Comprehensive Constitutional Genetic and Epigenetic Characterization of Lynch-Like Individuals

 , , , , ,
, , , , ,  , , and
, , and
Abstract
:
1. Introduction
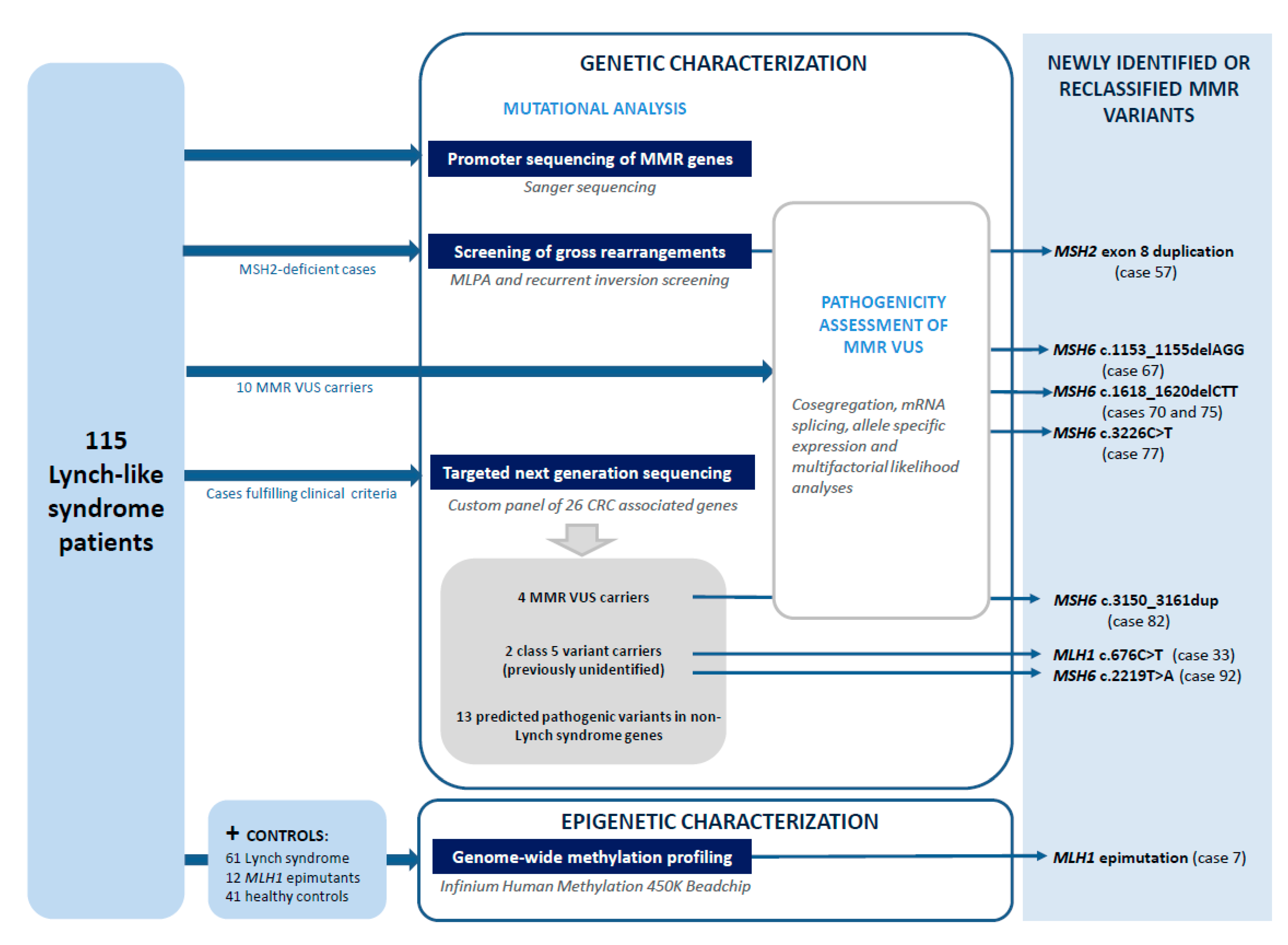
2. Materials and Methods
2.1. Patients
2.2. Samples
2.3. Mismatch Repair Genes Mutational Analysis
2.3.1. Mutational Analysis of Coding Regions of MMR Genes
2.3.2. Direct Sequencing of MMR Promoter Regions and 3′UTR of the EPCAM Gene
2.4. Targeted Next Generation Sequencing
2.5. Pathogenicity Assessment of Genetic Variants
2.5.1. Variant Frequency and Cosegregation Analysis
2.5.2. In Silico Prediction of the Functional Impact
2.5.3. Multifactorial Likelihood Analysis
2.5.4. mRNA Splicing Analysis and Allele Specific Expression Analysis
2.6. Tumor Analysis
2.7. Genome-Wide Methylation Profiling
2.8. Availability of Data
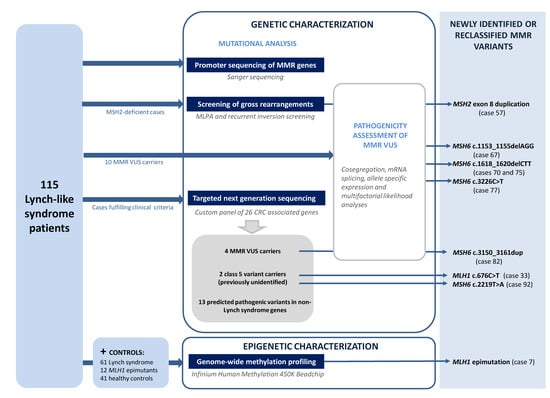
3. Results
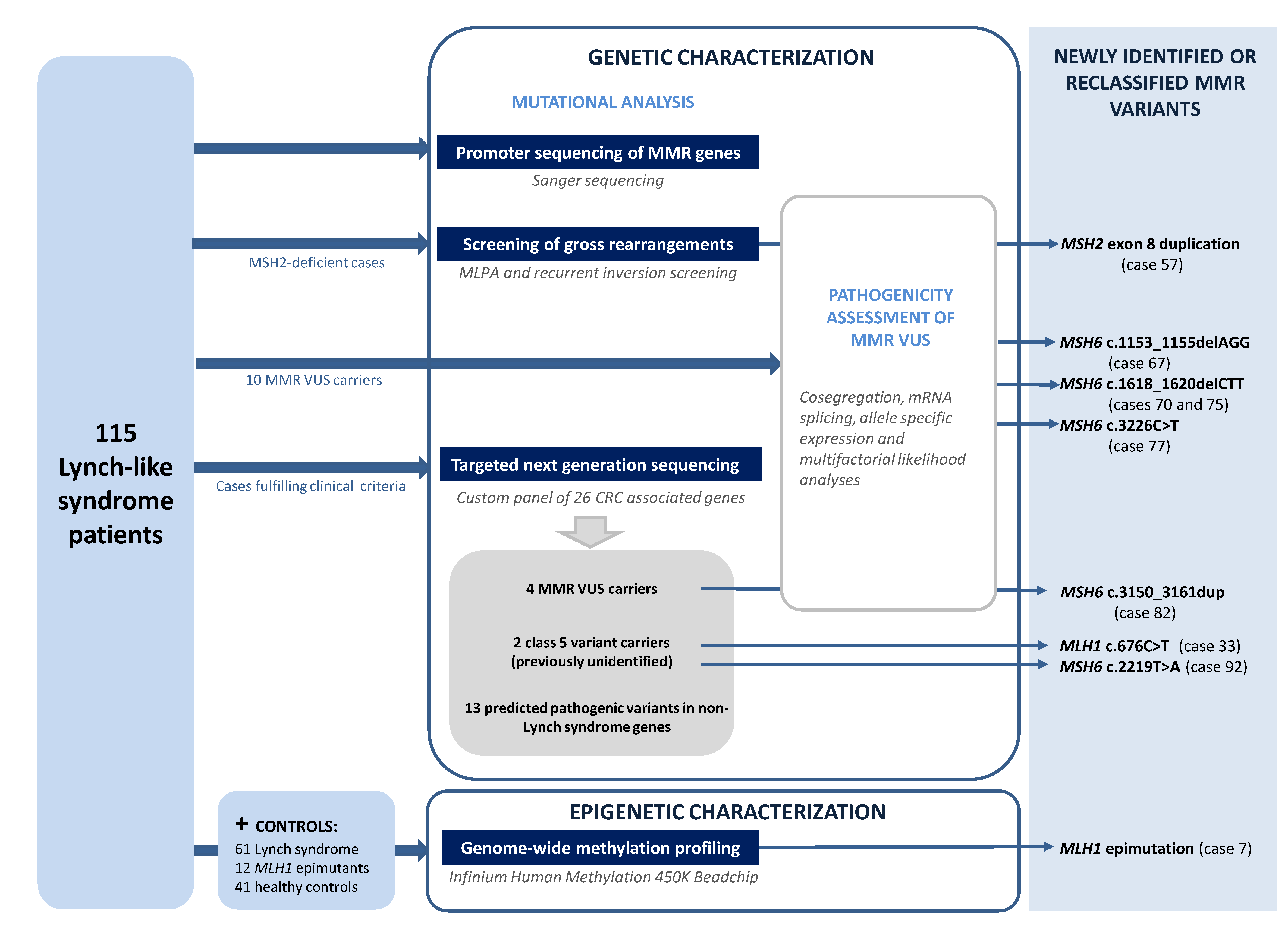
3.1. Reassessment of Germline Genetic Variants in the MMR Genes
3.2. Pathogenicity Assessment of MMR Variants
3.3. Identification of Variants in Other CRC-Predisposing Genes
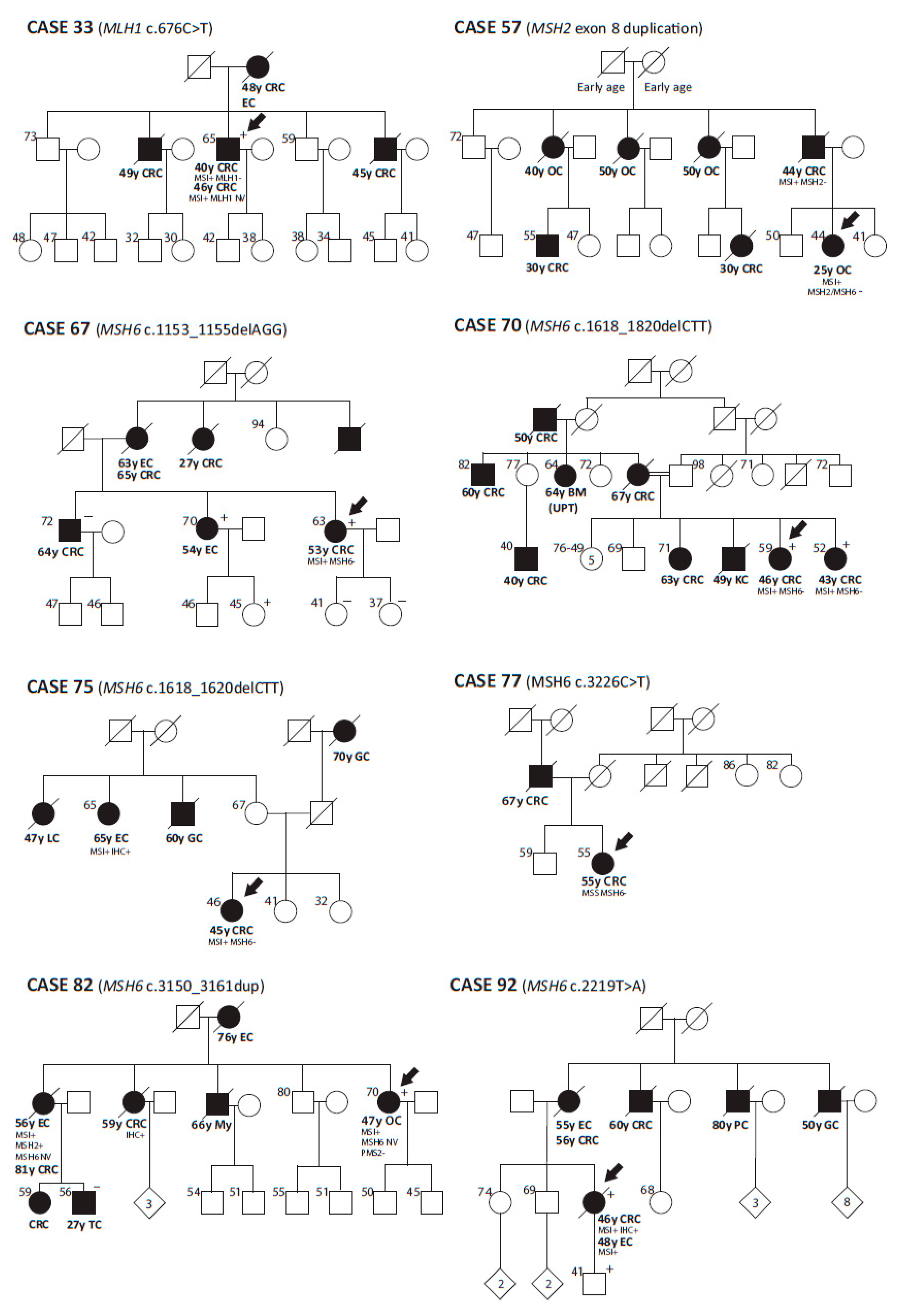
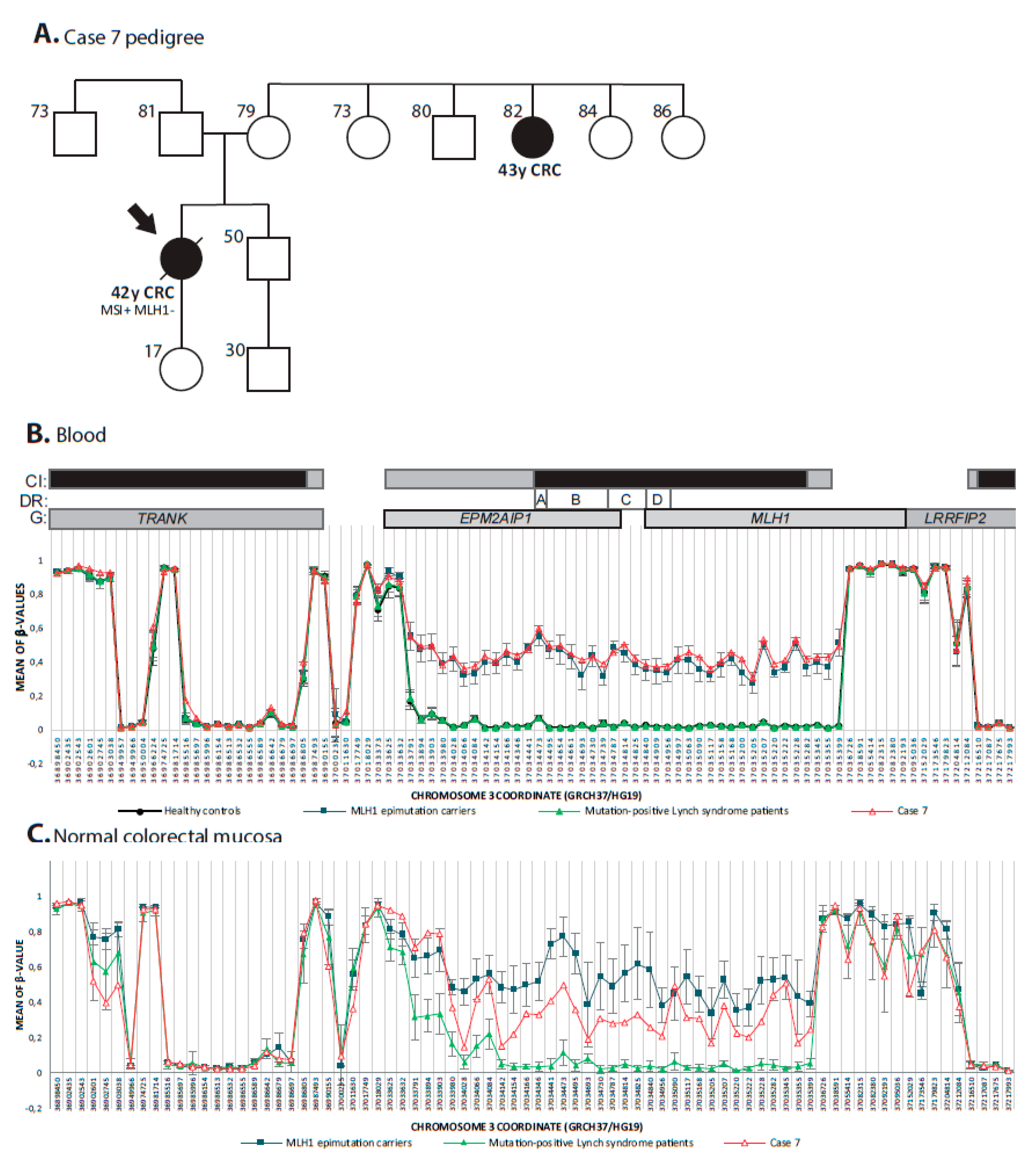
3.4. Constitutional Epigenetic Alterations in MMR Genes
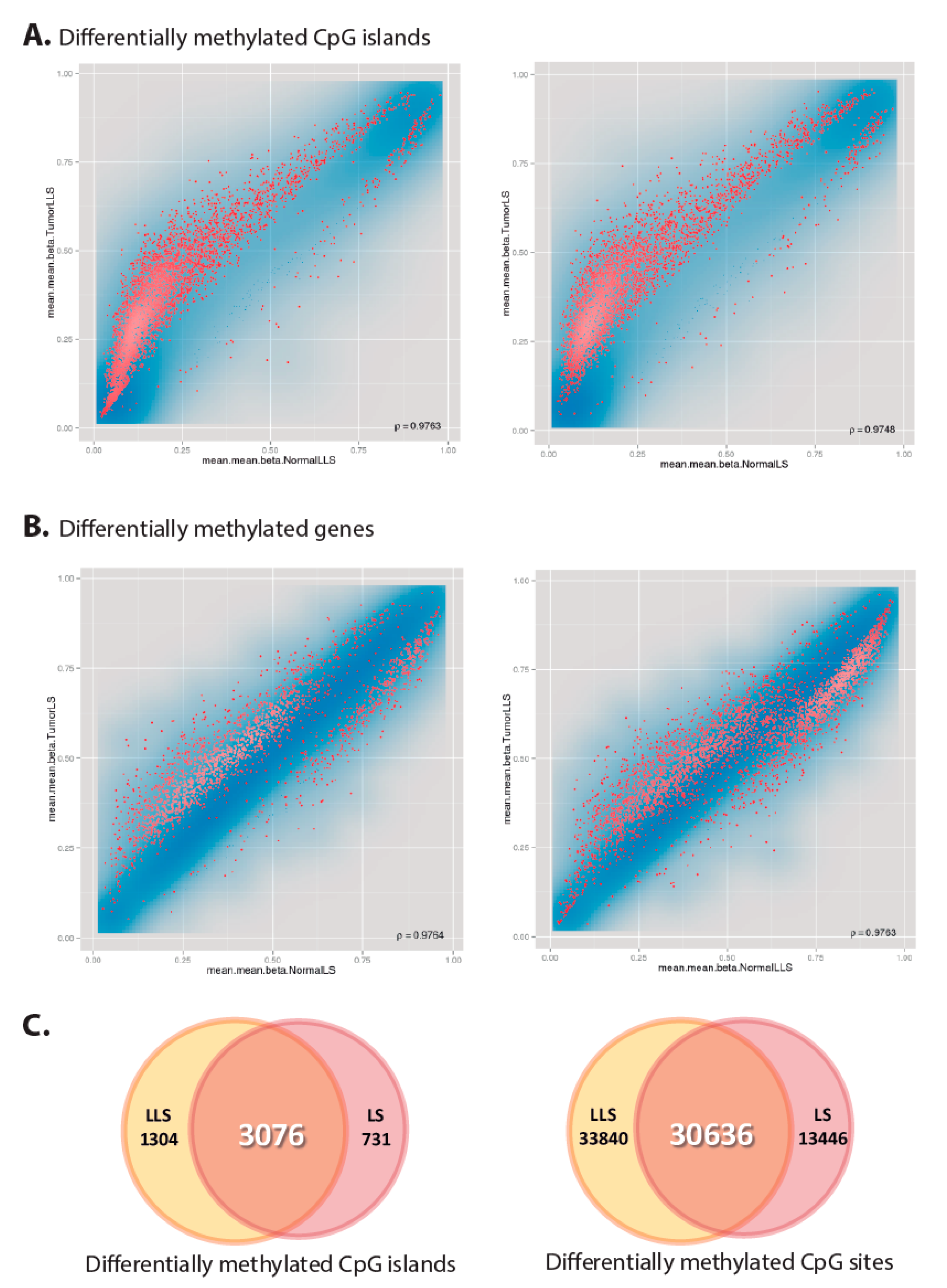
3.5. Global Epigenetic Characterization of Lynch-Like Cases
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ASE | Allele Specific expression |
| cDNA | complementary DNA |
| CNV | Copy number variation |
| CRC | Colorectal cancer |
| DM | Differentially methylated |
| DMR | Differentially methylated region |
| DNA | Deoxyribonucleic acid |
| EMAST | Elevated Microsatellite instability at selected tetranucleotide repeats |
| FFPE | Formalin-fixed, Paraffin-embedded (tissue) |
| gDNA | genomic DNA |
| IHC | Immunohistochemistry |
| LLS | Lynch-like syndrome |
| LR | Likelihood ratio |
| LS | Lynch syndrome |
| MLPA | Multiplex Ligation-dependent Probe Amplification |
| MMR | Mismatch repair |
| mRNA | Messenger Ribonucleic acid |
| MSI | Microsatellite instability |
| MS-MLPA | Methylation- Specific Multiplex Ligation-dependent Probe Amplification |
| PBL | Peripheral blood lymphocytes |
| PC | Pancreatic cancer |
| QC | Quality Control |
| SNuPE | Single-nucleotide primer extension |
| SSCP | Single Strand Conformation Polymorphism |
| TSS | Transcriptional start site |
| VUS | Variant of Unknown significance |
Appendix A

References
- Lynch, H.T.; Snyder, C.L.; Shaw, T.G.; Heinen, C.D.; Hitchins, M.P. Milestones of Lynch syndrome: 1895–2015. Nat. Rev. Cancer 2015, 15, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Imai, K. Microsatellite instability: An update. Arch. Toxicol. 2015, 89, 899–921. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, D.D.; Rosty, C.; Clendenning, M.; Spurdle, A.B.; Win, A.K. Clinical problems of colorectal cancer and endometrial cancer cases with unknown cause of tumor mismatch repair deficiency (suspected Lynch syndrome). Appl. Clin. Genet. 2014, 7, 183–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Soler, M.; Pérez-Carbonell, L.; Guarinos, C.; Zapater, P.; Castillejo, A.; Barberá, V.M.; Juárez, M.; Bessa, X.; Xicola, R.M.; Clofent, J.; et al. Risk of Cancer in Cases of Suspected Lynch Syndrome Without Germline Mutation. Gastroenterology 2013, 144, 926–932.e1. [Google Scholar] [CrossRef] [Green Version]
- Win, A.K.; Buchanan, D.D.; Rosty, C.; MacInnis, R.J.; Dowty, J.G.; Dite, G.S.; Giles, G.G.; Southey, M.C.; Young, J.P.; Clendenning, M.; et al. Role of tumour molecular and pathology features to estimate colorectal cancer risk for first-degree relatives. Gut 2015, 64, 101–110. [Google Scholar] [CrossRef]
