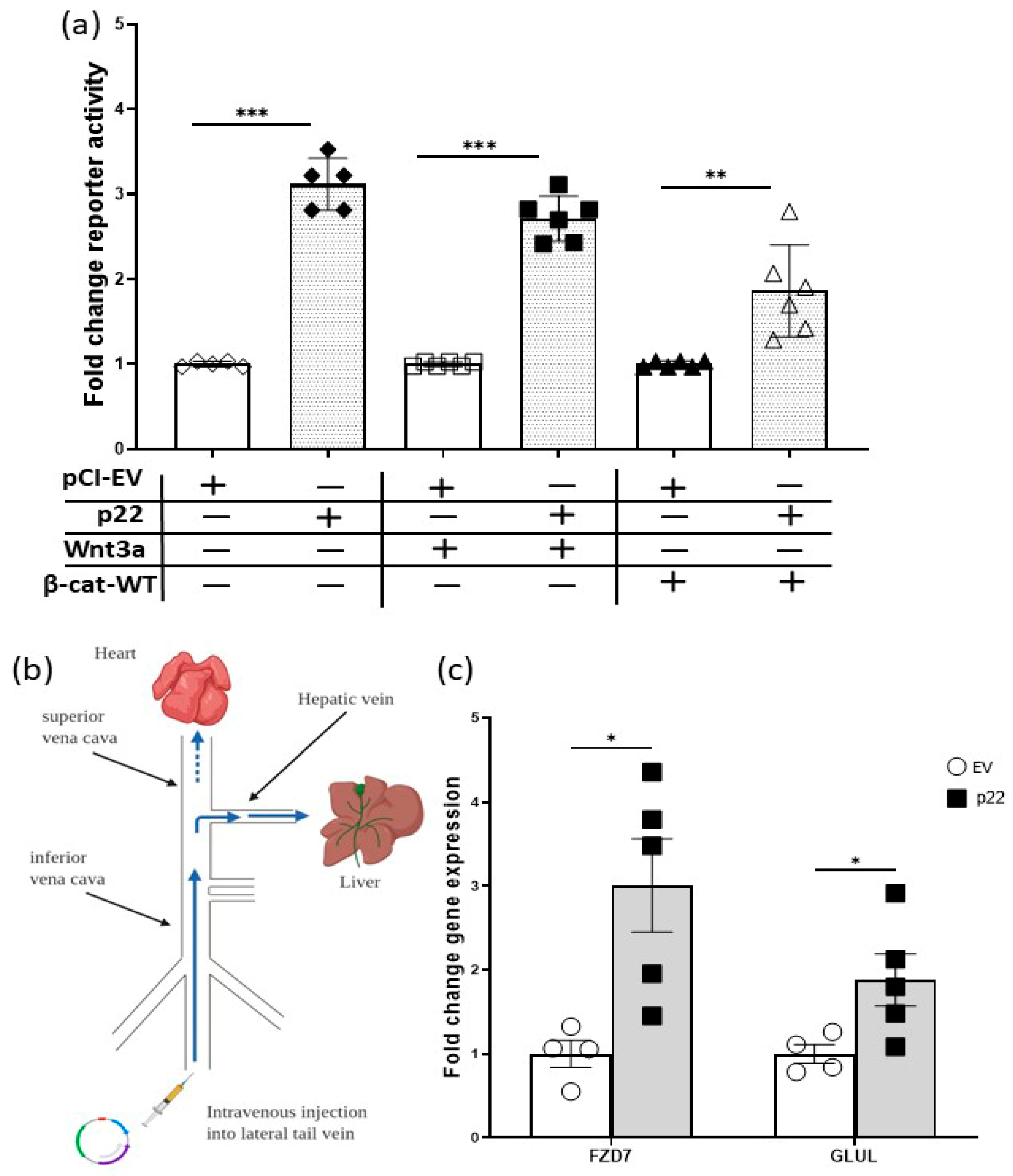
2.4. HBV p22 Activates TCF/β-Catenin Transcription in Addition to a Mutation to Downstream Wnt Pathway Components
3. Discussion
4. Materials and Methods
4.1. Hydrodynamic Injection of Mice
4.2. RNA Extraction and Quantitative RT-PCR (qRT-PCR)
4.3. Cell Lines and Wnt3a Conditioned Medium
4.4. Transfection and Reporter Assays
4.5. Immunoblot Analysis
4.6. Immunofluorescenc Confocal Microscopy
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cancer Research UK. Liver Cancer Statisitcs. Available online: https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/liver-cancer (accessed on 10 September 2019).
