Restoration of MHC-I on Tumor Cells by Fhit Transfection Promotes Immune Rejection and Acts as an Individualized Immunotherapeutic Vaccine


Abstract
:1. Introduction
2. Results
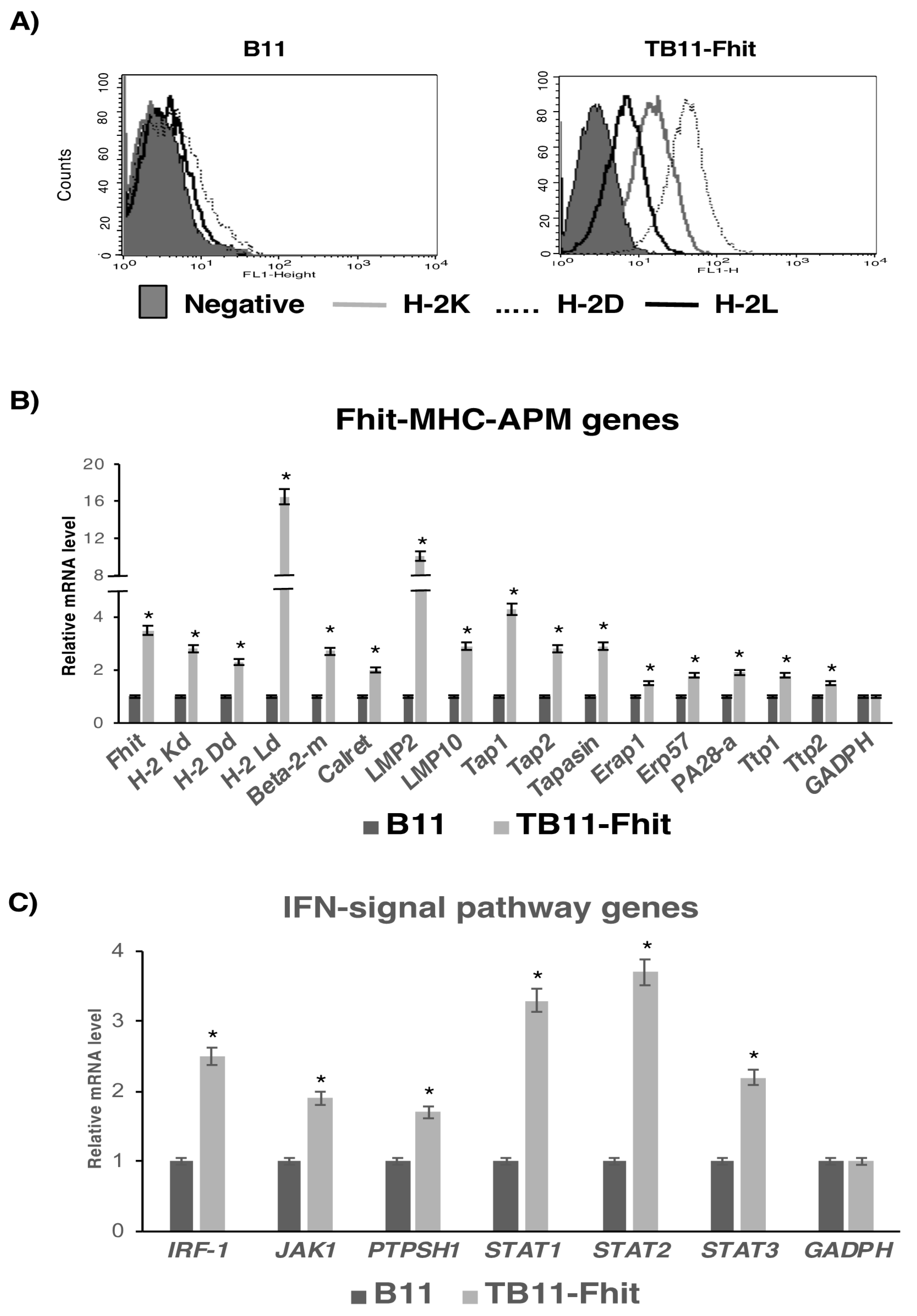
2.1. Stable Fhit Gene Transfection Recovers MHC-I Expression on B9 and B11 Tumor Cells
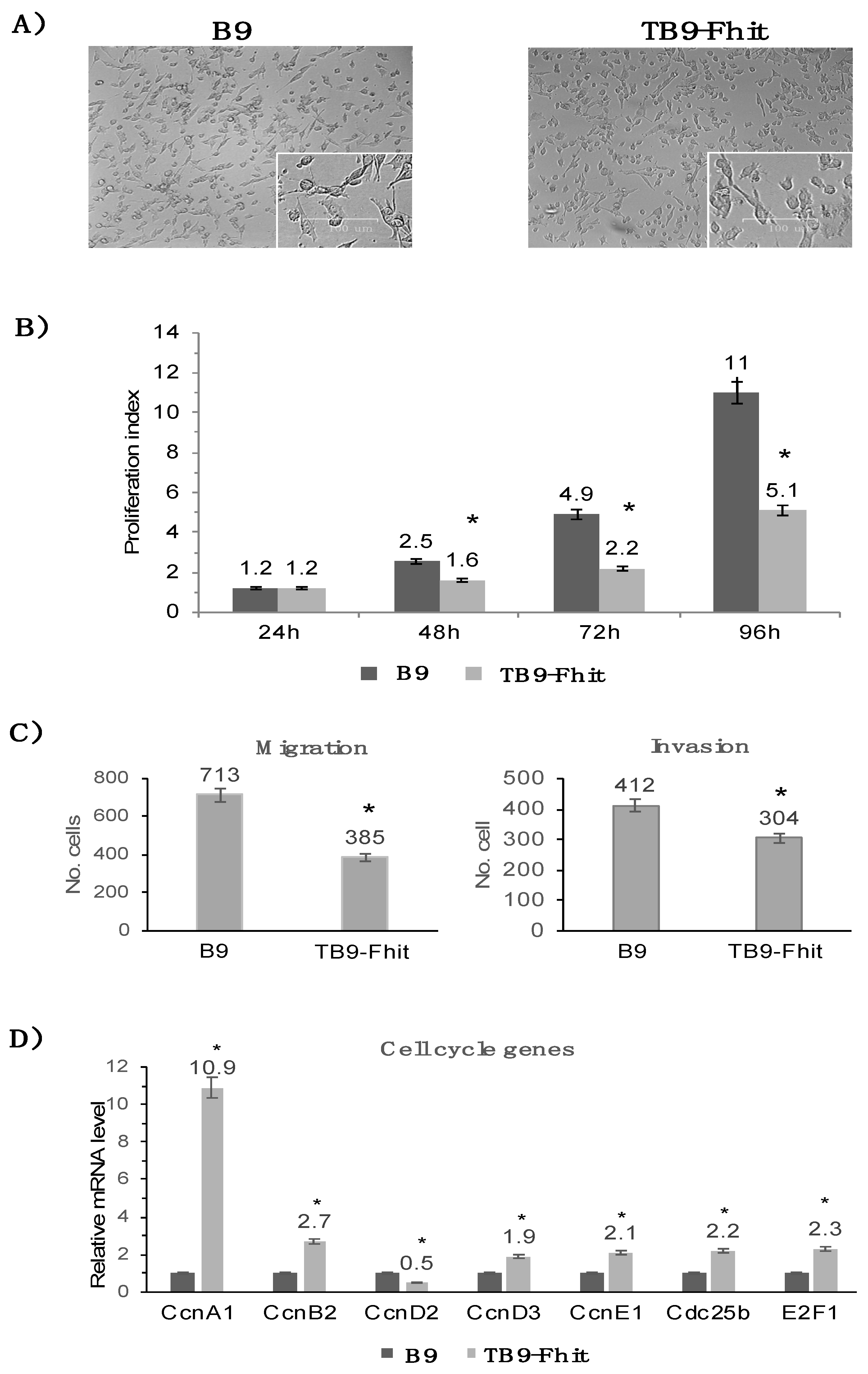
2.2. Fhit Gene Transfection Decreased the In Vitro Proliferation, Migration, and Invasion of Tumor Cells
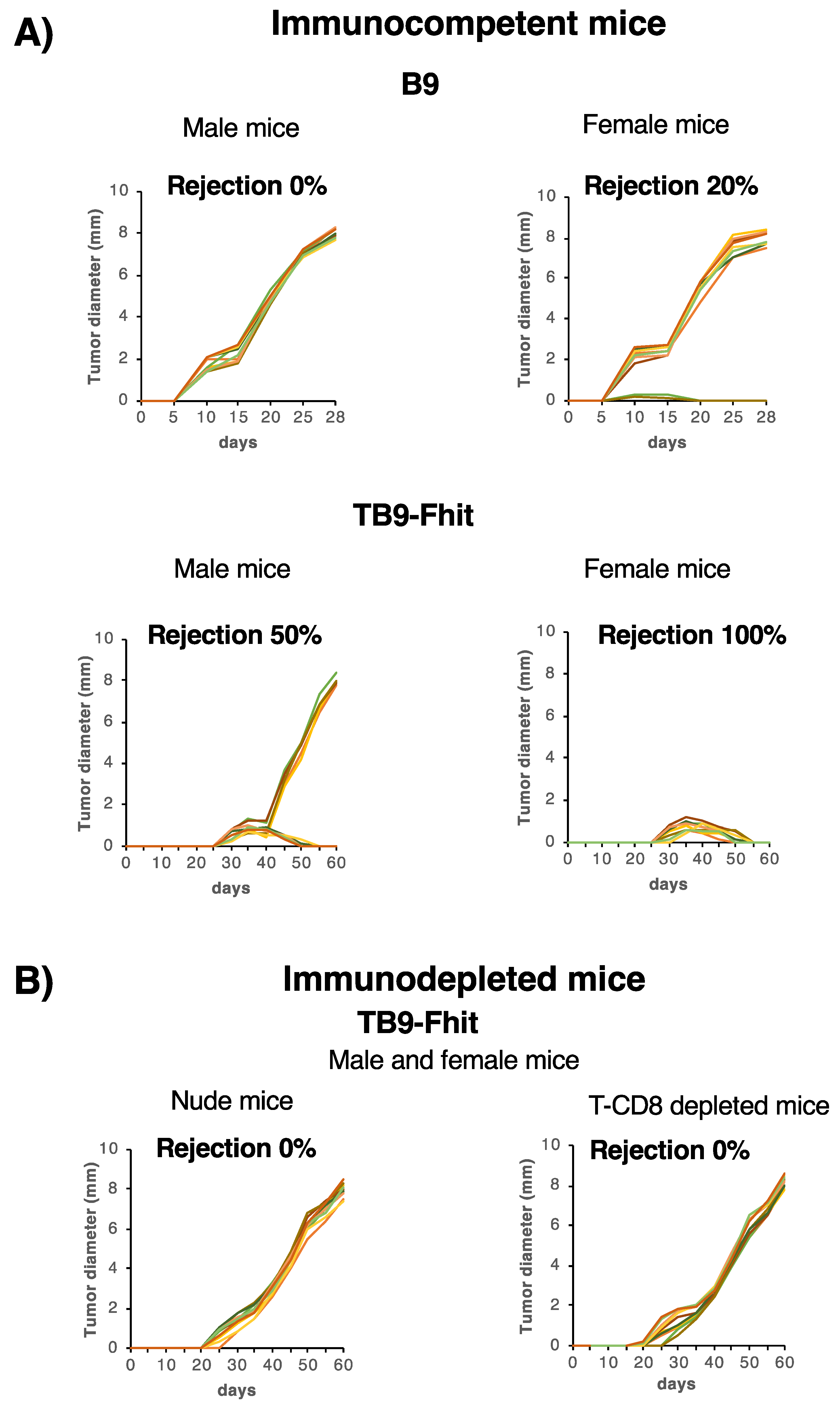
2.3. Fhit-Transfected Tumor Cells Were Immune-Rejected In Vivo by Syngeneic Immunocompetent Hosts, with Differences between the Sexes
2.4. Changes in Immune Cell Subpopulations Produced by Fhit-Transfected Tumor Cells in Female and Male Immunocompetent Mice
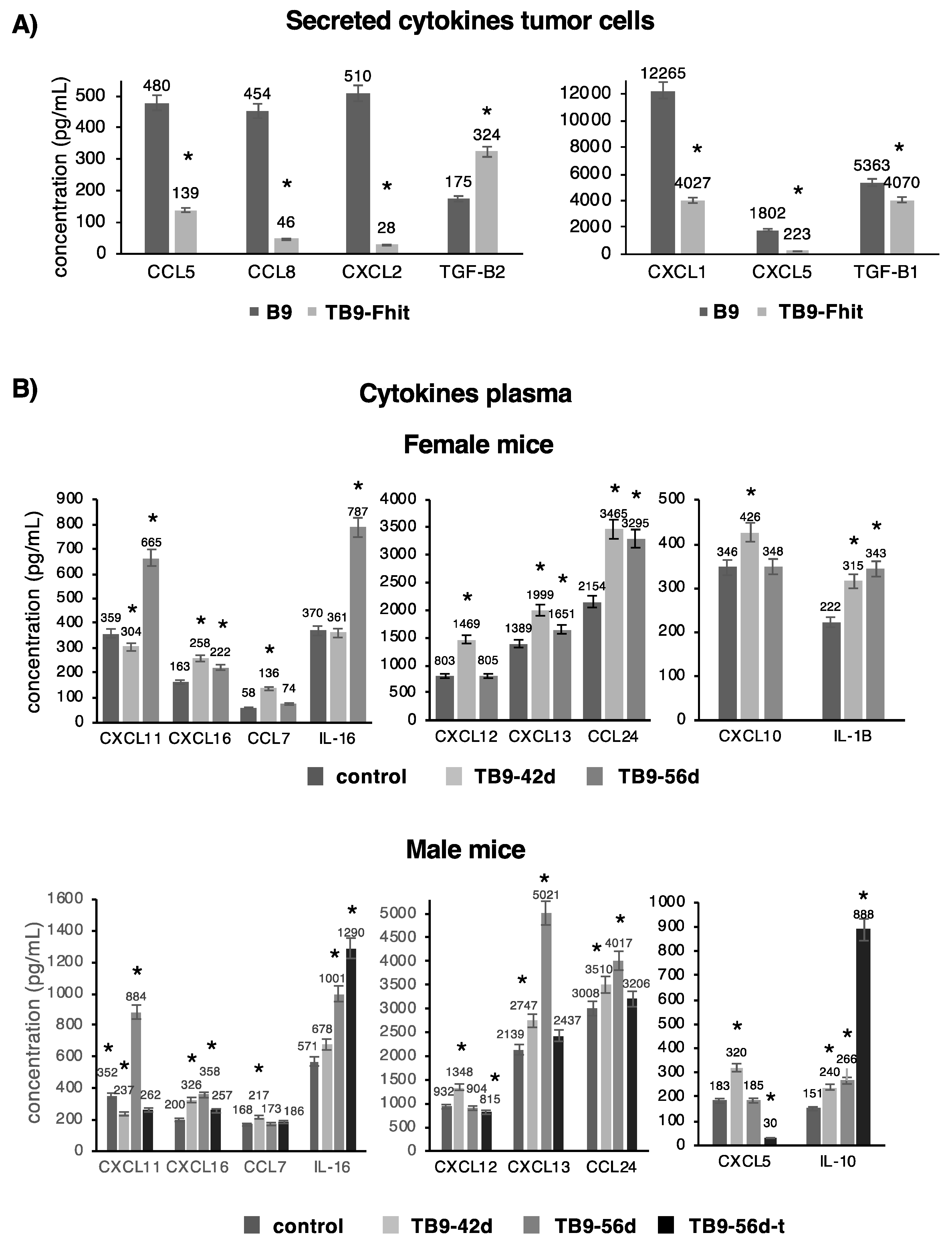
2.5. Changes in the Secretion of Chemokines/Cytokines by the Tumor Cells and Their Presence in the Plasma of Mice Inoculated with Fhit-Transfected Tumor Cells
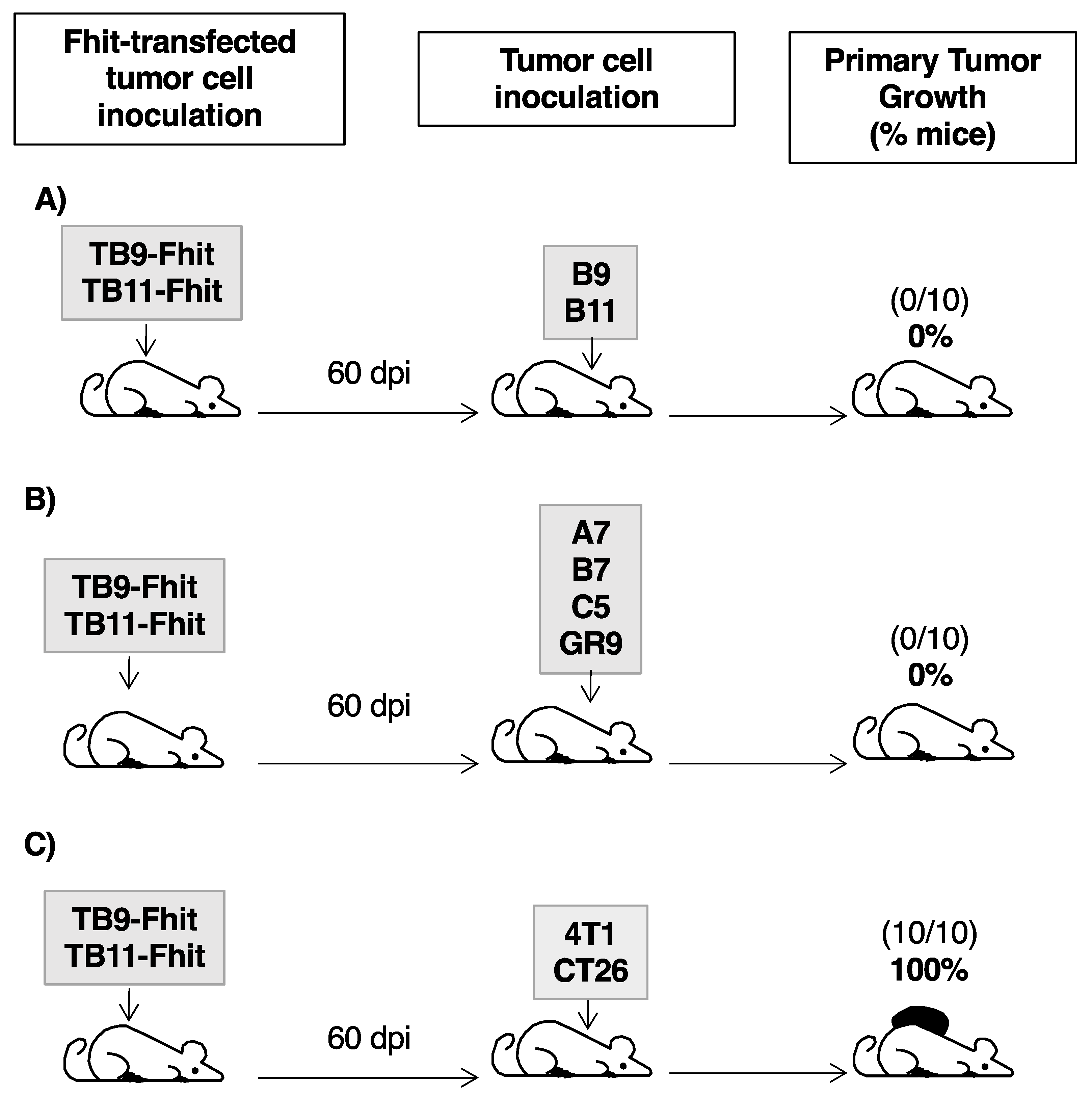
2.6. Fhit-Transfected Tumor Cells Act as Individualized Immunotherapeutic Vaccine against GR9 Tumor Cell Clones
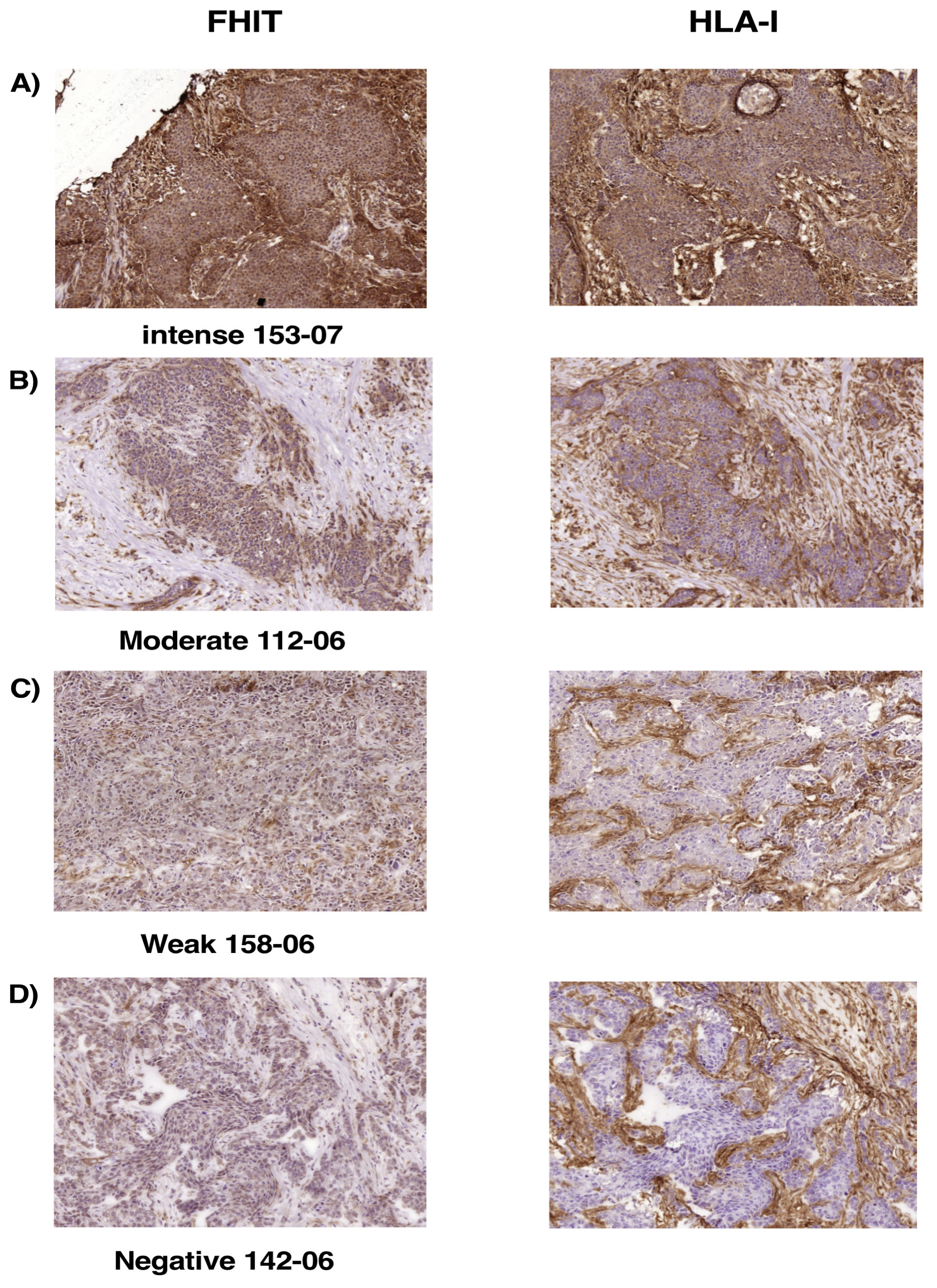
2.7. Loss of FHIT Expression in Human Breast Tumors Is Associated with Complete Loss or Downregulation of MHC-I Expression
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Cell Lines and Stimulation
4.3. Cloning of Fhit cDNA and Transfection into B11 Cells
4.4. MHC Class I and MHC Class II Cell Surface Expression
4.5. RT and Quantitative Real-Time PCR
4.6. Cell Proliferation, Migration, and Invasion Assays
4.7. In Vitro Assessment of Apoptosis
4.8. In Vivo Local Tumor Growth Assay
4.9. Spectral Kariotyping
4.10. Determination of Cytokine Levels
4.11. Isolation of Splenic Leukocytes
4.12. Flow Cytometry Analysis of Immune Cell Subsets
4.13. Assays of Immune Protection
4.14. Immunohistochemistry Analysis of HLA-I and FHIT Expression in Human Breast Cancers
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Finn, O.J. A Believer’s Overview of Cancer Immunosurveillance and Immunotherapy. J. Immunol. 2018, 200, 385–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanitis, E.; Dangaj, D.; Irving, M.; Coukos, G. Mechanisms regulating T-cell infiltration and activity in solid tumors. Ann. Oncol. 2017, 28, xii18–xii32. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Shalabi, A.; Hubbard-Lucey, V.M. Comprehensive analysis of the clinical immuno-oncology landscape. Ann. Oncol. 2018, 29, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Goedegebuure, S.P.; Gillanders, W.E. Preclinical and clinical development of neoantigen vaccines. Ann. Oncol. 2017, 28, xii11–xii17. [Google Scholar] [CrossRef]