- Liu, Q.; Hesson, L.B.; Nunez, A.C.; Packham, D.; Williams, R.; Ward, R.L.; Sloane, M.A. A cryptic paracentric inversion of MSH2 exons 2–6 causes Lynch syndrome. Carcinogenesis 2016, 37, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Meyer, C.; Brieger, A.; Plotz, G.; Weber, N.; Passmann, S.; Dingermann, T.; Zeuzem, S.; Trojan, J.; Marschalek, R. An Interstitial Deletion at 3p21.3 Results in the Genetic Fusion of MLH1 and ITGA9 in a Lynch Syndrome Family. Clin. Cancer Res. 2009, 15, 762–769. [Google Scholar] [CrossRef] [Green Version]
- Morak, M.; Koehler, U.; Schackert, H.K.; Steinke, V.; Royer-Pokora, B.; Schulmann, K.; Kloor, M.; Höchter, W.; Weingart, J.; Keiling, C.; et al. Biallelic MLH1 SNP cDNA expression or constitutional promoter methylation can hide genomic rearrangements causing Lynch syndrome. J. Med. Genet. 2011, 48, 513–519. [Google Scholar] [CrossRef] [Green Version]
- Sourrouille, I.; Coulet, F.; Lefevre, J.H.; Colas, C.; Eyries, M.; Svrcek, M.; Bardier-Dupas, A.; Parc, Y.; Soubrier, F. Somatic mosaicism and double somatic hits can lead to MSI colorectal tumors. Fam. Cancer 2013, 12, 27–33. [Google Scholar] [CrossRef]
- Vargas-Parra, G.M.; González-Acosta, M.; Thompson, B.A.; Gómez, C.; Fernández, A.; Dámaso, E.; Pons, T.; Morak, M.; del Valle, J.; Iglesias, S.; et al. Elucidating the molecular basis of MSH2-deficient tumors by combined germline and somatic analysis. Int. J. Cancer 2017, 141, 1365–1380. [Google Scholar] [CrossRef]
- Wagner, A.; van der Klift, H.; Franken, P.; Wijnen, J.; Breukel, C.; Bezrookove, V.; Smits, R.; Kinarsky, Y.; Barrows, A.; Franklin, B.; et al. A 10-Mb paracentric inversion of chromosome arm 2p inactivates MSH2 and is responsible for hereditary nonpolyposis colorectal cancer in a North-American kindred. Genes Chromosom. Cancer 2002, 35, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Morak, M.; Steinke-Lange, V.; Massdorf, T.; Benet-Pages, A.; Locher, M.; Laner, A.; Kayser, K.; Aretz, S.; Holinski-Feder, E. Prevalence of CNV-neutral structural genomic rearrangements in MLH1, MSH2, and PMS2 not detectable in routine NGS diagnostics. Fam. Cancer 2020, 19, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Geurts-Giele, W.R.; Leenen, C.H.; Dubbink, H.J.; Meijssen, I.C.; Post, E.; Sleddens, H.F.; Kuipers, E.J.; Goverde, A.; van den Ouweland, A.M.; van Lier, M.G.; et al. Somatic aberrations of mismatch repair genes as a cause of microsatellite-unstable cancers. J. Pathol. 2014, 234, 548–559. [Google Scholar] [CrossRef] [PubMed]
- Haraldsdottir, S.; Hampel, H.; Tomsic, J.; Frankel, W.L.; Pearlman, R.; de la Chapelle, A.; Pritchard, C.C. Colon and Endometrial Cancers With Mismatch Repair Deficiency Can Arise From Somatic, Rather Than Germline, Mutations. Gastroenterology 2014, 147, 1308–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, A.M.L.; van Wezel, T.; van den Akker, B.E.W.M.; Ventayol Garcia, M.; Ruano, D.; Tops, C.M.J.; Wagner, A.; Letteboer, T.G.W.; Gómez-García, E.B.; Devilee, P.; et al. Combined mismatch repair and POLE/POLD1 defects explain unresolved suspected Lynch syndrome cancers. Eur. J. Hum. Genet. 2016, 24, 1089. [Google Scholar] [CrossRef]
- Mensenkamp, A.R.; Vogelaar, I.P.; van Zelst–Stams, W.A.G.; Goossens, M.; Ouchene, H.; Hendriks–Cornelissen, S.J.B.; Kwint, M.P.; Hoogerbrugge, N.; Nagtegaal, I.D.; Ligtenberg, M.J.L. Somatic Mutations in MLH1 and MSH2 Are a Frequent Cause of Mismatch-Repair Deficiency in Lynch Syndrome-Like Tumors. Gastroenterology 2014, 146, 643–646e8. [Google Scholar] [CrossRef]