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; De Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Schulze, K.; Imbeaud, S.; Letouzé, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet 2015, 47, 505–511. [Google Scholar] [CrossRef] [PubMed]
- De La Coste, A.; Romagnolo, B.; Billuart, P.; Renard, C.A.; Buendia, M.A.; Soubrane, O.; Fabre, M.; Chelly, J.; Beldjord, C.; Kahn, A.; et al. Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc. Natl. Acad. Sci. USA 1998, 95, 8847–8851. [Google Scholar] [CrossRef] [Green Version]
- Nusse, R.; Clevers, H. Wnt/beta-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef] [PubMed]
- Van Noort, M.; van de Wetering, M.; Clevers, H. Identification of Two Novel Regulated Serines in the N Terminus of beta-Catenin. Exp. Cell Res. 2002, 276, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Dale, T. Wnt signal transduction: Kinase cogs in a nano-machine? Trends Biochem. Sci. 2002, 27, 327–329. [Google Scholar] [CrossRef]
- Levrero, M.; Zucman-Rossi, J. Mechanisms of HBV-induced hepatocellular carcinoma. J. Hepatol. 2016, 64 (Suppl. 1), S84–S101. [Google Scholar] [CrossRef]
- Lyou, Y.; Habowski, A.N.; Chen, G.T.; Waterman, M.L. Inhibition of nuclear Wnt signalling: Challenges of an elusive target for cancer therapy. Br. J. Pharmacol. 2017, 174, 4589–4599. [Google Scholar] [CrossRef]
- Kim, S.S.; Cho, H.J.; Lee, H.Y.; Park, J.H.; Noh, C.K.; Shin, S.J.; Lee, K.M.; Yoo, B.M.; Lee, K.J.; Cho, S.W.; et al. Genetic polymorphisms in the Wnt/beta-catenin pathway genes as predictors of tumor development and survival in patients with hepatitis B virus-associated hepatocellular carcinoma. Clin. Biochem. 2016, 49, 792–801. [Google Scholar] [CrossRef]
- MacLachlan, J.H.; Locarnini, S.; Cowie, B.C. Estimating the global prevalence of hepatitis B. Lancet 2015, 386, 1515–1517. [Google Scholar] [CrossRef]
- Garcia, P.D.; Ou, J.H.; Rutter, W.J.; Walter, P. Targeting of the hepatitis B virus precore protein to the endoplasmic reticulum membrane: After signal peptide cleavage translocation can be aborted and the product released into the cytoplasm. J. Cell Biol. 1988, 106, 1093–1104. [Google Scholar] [CrossRef] [PubMed]
- Geng, M.; Xin, X.; Bi, L.Q.; Zhou, L.T.; Liu, X.H. Molecular mechanism of hepatitis B virus X protein function in hepatocarcinogenesis. World J. Gastroenterol. 2015, 21, 10732–10738. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, D.J.; Vincan, E.; Phesse, T.J. Winding back Wnt signalling: Potential therapeutic targets for treating gastric cancers. Br. J. Pharmacol. 2017, 174, 4666–4683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincan, E.; Barker, N. The upstream components of the Wnt signalling pathway in the dynamic EMT and MET associated with colorectal cancer progression. Clin. Exp. Metastasis 2008, 25, 657–663. [Google Scholar] [CrossRef]
- Flanagan, D.J.; Vincan, E.; Phesse, T.J. Wnt Signaling in Cancer: Not a Binary ON: OFF Switch. Cancer Res. 2019, 79, 5901–5906. [Google Scholar] [CrossRef]
- Biechele, T.L.; Moon, R.T. Assaying beta-catenin/TCF transcription with beta-catenin/TCF transcription-based reporter constructs. Methods Mol. Biol. 2008, 468, 99–110. [Google Scholar]
- Korinek, V.; Barker, N.; Morin, P.J.; Van Wichen, D.; De Weger, R.; Kinzler, K.W.; Vogelstein, B.; Clevers, H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/- colon carcinoma. Science 1997, 275, 1784–1787. [Google Scholar] [CrossRef] [Green Version]