- Sanchez-Paulete, A.R.; Teijeira, A.; Cueto, F.J.; Garasa, S.; Perez-Gracia, J.L.; Sanchez-Arraez, A.; Sancho, D.; Melero, I. Antigen cross-presentation and T-cell cross-priming in cancer immunology and immunotherapy. Ann. Oncol. 2017, 28, xii74. [Google Scholar] [CrossRef]
- Houot, R.; Schultz, L.M.; Marabelle, A.; Kohrt, H. T-cell-based Immunotherapy: Adoptive Cell Transfer and Checkpoint Inhibition. Cancer Immunol. Res. 2015, 3, 1115–1122. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.C.; Rosenberg, S.A. Adoptive T-Cell Therapy for Cancer. Adv. Immunol. 2016, 130, 279–294. [Google Scholar] [CrossRef]
- Garrido, F.; Ruiz-Cabello, F.; Cabrera, T.; Perez-Villar, J.J.; Lopez-Botet, M.; Duggan-Keen, M.; Stern, P.L. Implications for immunosurveillance of altered HLA class I phenotypes in human tumours. Immunol. Today 1997, 18, 89–95. [Google Scholar] [CrossRef]
- Gettinger, S.; Choi, J.; Hastings, K.; Truini, A.; Datar, I.; Sowell, R.; Wurtz, A.; Dong, W.; Cai, G.; Melnick, M.A.; et al. Impaired HLA Class I Antigen Processing and Presentation as a Mechanism of Acquired Resistance to Immune Checkpoint Inhibitors in Lung Cancer. Cancer Discov. 2017, 7, 1420–1435. [Google Scholar] [CrossRef] [Green Version]
- Paulson, K.G.; Voillet, V.; McAfee, M.S.; Hunter, D.S.; Wagener, F.D.; Perdicchio, M.; Valente, W.J.; Koelle, S.J.; Church, C.D.; Vandeven, N.; et al. Acquired cancer resistance to combination immunotherapy from transcriptional loss of class I HLA. Nat. Commun. 2018, 9, 3868. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Lora, A.; Algarra, I.; Garrido, F. MHC class I antigens, immune surveillance, and tumor immune escape. J. Cell. Physiol. 2003, 195, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Garrido, C.; Algarra, I.; Maleno, I.; Stefanski, J.; Collado, A.; Garrido, F.; Garcia-Lora, A.M. Alterations of HLA class I expression in human melanoma xenografts in immunodeficient mice occur frequently and are associated with higher tumorigenicity. Cancer Immunol. Immunother. 2010, 59, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Garrido, F.; Cabrera, T.; Aptsiauri, N. “Hard” and “soft” lesions underlying the HLA class I alterations in cancer cells: Implications for immunotherapy. Int. J. Cancer 2010, 127, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Ennishi, D.; Takata, K.; Beguelin, W.; Duns, G.; Mottok, A.; Farinha, P.; Bashashati, A.; Saberi, S.; Boyle, M.; Meissner, B.; et al. Molecular and Genetic Characterization of MHC Deficiency Identifies EZH2 as Therapeutic Target for Enhancing Immune Recognition. Cancer Discov. 2019, 9, 546–563. [Google Scholar] [CrossRef] [Green Version]