- Hemminger, J.A.; Pearlman, R.; Haraldsdottir, S.; Knight, D.; Jonasson, J.G.; Pritchard, C.C.; Hampel, H.; Frankel, W.L. Histology of colorectal adenocarcinoma with double somatic mismatch-repair mutations is indistinguishable from those caused by Lynch syndrome. Hum. Pathol. 2018, 78, 125–130. [Google Scholar] [CrossRef]
- Morak, M.; Käsbauer, S.; Kerscher, M.; Laner, A.; Nissen, A.M.; Benet-Pagès, A.; Schackert, H.K.; Keller, G.; Massdorf, T.; Holinski-Feder, E. Loss of MSH2 and MSH6 due to heterozygous germline defects in MSH3 and MSH6. Fam. Cancer 2017, 16, 491–500. [Google Scholar] [CrossRef]
- Castillejo, A.; Vargas, G.; Castillejo, M.I.; Navarro, M.; Barberá, V.M.; González, S.; Hernández-Illán, E.; Brunet, J.; Ramón y Cajal, T.; Balmaña, J.; et al. Prevalence of germline MUTYH mutations among Lynch-like syndrome patients. Eur. J. Cancer 2014, 50, 2241–2250. [Google Scholar] [CrossRef]
- Morak, M.; Heidenreich, B.; Keller, G.; Hampel, H.; Laner, A.; de la Chapelle, A.; Holinski-Feder, E. Biallelic MUTYH mutations can mimic Lynch syndrome. Eur. J. Hum. Genet. 2014, 22, 1334. [Google Scholar] [CrossRef] [Green Version]
- Sutcliffe, E.G.; Bartenbaker Thompson, A.; Stettner, A.R.; Marshall, M.L.; Roberts, M.E.; Susswein, L.R.; Wang, Y.; Klein, R.T.; Hruska, K.S.; Solomon, B.D. Multi-gene panel testing confirms phenotypic variability in MUTYH-Associated Polyposis. Fam. Cancer 2019, 18, 203–209. [Google Scholar] [CrossRef]
- Elsayed, F.A.; Kets, C.M.; Ruano, D.; van den Akker, B.; Mensenkamp, A.R.; Schrumpf, M.; Nielsen, M.; Wijnen, J.T.; Tops, C.M.; Ligtenberg, M.J.; et al. Germline variants in POLE are associated with early onset mismatch repair deficient colorectal cancer. Eur. J. Hum. Genet. 2014, 23, 1080. [Google Scholar] [CrossRef] [Green Version]
- De Voer, R.M.; Geurts van Kessel, A.; Weren, R.D.A.; Ligtenberg, M.J.L.; Smeets, D.; Fu, L.; Vreede, L.; Kamping, E.J.; Verwiel, E.T.P.; Hahn, M.M.; et al. Germline Mutations in the Spindle Assembly Checkpoint Genes BUB1 and BUB3 Are Risk Factors for Colorectal Cancer. Gastroenterology 2013, 145, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, Y.; Halpern, N.; Hubert, A.; Adler, S.N.; Cohen, S.; Plesser-Duvdevani, M.; Pappo, O.; Shaag, A.; Meiner, V. Mutated MCM9 is associated with predisposition to hereditary mixed polyposis and colorectal cancer in addition to primary ovarian failure. Cancer Genet. 2015, 208, 621–624. [Google Scholar] [CrossRef] [PubMed]
- Seguí, N.; Mina, L.B.; Lázaro, C.; Sanz-Pamplona, R.; Pons, T.; Navarro, M.; Bellido, F.; López-Doriga, A.; Valdés-Mas, R.; Pineda, M.; et al. Germline Mutations in FAN1 Cause Hereditary Colorectal Cancer by Impairing DNA Repair. Gastroenterology 2015, 149, 563–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xavier, A.; Olsen, M.F.; Lavik, L.A.; Johansen, J.; Singh, A.K.; Sjursen, W.; Scott, R.J.; Talseth-Palmer, B.A. Comprehensive mismatch repair gene panel identifies variants in patients with Lynch-like syndrome. Mol. Genet. Genom. Med. 2019, 7, e850. [Google Scholar] [CrossRef] [Green Version]
- Hitchins, M.P. Constitutional epimutation as a mechanism for cancer causality and heritability? Nat. Rev. Cancer 2015, 15, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Hansmann, T.; Pliushch, G.; Leubner, M.; Kroll, P.; Endt, D.; Gehrig, A.; Preisler-Adams, S.; Wieacker, P.; Haaf, T. Constitutive promoter methylation of BRCA1 and RAD51C in patients with familial ovarian cancer and early-onset sporadic breast cancer. Hum. Mol. Genet. 2012, 21, 4669–4679. [Google Scholar] [CrossRef]
- Bennett, K.L.; Mester, J.; Eng, C. Germline epigenetic regulation of killin in cowden and cowden-like syndrome. JAMA 2010, 304, 2724–2731. [Google Scholar] [CrossRef]