- Mitra, B.; Wang, J.; Kim, E.S.; Mao, R.; Dong, M.; Liu, Y.; Zhang, J.; Guo, H. Hepatitis B Virus Precore Protein p22 Inhibits Alpha Interferon Signaling by Blocking STAT Nuclear Translocation. J. Virol. 2019, 93, e00196-19. [Google Scholar] [CrossRef] [Green Version]
- Gammons, M.; Bienz, M. Multiprotein complexes governing Wnt signal transduction. Curr. Opin. Cell Biol. 2018, 51, 42–49. [Google Scholar] [CrossRef]
- Van Noort, M.; Meeldijk, J.; van der Zee, R.; Destree, O.; Clevers, H. Wnt signaling controls the phosphorylation status of beta-catenin. J. Biol. Chem. 2002, 277, 17901–17905. [Google Scholar] [CrossRef] [Green Version]
- Van Noort, M.; Weerkamp, F.; Clevers, H.C.; Staal, F.J. Wnt signaling and phosphorylation status of beta-catenin: Importance of the correct antibody tools. Blood 2007, 110, 2778–2779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincan, E.; Flanagan, D.J.; Pouliot, N.; Brabletz, T.; Spaderna, S. Variable FZD7 expression in colorectal cancers indicates regulation by the tumour microenvironment. Dev. Dyn. 2010, 239, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Willert, J.; Epping, M.; Pollack, J.R.; Brown, P.O.; Nusse, R. A transcriptional response to Wnt protein in human embryonic carcinoma cells. BMC Dev. Biol. 2002, 2, 8–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phesse, T.; Flanagan, D.; Vincan, E. Frizzled7: A Promising Achilles’ Heel for Targeting the Wnt Receptor Complex to Treat Cancer. Cancers 2016, 8, 50. [Google Scholar] [CrossRef] [Green Version]
- Merle, P.; Kim, M.; Herrmann, M.; Gupte, A.; Lefrançois, L.; Califano, S.; Tre, C.; Tanaka, S.; Vitvitski, L.; de la Monte, S.; et al. Oncogenic role of the frizzled-7/beta-catenin pathway in hepatocellular carcinoma. J. Hepatol. 2005, 43, 854–862. [Google Scholar] [CrossRef]
- Torresi, J.; Tran, B.M.; Christiansen, D.; Earnest-Silveira, L.; Schwab, R.H.M.; Vincan, E. HBV-related hepatocarcinogenesis: The role of signalling pathways and innovative ex vivo research models. BMC Cancer 2019, 19, 707. [Google Scholar] [CrossRef] [Green Version]
- Van Amerongen, R.; Nusse, R. Towards an integrated view of Wnt signaling in development. Development 2009, 136, 3205–3214. [Google Scholar] [CrossRef] [Green Version]
- Ebert, G.; Preston, S.; Allison, C.; Cooney, J.; Toe, J.G.; Stutz, M.D.; Ojaimi, S.; Scott, H.W.; Baschuk, N.; Nachbur, U.; et al. Cellular inhibitor of apoptosis proteins prevent clearance of hepatitis B virus. Proc. Natl. Acad. Sci. USA 2015, 112, 5797–5802. [Google Scholar] [CrossRef] [Green Version]
- Cole, A.M.; Myant, K.; Reed, K.R.; Ridgway, R.A.; Athineos, D.; Van den Brink, G.R.; Muncan, V.; Clevers, H.; Clarke, A.R.; Sicinski, P.; et al. Cyclin D2-cyclin-dependent kinase 4/6 is required for efficient proliferation and tumorigenesis following Apc loss. Cancer Res. 2010, 70, 8149–8158. [Google Scholar] [CrossRef] [Green Version]
- Casari, C.; Lenting, P.J.; Christophe, O.D.; Denis, C.V. Von Willebrand Factor Abnormalities Studied in the Mouse Model: What We Learned about VWF Functions. Mediterr. J. Hematol. Infect. Dis. 2013, 5, e2013047. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Morin, P.J.; Sparks, A.B.; Korinek, V.; Barker, N.; Clevers, H.; Vogelstein, B.; Kinzler, K.W. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 1997, 275, 1787–1790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willert, K.; Brown, J.D.; Danenberg, E.; Duncan, A.W.; Weissman, I.L.; Reya, T.; Yates, J.R.; Nusse, R. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature 2003, 423, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensivemolecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Harada, N.; Tamai, Y.; Ishikawa, T.O.; Sauer, B.; Takaku, K.; Oshima, M.; Taketo, M.M. Intestinal polyposis in mice with a dominant stable mutation of the beta-catenin gene. EMBO J. 1999, 18, 5931–5942. [Google Scholar] [CrossRef]