- Romero, I.; Martinez, M.; Garrido, C.; Collado, A.; Algarra, I.; Garrido, F.; Garcia-Lora, A.M. The tumour suppressor Fhit positively regulates MHC class I expression on cancer cells. J. Pathol. 2012, 227, 367–379. [Google Scholar] [CrossRef]
- Garcia-Lora, A.; Martinez, M.; Algarra, I.; Gaforio, J.J.; Garrido, F. MHC class I-deficient metastatic tumor variants immunoselected by T lymphocytes originate from the coordinated downregulation of APM components. Int. J. Cancer 2003, 106, 521–527. [Google Scholar] [CrossRef]
- Ohta, M.; Inoue, H.; Cotticelli, M.G.; Kastury, K.; Baffa, R.; Palazzo, J.; Siprashvili, Z.; Mori, M.; McCue, P.; Druck, T.; et al. The FHIT gene, spanning the chromosome 3p14.2 fragile site and renal carcinoma-associated t(3;8) breakpoint, is abnormal in digestive tract cancers. Cell 1996, 84, 587–597. [Google Scholar] [CrossRef] [Green Version]
- Campiglio, M.; Pekarsky, Y.; Menard, S.; Tagliabue, E.; Pilotti, S.; Croce, C.M. FHIT loss of function in human primary breast cancer correlates with advanced stage of the disease. Cancer Res. 1999, 59, 3866–3869. [Google Scholar]
- Verri, C.; Roz, L.; Conte, D.; Liloglou, T.; Livio, A.; Vesin, A.; Fabbri, A.; Andriani, F.; Brambilla, C.; Tavecchio, L.; et al. Fragile histidine triad gene inactivation in lung cancer: The European Early Lung Cancer project. Am. J. Respir. Crit. Care Med. 2009, 179, 396–401. [Google Scholar] [CrossRef] [Green Version]
- Fouts, R.L.; Sandusky, G.E.; Zhang, S.; Eckert, G.J.; Koch, M.O.; Ulbright, T.M.; Eble, J.N.; Cheng, L. Down-regulation of fragile histidine triad expression in prostate carcinoma. Cancer 2003, 97, 1447–1452. [Google Scholar] [CrossRef]
- Tseng, J.E.; Kemp, B.L.; Khuri, F.R.; Kurie, J.M.; Lee, J.S.; Zhou, X.; Liu, D.; Hong, W.K.; Mao, L. Loss of Fhit is frequent in stage I non-small cell lung cancer and in the lungs of chronic smokers. Cancer Res. 1999, 59, 4798–4803. [Google Scholar] [PubMed]
- Rohr, U.P.; Rehfeld, N.; Geddert, H.; Pflugfelder, L.; Bruns, I.; Neukirch, J.; Rohrbeck, A.; Grote, H.J.; Steidl, U.; Fenk, R.; et al. Prognostic relevance of fragile histidine triad protein expression in patients with small cell lung cancer. Clin. Cancer Res. 2005, 11, 180–185. [Google Scholar] [PubMed]