- Raval, A.; Tanner, S.M.; Byrd, J.C.; Angerman, E.B.; Perko, J.D.; Chen, S.-S.; Hackanson, B.; Grever, M.R.; Lucas, D.M.; Matkovic, J.J.; et al. Downregulation of Death-Associated Protein Kinase 1 (DAPK1) in Chronic Lymphocytic Leukemia. Cell 2007, 129, 879–890. [Google Scholar] [CrossRef] [Green Version]
- Seguí, N.; Navarro, M.; Pineda, M.; Köger, N.; Bellido, F.; González, S.; Campos, O.; Iglesias, S.; Valdés-Mas, R.; López-Doriga, A.; et al. Exome sequencing identifies MUTYH mutations in a family with colorectal cancer and an atypical phenotype. Gut 2015, 64, 355–356. [Google Scholar] [CrossRef] [PubMed]
- Dámaso, E.; Castillejo, A.; Arias, M.d.M.; Canet-Hermida, J.; Navarro, M.; del Valle, J.; Campos, O.; Fernández, A.; Marín, F.; Turchetti, D.; et al. Primary constitutional MLH1 epimutations: A focal epigenetic event. Br. J. Cancer 2018. [Google Scholar] [CrossRef] [Green Version]
- Thompson, B.A.; Spurdle, A.B.; Plazzer, J.-P.; Greenblatt, M.S.; Akagi, K.; Al-Mulla, F.; Bapat, B.; Bernstein, I.; Capellá, G.; den Dunnen, J.T.; et al. Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat. Genet. 2013, 46, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Qian, D.; Thompson, B.A.; Gutierrez, S.; Wu, S.; Pesaran, T.; LaDuca, H.; Lu, H.-M.; Chao, E.C.; Black, M.H. Tumour characteristics provide evidence for germline mismatch repair missense variant pathogenicity. J. Med Genet. 2020, 57, 62–69. [Google Scholar] [CrossRef]
- Thompson, B.A.; Goldgar, D.E.; Paterson, C.; Clendenning, M.; Walters, R.; Arnold, S.; Parsons, M.T.; Michael, W.D.; Gallinger, S.; Haile, R.W.; et al. A Multifactorial Likelihood Model for MMR Gene Variant Classification Incorporating Probabilities Based on Sequence Bioinformatics and Tumor Characteristics: A Report from the Colon Cancer Family Registry. Hum. Mutat. 2013, 34, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Pineda, M.; Mur, P.; Iniesta, M.D.; Borras, E.; Campos, O.; Vargas, G.; Iglesias, S.; Fernandez, A.; Gruber, S.B.; Lazaro, C.; et al. MLH1 methylation screening is effective in identifying epimutation carriers. Eur. J. Hum. Genet. 2012, 20, 1256–1264. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Ng, A.W.T.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.; Lopez-Bigas, N.; et al. The Repertoire of Mutational Signatures in Human Cancer. bioRxiv 2018. [CrossRef] [Green Version]
- Rosenthal, R.; McGranahan, N.; Herrero, J.; Taylor, B.S.; Swanton, C. DeconstructSigs: Delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016, 17, 31. [Google Scholar] [CrossRef] [Green Version]
- Adam, R.; Spier, I.; Zhao, B.; Kloth, M.; Marquez, J.; Hinrichsen, I.; Kirfel, J.; Tafazzoli, A.; Horpaopan, S.; Uhlhaas, S.; et al. Exome Sequencing Identifies Biallelic MSH3 Germline Mutations as a Recessive Subtype of Colorectal Adenomatous Polyposis. Am. J. Hum. Genet. 2016, 99, 337–351. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.; Akopyan, G.; Garban, H.; Bonavida, B. Transcription factor YY1: Structure, function, and therapeutic implications in cancer biology. Oncogene 2005, 25, 1125. [Google Scholar] [CrossRef] [Green Version]
- Plaschke, J.; Linnebacher, M.; Kloor, M.; Gebert, J.; Cremer, F.W.; Tinschert, S.; Aust, D.E.; von Knebel Doeberitz, M.; Schackert, H.K. Compound heterozygosity for two MSH6 mutations in a patient with early onset of HNPCC-associated cancers, but without hematological malignancy and brain tumor. Eur. J. Hum. Genet. 2006, 14, 561. [Google Scholar] [CrossRef] [PubMed]