- Su, L.K.; Kinzler, K.W.; Vogelstein, B.; Preisinger, A.C.; Moser, A.R.; Luongo, C.; Gould, K.A.; Dove, W.F. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science 1992, 256, 668–670. [Google Scholar] [CrossRef]
- Flanagan, D.J.; Barker, N.; Di Costanzo, N.S.; Mason, E.A.; Gurney, A.; Meniel, V.S.; Koushyar, S.; Austin, C.R.; Ernst, M.; Pearson, H.B.; et al. Frizzled-7 Is Required for Wnt Signaling in Gastric Tumors with and Without Apc Mutations. Cancer Res. 2019, 79, 970–981. [Google Scholar] [CrossRef] [Green Version]
- Bengochea, A.; De Souza, M.M.; Lefrancois, L.; Le Roux, E.; Galy, O.; Chemin, I.; Kim, M.; Wands, J.R.; Trepo, C.; Hainaut, P.; et al. Common dysregulation of Wnt/Frizzled receptor elements in human hepatocellular carcinoma. Br. J. Cancer 2008, 99, 143–150. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.I.; Lu, S.N.; Liaw, Y.F.; You, S.L.; Sun, C.A.; Wang, L.Y.; Hsiao, C.K.; Chen, P.J.; Chen, D.S.; Chen, C.J. Hepatitis B e antigen and the risk of hepatocellular carcinoma. N. Engl. J. Med. 2002, 347, 168–174. [Google Scholar] [CrossRef] [Green Version]
- You, S.L.; Yang, H.I.; Chen, C.J. Seropositivity of hepatitis B e antigen and hepatocellular carcinoma. Ann. Med. 2004, 36, 215–224. [Google Scholar] [CrossRef]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 2012, 1, e00049. [Google Scholar] [CrossRef] [PubMed]
- Burke, Z.D.; Reed, K.R.; Phesse, T.J.; Sansom, O.J.; Clarke, A.R.; Tosh, D. Liver zonation occurs through a beta-catenin-dependent, c-Myc-independent mechanism. Gastroenterology 2009, 136, e1–e3. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T.; Jung, A.; Hermann, K.; Günther, K.; Hohenberger, W.; Kirchner, T. Nuclear overexpression of the oncoprotein beta-catenin in colorectal cancer is localized predominantly at the invasion front. Pathol. Res. Pract. 1998, 194, 701–704. [Google Scholar] [CrossRef]
- Brabletz, T.; Jung, A.; Reu, S.; Porzner, M.; Hlubek, F.; Kunz-Schughart, L.A.; Knuechel, R.; Kirchner, T. Variable beta-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc. Natl. Acad. Sci. USA 2001, 98, 10356–10361. [Google Scholar] [CrossRef] [Green Version]
- Vincan, E.; Darcy, P.K.; Farrelly, C.A.; Faux, M.C.; Brabletz, T.; Ramsay, R.G. Frizzled-7 dictates three-dimensional organization of colorectal cancer cell carcinoids. Oncogene 2007, 26, 2340–2352. [Google Scholar] [CrossRef] [Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tran, B.M.; Flanagan, D.J.; Ebert, G.; Warner, N.; Tran, H.; Fifis, T.; Kastrappis, G.; Christophi, C.; Pellegrini, M.; Torresi, J.; et al. The Hepatitis B Virus Pre-Core Protein p22 Activates Wnt Signaling. Cancers 2020, 12, 1435. https://doi.org/10.3390/cancers12061435
Tran BM, Flanagan DJ, Ebert G, Warner N, Tran H, Fifis T, Kastrappis G, Christophi C, Pellegrini M, Torresi J, et al. The Hepatitis B Virus Pre-Core Protein p22 Activates Wnt Signaling. Cancers. 2020; 12(6):1435. https://doi.org/10.3390/cancers12061435
Chicago/Turabian StyleTran, Bang Manh, Dustin James Flanagan, Gregor Ebert, Nadia Warner, Hoanh Tran, Theodora Fifis, Georgios Kastrappis, Christopher Christophi, Marc Pellegrini, Joseph Torresi, and et al. 2020. "The Hepatitis B Virus Pre-Core Protein p22 Activates Wnt Signaling" Cancers 12, no. 6: 1435. https://doi.org/10.3390/cancers12061435