- Yang, Q.; Yoshimura, G.; Suzuma, T.; Tamaki, T.; Umemura, T.; Nakamura, M.; Nakamura, Y.; Wang, X.; Mori, I.; Sakurai, T.; et al. Clinicopathological significance of fragile histidine triad transcription protein expression in breast carcinoma. Clin. Cancer Res. 2001, 7, 3869–3873. [Google Scholar] [PubMed]
- Krivak, T.C.; McBroom, J.W.; Seidman, J.; Venzon, D.; Crothers, B.; MacKoul, P.J.; Rose, G.S.; Carlson, J.W.; Birrer, M.J. Abnormal fragile histidine triad (FHIT) expression in advanced cervical carcinoma: A poor prognostic factor. Cancer Res. 2001, 61, 4382–4385. [Google Scholar]
- Joannes, A.; Bonnomet, A.; Bindels, S.; Polette, M.; Gilles, C.; Burlet, H.; Cutrona, J.; Zahm, J.M.; Birembaut, P.; Nawrocki-Raby, B. Fhit regulates invasion of lung tumor cells. Oncogene 2010, 29, 1203–1213. [Google Scholar] [CrossRef] [Green Version]
- Ishii, H.; Mimori, K.; Inoue, H.; Inageta, T.; Ishikawa, K.; Semba, S.; Druck, T.; Trapasso, F.; Tani, K.; Vecchione, A.; et al. Fhit modulates the DNA damage checkpoint response. Cancer Res. 2006, 66, 11287–11292. [Google Scholar] [CrossRef] [Green Version]
- Otterson, G.A.; Xiao, G.H.; Geradts, J.; Jin, F.; Chen, W.D.; Niklinska, W.; Kaye, F.J.; Yeung, R.S. Protein expression and functional analysis of the FHIT gene in human tumor cells. J. Natl. Cancer Inst. 1998, 90, 426–432. [Google Scholar] [CrossRef] [Green Version]
- Muller, C.Y.; O’Boyle, J.D.; Fong, K.M.; Wistuba, I.I.; Biesterveld, E.; Ahmadian, M.; Miller, D.S.; Gazdar, A.F.; Minna, J.D. Abnormalities of fragile histidine triad genomic and complementary DNAs in cervical cancer: Association with human papillomavirus type. J. Natl. Cancer Inst. 1998, 90, 433–439. [Google Scholar] [CrossRef] [Green Version]
- Wu, R.; Connolly, D.C.; Dunn, R.L.; Cho, K.R. Restored expression of fragile histidine triad protein and tumorigenicity of cervical carcinoma cells. J. Natl. Cancer Inst. 2000, 92, 338–344. [Google Scholar] [CrossRef] [Green Version]
- Romero, I.; Garrido, C.; Algarra, I.; Collado, A.; Garrido, F.; Garcia-Lora, A.M. T lymphocytes restrain spontaneous metastases in permanent dormancy. Cancer Res. 2014, 74, 1958–1968. [Google Scholar] [CrossRef] [Green Version]
- Cabrera, T.; Angustias Fernandez, M.; Sierra, A.; Garrido, A.; Herruzo, A.; Escobedo, A.; Fabra, A.; Garrido, F. High frequency of altered HLA class I phenotypes in invasive breast carcinomas. Hum. Immunol. 1996, 50, 127–134. [Google Scholar] [CrossRef]