- Rahner, N.; Höefler, G.; Högenauer, C.; Lackner, C.; Steinke, V.; Sengteller, M.; Friedl, W.; Aretz, S.; Propping, P.; Mangold, E.; et al. Compound heterozygosity for two MSH6 mutations in a patient with early onset colorectal cancer, vitiligo and systemic lupus erythematosus. Am. J. Med. Genet. Part A 2008, 146A, 1314–1319. [Google Scholar] [CrossRef] [PubMed]
- Betz, B.; Theiss, S.; Aktas, M.; Konermann, C.; Goecke, T.O.; Möslein, G.; Schaal, H.; Royer-Pokora, B. Comparative in silico analyses and experimental validation of novel splice site and missense mutations in the genes MLH1 and MSH2. J. Cancer Res. Clin. Oncol. 2009, 136, 123. [Google Scholar] [CrossRef]
- Obmolova, G.; Ban, C.; Hsieh, P.; Yang, W. Crystal structures of mismatch repair protein MutS and its complex with a substrate DNA. Nature 2000, 407, 703. [Google Scholar] [CrossRef] [PubMed]
- Borràs, E.; Pineda, M.; Brieger, A.; Hinrichsen, I.; Gómez, C.; Navarro, M.; Balmaña, J.; Ramón y Cajal, T.; Torres, A.; Brunet, J.; et al. Comprehensive functional assessment of MLH1 variants of unknown significance. Hum. Mut. 2012, 33, 1576–1588. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Lasset, C.; Desseigne, F.; Saurin, J.C.; Maugard, C.; Navarro, C.; Ruano, E.; Descos, L.; Trillet-Lenoir, V.; Bosset, J.F.; et al. Prevalence of germline mutations of hMLH1, hMSH2, hPMS1, hPMS2, and hMSH6 genes in 75 French kindreds with nonpolyposis colorectal cancer. Hum. Genet. 1999, 105, 79–85. [Google Scholar] [CrossRef]
- Smith, A.L.; Alirezaie, N.; Connor, A.; Chan-Seng-Yue, M.; Grant, R.; Selander, I.; Bascuñana, C.; Borgida, A.; Hall, A.; Whelan, T.; et al. Candidate DNA repair susceptibility genes identified by exome sequencing in high-risk pancreatic cancer. Cancer Lett. 2016, 370, 302–312. [Google Scholar] [CrossRef] [Green Version]
- Lachaud, C.; Moreno, A.; Marchesi, F.; Toth, R.; Blow, J.J.; Rouse, J. Ubiquitinated Fancd2 recruits Fan1 to stalled replication forks to prevent genome instability. Science 2016, 351, 846–849. [Google Scholar] [CrossRef] [Green Version]
- Pfeifer, G. Defining Driver DNA Methylation Changes in Human Cancer. Int. J. Mol. Sci. 2018, 19, 1166. [Google Scholar] [CrossRef] [Green Version]
- Monahan, K.J.; Bradshaw, N.; Dolwani, S.; Desouza, B.; Dunlop, M.G.; East, J.E.; Ilyas, M.; Kaur, A.; Lalloo, F.; Latchford, A.; et al. Guidelines for the management of hereditary colorectal cancer from the British Society of Gastroenterology (BSG)/Association of Coloproctology of Great Britain and Ireland (ACPGBI)/United Kingdom Cancer Genetics Group (UKCGG). Gut 2020, 69, 411–444. [Google Scholar] [CrossRef]
- Schmidt, A.Y.; Hansen, T.V.O.; Ahlborn, L.B.; Jønson, L.; Yde, C.W.; Nielsen, F.C. Next-Generation Sequencing-Based Detection ofmline Copy Number Variations in BRCA1/BRCA2. J. Mol. Diagn. 2017, 19, 809–816. [Google Scholar] [CrossRef] [Green Version]
- Katona, B.W.; Yurgelun, M.B.; Garber, J.E.; Offit, K.; Domchek, S.M.; Robson, M.E.; Stadler, Z.K. A counseling framework for moderate-penetrance colorectal cancer susceptibility genes. Genet. Med. 2018. [CrossRef] [PubMed] [Green Version]
- Jagmohan-Changur, S.; Poikonen, T.; Vilkki, S.; Launonen, V.; Wikman, F.; Orntoft, T.F.; Møller, P.; Vasen, H.; Tops, C.; Kolodner, R.D.; et al. EXO1 Variants Occur Commonly in Normal Population. Evidence against a Role in Hereditary Nonpolyposis Colorectal Cancer. Cancer Res. 2003, 63, 154–158. [Google Scholar] [PubMed]
- Peltomäki, P. Role of DNA Mismatch Repair Defects in the Pathogenesis of Human Cancer. J. Clin. Oncol. 2003, 21, 1174–1179. [Google Scholar] [CrossRef] [PubMed]
- Duraturo, F.; Liccardo, R.; Cavallo, A.; Rosa, M.D.; Grosso, M.; Izzo, P. Association of low-risk MSH3 and MSH2 variant alleles with Lynch syndrome: Probability of synergistic effects. Int. J. Cancer 2011, 129, 1643–1650. [Google Scholar] [CrossRef] [PubMed]
- Broderick, P.; Dobbins, S.E.; Chubb, D.; Kinnersley, B.; Dunlop, M.G.; Tomlinson, I.; Houlston, R.S. Validation of Recently Proposed Colorectal Cancer Susceptibility Gene Variants in an Analysis of Families and Patients: A Systematic Review. Gastroenterology 2017, 152, 75–77e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fievet, A.; Mouret-Fourme, E.; Colas, C.; de Pauw, A.; Stoppa-Lyonnet, D.; Buecher, B. Prevalence of Pathogenic Variants of FAN1 in More Than 5000 Patients Assessed for Genetic Predisposition to Colorectal, Breast, Ovarian, or Other Cancers. Gastroenterology 2019, 156, 1919–1920. [Google Scholar] [CrossRef] [PubMed]
- Ligtenberg, M.J.L.; Kuiper, R.P.; Chan, T.L.; Goossens, M.; Hebeda, K.M.; Voorendt, M.; Lee, T.Y.H.; Bodmer, D.; Hoenselaar, E.; Hendriks-Cornelissen, S.J.B.; et al. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3[prime] exons of TACSTD1. Nat. Genet. 2009, 41, 112–117. [Google Scholar] [CrossRef]
- Hinoue, T.; Weisenberger, D.; Lange, C.; Shen, H.; Byun, H.-M.; Van Den Berg, D.; Malik, S.; Pan, F.; Noushmehr, H.; van Dijk, C.; et al. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res. 2012, 22, 271–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Cancer Genome Atlas, N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case ID | Results from Previous MMR Mutational Analysis by Sanger Sequencing/SSCP (#) | Results from the Analysis of CRC-Associated Genes (This Study) | Pathogenicity Assessment of MMR VUS | Final Case Classification | |||
|---|---|---|---|---|---|---|---|
| Variants in LS-Associated Genes (Insight Classification) | Variants in LS-Associated Genes (Insight Classification) | Predicted Pathogenic Variants in Other Predisposing Genes (ClinVar Classification) | VUS Assessment | Final Variant Classification | |||
| A. Results obtained in the analysis of 42 samples by using an NGS subexome panel of CRC-associated genes. Only the MMR variants and the predicted pathogenic variants in other genes are shown (see Table S1). | |||||||
| 5 | - | MSH6 c.2092C>G, p.Gln698Glu (Class 3) | - | LLS (MMR VUS carrier) | |||
| 6 | - | - | - | LLS | |||
| 7 | - | MLH1epimutation | - | Confirmation by MS-MLPA (48%) | LS (MLH1 epimutation carrier) | ||
| 8 | - | - | - | LLS | |||
| 10 | - | - | EPCAM^ c.811G>T, p.Val271Phe (not reported) | LLS (VUS carrier) | |||
| 13 | - | MLH1 c.-574T>C, p.? (Class 3*) | - | LLS (MMR VUS carrier) | |||
| 28 | - | - | - | LLS | |||
| 29 | - | - | POLD1 c.2275G>A, p.Val759Ile (Class 1,2,3) | LLS (VUS carrier) | |||
| 30 | - | - | APC c.7936C>G, p.Gln2646Glu (Class 3) | LLS (VUS carrier) | |||
| 33 | - | MLH1 c.676C>T, p.Arg226* (Class 5) | - | LS | |||
| 39 | MSH2 c.1787A>G; p.Asn596Ser (Class 3) | MSH2 c.1787A>G; p.Asn596Ser (Class 3) | FAN1 c.149T>G, p.Met50Arg (not reported) | LLS (MMR VUS carrier) | |||
| 42 | - | - | - | LLS | |||
| 44 | - | - | - | LLS (VUS carrier) | |||
| 45 | - | - | - | LLS | |||
| 48 | - | - | - | LLS | |||
| 53 | - | - | - | LLS | |||
| 55 | - | - | PMS1 c.497A>C, p.Lys166Thr (not reported) | LLS (VUS carrier) | |||
| 56 | - | - | - | LLS | |||
| 57 | - | MSH2 E8 duplication (Class 3*) | - | Aberrant splicing | MSH2E8 duplication (Class 5) | LS | |
| 58 | MSH2 c.2045C>G; p.Thr682Ser (Class 3*) | MSH2 c.2045C>G; p.Thr682Ser (Class 3*) | EXO1 c.2212-1G>A (Class 3) | LLS (MMR VUS carrier) | |||
| 59 | - | - | APC c.1966C>G, p.Leu656Val (Class 3) | LLS (VUS carrier) | |||
| 61 | - | - | - | LLS | |||