- Garrido, M.A.; Rodriguez, T.; Zinchenko, S.; Maleno, I.; Ruiz-Cabello, F.; Concha, A.; Olea, N.; Garrido, F.; Aptsiauri, N. HLA class I alterations in breast carcinoma are associated with a high frequency of the loss of heterozygosity at chromosomes 6 and 15. Immunogenetics 2018, 70, 647–659. [Google Scholar] [CrossRef] [PubMed]
- Garrido, F.; Aptsiauri, N.; Doorduijn, E.M.; Lora, A.M.G.; van Hall, T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr. Opin. Immunol. 2016, 39, 44–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiske, J.; Albring, K.F.; Huber, O. The tumor suppressor Fhit acts as a repressor of beta-catenin transcriptional activity. Proc. Natl. Acad. Sci. USA 2007, 104, 20344–20349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.W.; Lee, M.C.; Hsu, N.Y.; Wu, T.C.; Wu, J.Y.; Wang, Y.C.; Cheng, Y.W.; Chen, C.Y.; Lee, H. FHIT loss confers cisplatin resistance in lung cancer via the AKT/NF-κB/Slug-mediated PUMA reduction. Oncogene 2015, 34, 2505–2515. [Google Scholar] [CrossRef]
- Druck, T.; Cheung, D.G.; Park, D.; Trapasso, F.; Pichiorri, F.; Gaspari, M.; Palumbo, T.; Aqeilan, R.I.; Gaudio, E.; Okumura, H.; et al. Fhit-Fdxr interaction in the mitochondria: Modulation of reactive oxygen species generation and apoptosis in cancer cells. Cell Death Dis. 2019, 10, 147. [Google Scholar] [CrossRef]
- Kiss, D.L.; Baez, W.; Huebner, K.; Bundschuh, R.; Schoenberg, D.R. Impact of FHIT loss on the translation of cancer-associated mRNAs. Mol. Cancer 2017, 16, 179. [Google Scholar] [CrossRef] [Green Version]
- Garrido, C.; Paco, L.; Romero, I.; Berruguilla, E.; Stefansky, J.; Collado, A.; Algarra, I.; Garrido, F.; Garcia-Lora, A.M. MHC class I molecules act as tumor suppressor genes regulating the cell cycle gene expression, invasion and intrinsic tumorigenicity of melanoma cells. Carcinogenesis 2012, 33, 687–693. [Google Scholar] [CrossRef] [Green Version]
- Lin, P.Y.; Sun, L.; Thibodeaux, S.R.; Ludwig, S.M.; Vadlamudi, R.K.; Hurez, V.J.; Bahar, R.; Kious, M.J.; Livi, C.B.; Wall, S.R.; et al. B7-H1-dependent sex-related differences in tumor immunity and immunotherapy responses. J. Immunol. 2010, 185, 2747–2753. [Google Scholar] [CrossRef]
- Ozdemir, B.C.; Dotto, G.P. Sex Hormones and Anticancer Immunity. Clin. Cancer Res. 2019. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Zhang, J.; He, Z.; Wu, K.; Liu, X.S. The predictive power of tumor mutational burden in lung cancer immunotherapy response is influenced by patients’ sex. Int. J. Cancer 2019. [Google Scholar] [CrossRef] [PubMed]