| 62 | - | - | MSH3 c.2732T>G, p.Leu911Trp (not reported) | LLS (VUS carrier) | |||
| 63 | MSH2 c.2702A>T; p.Glu901Val (Class 3*) | MSH2 c.2702A>T; p.Glu901Val (Class 3*) | - | LLS (MMR VUS carrier) | |||
| 64 | - | - | - | LLS | |||
| 65 | - | - | MUTYH c.1437_1439delGGA, p.Glu480del (Class 5) | LLS (monoallelic MUTYH carrier) | |||
| 66 | - | - | - | LLS | |||
| 74 | - | - | MSH3 c.685T>C, p.Tyr229His (not reported); MSH3 c.2732T>G, p.Leu911Trp (not reported) | MSH3 conserved expression/EMAST/in cis | MSH3 c.685T>C, p.Tyr229His (VUS); MSH3 c.2732T>G, p.Leu911Trp (VUS) | LLS (VUS carrier) | |
| 76 | - | - | - | LLS | |||
| 78 | - | - | - | LLS | |||
| 79 | - | - | - | LLS | |||
| 81 | - | - | BUB1 c.2473C>T, p.Pro825Ser (not reported) | LLS (VUS carrier) | |||
| 82 | - | MSH6 c.3150_3161dup, p.(Val1051_Ile1054dup) (Class 3*) | - | Multifactorial (>0.99)/normal splicing | MSH6 c.3150_3161dup, p.(Val1051_Ile1054dup) (Class 5) | LS | |
| 85 | - | PMS2 c.1320A>G, p.Pro440= (Class 3*) | MSH3 c.3072G>C, p.Gln1024His (not reported) | LLS (MMR VUS carrier) | |||
| 87 | - | - | - | LLS | |||
| 92 | - | MSH6 c.2219T>A, p.Leu740* (Class 5) | - | LS | |||
| 93 | - | - | - | LLS | |||
| 94 | - | - | - | LLS | |||
| 95 | - | - | - | LLS | |||
| 96 | - | - | APC c.7514G>A, p.Arg2505Gln (Class 1,2) | LLS (VUS carrier) | |||
| 97 | - | - | - | LLS | |||
| 98 | - | MSH2 c.2802G>A, p.Thr934Thr (Class 3) | - | LLS (MMR VUS carrier) | |||
| B. Results obtained in 7 additional cases harboring MMR variants identified by previous Sanger sequencing | |||||||
| 35 | MLH1 c.25C>T, p.Arg9Trp, (Class 3) APC c.1958+3A>G (Class 5) (Borrás et al. 2012) | - | - | FAP (MMR VUS carrier) | |||
| 67 | MSH6 c.1153_1155del, p.Arg385del (Class 3 *) | - | Multifactorial (0.98)/normal splicing | MSH6 c.1153_1155delAGG p.Arg385del (Class 4*) | LS | ||
| 70 | MSH6 c.1618_1620delCTT; p.Leu540del (Class 3 *) | - | - | Multifactorial (>0.99)/aberrant splicing at low proportion | MSH6 c.1618_1620delCTT; p.Leu540del (Class 5*) | LS | |
| 72 | MSH6 c.1450G>A; p.Glu484Lys (Class 3 *) | - | - | LLS (MMR VUS carrier) | |||
| 73 | MSH6 c.3296T>A; p.Ile1099Asn (Class 3 *) | - | - | LLS (MMR VUS carrier) | |||
| 75 | MSH6 c.1618_1620del; p.Leu540del (Class 3 *) | - | - | Multifactorial (>0.99)/aberrant splicing at low proportion | MSH6 c.1618_1620delCTT; p.Leu540del (Class 5*) | LS | |
| 77 | MSH6 c.3226C>T, p.Arg1076Cys (Class 3) | - | - | Insight variant classification revision | MSH6 c.3226C>T, p.Arg1076Cys (Class 4, Insight March 2018) | LS | |
| 33 | - | MLH1 c.676C>T, p.Arg226* (Class 5) | - | LS | |||
| 39 | MSH2 c.1787A>G; p.Asn596Ser (Class 3) | MSH2 c.1787A>G; p.Asn596Ser (Class 3) | FAN1 c.149T>G, p.Met50Arg (not reported) | LLS (MMR VUS carrier) | |||
| 42 | - | - | - | LLS | |||
| 44 | - | - | - | LLS (VUS carrier) | |||
| 45 | - | - | - | LLS | |||
| 48 | - | - | - | LLS | |||
| 53 | - | - | - | LLS | |||
| 55 | - | - | PMS1 c.497A>C, p.Lys166Thr (not reported) | LLS (VUS carrier) | |||
| 56 | - | - | - | LLS | |||
| 57 | - | MSH2 E8 duplication (Class 3*) | - | Aberrant splicing | MSH2E8 duplication (Class 5) | LS | |
| 58 | MSH2 c.2045C>G; p.Thr682Ser (Class 3*) | MSH2 c.2045C>G; p.Thr682Ser (Class 3*) | EXO1 c.2212-1G>A (Class 3) | LLS (MMR VUS carrier) | |||
| 59 | - | - | APC c.1966C>G, p.Leu656Val (Class 3) | LLS (VUS carrier) | |||
| 61 | - | - | - | LLS | |||
| 62 | - | - | MSH3 c.2732T>G, p.Leu911Trp (not reported) | LLS (VUS carrier) | |||
| 63 | MSH2 c.2702A>T; p.Glu901Val (Class 3*) | MSH2 c.2702A>T; p.Glu901Val (Class 3*) | - | LLS (MMR VUS carrier) | |||
| 64 | - | - | - | LLS | |||