- Capone, I.; Marchetti, P.; Ascierto, P.A.; Malorni, W.; Gabriele, L. Sexual Dimorphism of Immune Responses: A New Perspective in Cancer Immunotherapy. Front. Immunol. 2018, 9, 552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fink, A.L.; Engle, K.; Ursin, R.L.; Tang, W.Y.; Klein, S.L. Biological sex affects vaccine efficacy and protection against influenza in mice. Proc. Natl. Acad. Sci. USA 2018, 115, 12477–12482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero, I.; Garrido, C.; Algarra, I.; Chamorro, V.; Collado, A.; Garrido, F.; Garcia-Lora, A.M. MHC Intratumoral Heterogeneity May Predict Cancer Progression and Response to Immunotherapy. Front. Immunol. 2018, 9, 102. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Lora, A.; Algarra, I.; Gaforio, J.J.; Ruiz-Cabello, F.; Garrido, F. Immunoselection by T lymphocytes generates repeated MHC class I-deficient metastatic tumor variants. Int. J. Cancer 2001, 91, 109–119. [Google Scholar] [CrossRef]
- Garrido, C.; Romero, I.; Berruguilla, E.; Cancela, B.; Algarra, I.; Collado, A.; Garcia-Lora, A.; Garrido, F. Immunotherapy eradicates metastases with reversible defects in MHC class I expression. Cancer Immunol. Immunother. 2011, 60, 1257–1268. [Google Scholar] [CrossRef]
- Murchison, E.P.; Tovar, C.; Hsu, A.; Bender, H.S.; Kheradpour, P.; Rebbeck, C.A.; Obendorf, D.; Conlan, C.; Bahlo, M.; Blizzard, C.A.; et al. The Tasmanian devil transcriptome reveals Schwann cell origins of a clonally transmissible cancer. Science 2010, 327, 84–87. [Google Scholar] [CrossRef] [Green Version]
- Siddle, H.V.; Kreiss, A.; Tovar, C.; Yuen, C.K.; Cheng, Y.; Belov, K.; Swift, K.; Pearse, A.M.; Hamede, R.; Jones, M.E.; et al. Reversible epigenetic down-regulation of MHC molecules by devil facial tumour disease illustrates immune escape by a contagious cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 5103–5108. [Google Scholar] [CrossRef] [Green Version]
- Cromme, F.V.; Meijer, C.J.; Snijders, P.J.; Uyterlinde, A.; Kenemans, P.; Helmerhorst, T.; Stern, P.L.; van den Brule, A.J.; Walboomers, J.M. Analysis of MHC class I and II expression in relation to presence of HPV genotypes in premalignant and malignant cervical lesions. Br. J. Cancer 1993, 67, 1372–1380. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Zhu, D.; Wang, W.; Li, W.; Jia, W.; Zeng, X.; Ding, W.; Yu, L.; Wang, X.; Wang, L.; et al. Genome-wide profiling of HPV integration in cervical cancer identifies clustered genomic hot spots and a potential microhomology-mediated integration mechanism. Nat. Genet. 2015, 47, 158–163. [Google Scholar] [CrossRef]
- Meissner, T.B.; Li, A.; Biswas, A.; Lee, K.H.; Liu, Y.J.; Bayir, E.; Iliopoulos, D.; van den Elsen, P.J.; Kobayashi, K.S. NLR family member NLRC5 is a transcriptional regulator of MHC class I genes. Proc. Natl. Acad. Sci. USA 2010, 107, 13794–13799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, A.; Meissner, T.B.; Kawai, T.; Kobayashi, K.S. Cutting edge: Impaired MHC class I expression in mice deficient for Nlrc5/class I transactivator. J. Immunol. 2012, 189, 516–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshihama, S.; Roszik, J.; Downs, I.; Meissner, T.B.; Vijayan, S.; Chapuy, B.; Sidiq, T.; Shipp, M.A.; Lizee, G.A.; Kobayashi, K.S. NLRC5/MHC class I transactivator is a target for immune evasion in cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 5999–6004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melero, I.; Gaudernack, G.; Gerritsen, W.; Huber, C.; Parmiani, G.; Scholl, S.; Thatcher, N.; Wagstaff, J.; Zielinski, C.; Faulkner, I.; et al. Therapeutic vaccines for cancer: An overview of clinical trials. Nat. Rev. Clin. Oncol. 2014, 11, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Small, E.J.; Petrylak, D.P.; Quinn, D.I.; Kibel, A.S.; Chang, N.N.; Dearstyne, E.; Harmon, M.; Campogan, D.; Haynes, H.; et al. Antigen-Specific CD8 Lytic Phenotype Induced by Sipuleucel-T in Hormone-Sensitive or Castration-Resistant Prostate Cancer and Association with Overall Survival. Clin. Cancer Res. 2018, 24, 4662–4671. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Splenic Leukocyte Populations | |||||||
| Females | CD3+ | CD3+ CD8+ | CD3+ CD4+ | CD3+ CD49b+ | CD3− CD19+ | CD3− CD49b+ | a CD4+ CD25+ FoxP3+ |
| Control | 51.6 ± 1.2 | 21.3 ± 1.0 | 30.4 ± 0.7 | 3.6 ± 0.4 | 42.5 ± 1.0 | 5.9 ± 0.4 | 5.5 ± 0.4 |
| 14 dpi d | 43.6 ± 2.7 c | 17.3 ± 1.7 b | 26.2 ± 2.9 b | 3.4 ± 0.3 | 51.5 ± 2.0 c | 4.7 ± 0.7 | 5.9 ± 0.7 |
| 21 dpi | 56.6 ± 3.9 b | 27.0 ± 2.3 c | 29.6 ± 3.5 | 4.6 ± 0.5 | 37.0 ± 4.2 b | 5.4 ± 0.4 | 6.87 ± 0.5 |
| 28 dpi | 61.0 ± 3.2 c | 26.6 ± 1.8 c | 34.3 ± 2.5 b | 5.0 ± 0.2 b | 32.0 ± 2.9 c | 7.1 ± 1.0 | 4.7 ± 0.4 |
| Splenic Leukocyte Populations | |||||||
| Males | CD3+ | CD3+ CD8+ | CD3+ CD4+ | CD3+ CD49b+ | CD3− CD19+ | CD3− CD49b+ | a CD4+ CD25+ FoxP3 |
| Control | 44.0 ± 0.7 | 17.8 ± 1.2 | 26.3 ± 1.8 | 2.9 ± 0.3 | 47.7 ± 0.8 | 7.7 ± 1.2 | 7.3 ± 0.8 |
| 14 dpi | 44.1 ± 1.1 | 16.9 ± 0.4 | 27.2 ± 1.2 | 2.5 ± 0.2 | 51.6 ± 0.8 b | 4.3 ± 0.4 b | 6.5 ± 0.1 |
| 21 dpi | 45.0 ± 2.2 | 19.7 ± 0.8 | 25.3 ± 1.0 | 3.2 ± 0.3 | 50.2 ± 2.1 | 4.8 ± 0.2 b | 6.1 ± 0.3 |
| 28 dpi | 49.4 ± 1.8 c | 20.9 ± 1.3 b | 28.5 ± 2.5 | 4.0 ± 0.3 | 44.7 ± 1.4 b | 5.7 ± 0.5 | 5.4 ± 0.5 b |
| Tumor Sample | Score a | Expression b |
|---|---|---|
| Breast Cancer | FHIT/HLA-I | FHIT/HLA-I |
| 27–05 | 9/9 | +++/+++ |
| 44–07 | 9/9 | +++/+++ |
| 69–07 | 9/9 | +++/+++ |
| 75–05 | 9/9 | +++/+++ |
| 94–06 | 9/9 | +++/+++ |
| 105–05 | 9/9 | +++/+++ |
| 119–05 | 9/9 | +++/+++ |
| 120–04 | 9/9 | +++/+++ |
| 120–06 | 9/9 | +++/+++ |
| 123–07 | 9/9 | +++/+++ |
| 153–07 | 9/9 | +++/+++ |
| 163–07 | 9/9 | +++/+++ |
| 192–06 | 9/9 | +++/+++ |
| 201–06 | 9/9 | +++/+++ |
| 250–07 | 9/9 | +++/+++ |
| 148–04 | 9/6 | +++/++ |
| 205–06 | 6/9 | ++/+++ |
| 4–07 | 6/6 | ++/++ |
| 35–07 | 6/6 | ++/++ |
| 112–06 | 6/6 | ++/++ |
| 133–05 | 4/4 | ++/++ |
| 32–07 | 4/0 | ++/- |
| 56–06 | 3/3 | +/+ |
| 93–07 | 3/3 | +/+ |
| 191–05 | 3/3 | +/+ |
| 196–06 | 3/3 | +/+ |
| 203–06 | 3/3 | +/+ |
| 95–06 | 3/0 | +/− |
| 207–06 | 3/0 | +/− |
| 41–07 | 2/2 | +/+ |
| 125–07 | 2/2 | +/+ |
| 128–06 | 2/2 | +/+ |
| 158–06 | 2/2 | +/+ |
| 118–06 | 2/0 | +/− |
| 136–06 | 2/0 | +/− |
| 137–06 | 2/0 | +/− |
| 203–05 | 2/0 | +/− |
| 16–07 | 0/0 | −/− |
| 23–07 | 0/0 | −/− |
| 100–06 | 0/0 | −/− |
| 108–06 | 0/0 | −/− |
| 120–07 | 0/0 | −/− |
| 134–06 | 0/0 | −/− |
| 142–06 | 0/0 | −/− |
| 150–07 | 0/0 | −/− |
| 154–05 | 0/0 | −/− |
| 186–07 | 0/0 | −/− |
| 204–06 | 0/0 | −/− |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pulido, M.; Chamorro, V.; Romero, I.; Algarra, I.; S-Montalvo, A.; Collado, A.; Garrido, F.; Garcia-Lora, A.M. Restoration of MHC-I on Tumor Cells by Fhit Transfection Promotes Immune Rejection and Acts as an Individualized Immunotherapeutic Vaccine. Cancers 2020, 12, 1563. https://doi.org/10.3390/cancers12061563
Pulido M, Chamorro V, Romero I, Algarra I, S-Montalvo A, Collado A, Garrido F, Garcia-Lora AM. Restoration of MHC-I on Tumor Cells by Fhit Transfection Promotes Immune Rejection and Acts as an Individualized Immunotherapeutic Vaccine. Cancers. 2020; 12(6):1563. https://doi.org/10.3390/cancers12061563
Chicago/Turabian StylePulido, María, Virginia Chamorro, Irene Romero, Ignacio Algarra, Alba S-Montalvo, Antonia Collado, Federico Garrido, and Angel M. Garcia-Lora. 2020. "Restoration of MHC-I on Tumor Cells by Fhit Transfection Promotes Immune Rejection and Acts as an Individualized Immunotherapeutic Vaccine" Cancers 12, no. 6: 1563. https://doi.org/10.3390/cancers12061563