| 65 | - | - | MUTYH c.1437_1439delGGA, p.Glu480del (Class 5) | LLS (monoallelic MUTYH carrier) | |||
| 66 | - | - | - | LLS | |||
| 74 | - | - | MSH3 c.685T>C, p.Tyr229His (not reported); MSH3 c.2732T>G, p.Leu911Trp (not reported) | MSH3 conserved expression/EMAST/in cis | MSH3 c.685T>C, p.Tyr229His (VUS); MSH3 c.2732T>G, p.Leu911Trp (VUS) | LLS (VUS carrier) | |
| 76 | - | - | - | LLS | |||
| 78 | - | - | - | LLS | |||
| 79 | - | - | - | LLS | |||
| 81 | - | - | BUB1 c.2473C>T, p.Pro825Ser (not reported) | LLS (VUS carrier) | |||
| 82 | - | MSH6 c.3150_3161dup, p.(Val1051_Ile1054dup) (Class 3*) | - | Multifactorial (>0.99)/normal splicing | MSH6 c.3150_3161dup, p.(Val1051_Ile1054dup) (Class 5) | LS | |
| 85 | - | PMS2 c.1320A>G, p.Pro440= (Class 3*) | MSH3 c.3072G>C, p.Gln1024His (not reported) | LLS (MMR VUS carrier) | |||
| 87 | - | - | - | LLS | |||
| 92 | - | MSH6 c.2219T>A, p.Leu740* (Class 5) | - | LS | |||
| 93 | - | - | - | LLS | |||
| 94 | - | - | - | LLS | |||
| 95 | - | - | - | LLS | |||
| 96 | - | - | APC c.7514G>A, p.Arg2505Gln (Class 1,2) | LLS (VUS carrier) | |||
| 97 | - | - | - | LLS | |||
| 98 | - | MSH2 c.2802G>A, p.Thr934Thr (Class 3) | - | LLS (MMR VUS carrier) | |||
| B. Results obtained in 7 additional cases harboring MMR variants identified by previous Sanger sequencing | |||||||
| 35 | MLH1 c.25C>T, p.Arg9Trp, (Class 3) APC c.1958+3A>G (Class 5) (Borrás et al. 2012) | - | - | FAP (MMR VUS carrier) | |||
| 67 | MSH6 c.1153_1155del, p.Arg385del (Class 3 *) | - | Multifactorial (0.98)/normal splicing | MSH6 c.1153_1155delAGG p.Arg385del (Class 4*) | LS | ||
| 70 | MSH6 c.1618_1620delCTT; p.Leu540del (Class 3 *) | - | - | Multifactorial (>0.99)/aberrant splicing at low proportion | MSH6c.1618_1620delCTT; p.Leu540del (Class 5*) | LS | |
| 72 | MSH6 c.1450G>A; p.Glu484Lys (Class 3 *) | - | - | LLS (MMR VUS carrier) | |||
| 73 | MSH6 c.3296T>A; p.Ile1099Asn (Class 3 *) | - | - | LLS (MMR VUS carrier) | |||
| 75 | MSH6 c.1618_1620del; p.Leu540del (Class 3 *) | - | - | Multifactorial (>0.99)/aberrant splicing at low proportion | MSH6c.1618_1620delCTT; p.Leu540del (Class 5*) | LS | |
| 77 | MSH6 c.3226C>T, p.Arg1076Cys (Class 3) | - | - | Insight variant classification revision | MSH6 c.3226C>T, p.Arg1076Cys (Class 4, Insight March 2018) | LS | |
| Case ID | MMR Gene | MMR Variant | Predicted Protein Change | Insight Classification (2015) | ClinVar Classification | Frequency in Controls (ExAC/ESP) | RefSNP (rs) | In Silico Predictions | RNA Analyses | Multifactorial Calculations | Final Classification | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Splicing | Protein Function | cDNA Splicing Analysis | cDNA Stability Analysis (+/− Puromicin) | ||||||||||
| 13 | MLH1 | c.-574T>C | p.? | Class 3 | Not reported | 0,000084/NR | rs558088820 | NA | NA | NP | NP | NP | Class 3 |
| 35 | c.25C>T | p.(Arg9Trp) | Class 3 | VUS (2)/+++ | NR/NR | rs587779000 | No changes | Damaging | r.25C>T^; p.Arg9Trp | NP | NP | Class 3 | |
| 39 | MSH2 | c.1787A>G | p.(Asn596Ser) | Class 3 | VUS (3) vs Bening/Likely bening (3)/+++ | NR/0.0002 | rs41295288 | No changes | Benign | r.1787A>G; p.Asn596Ser | Biallelic expression (Sanger seq) | NP | Class 3 |
| 57 | exon 8 duplication | p.? | Not reported | Not reported | _ | _ | NA | NA | r.1277_1387dup; p.Val463Glufs*11 | NP | NP | Class 5 | |
| 58 | c.2045C>G | p.(Thr682Ser) | Not reported | Not reported | NR/NR | _ | No changes | Benign | NA | NA | NP | Class 3 | |
| 63 | c.2702A>T | p.(Glu901Val) | Not reported | Not reported | NR/NR | _ | No changes | Damaging | NA | NA | NP | Class 3 | |
| 98 | c.2802G>A | p.(Thr934=) | Class 3 | VUS (2) vs Bening/Likely Bening (5)/+++ | 0.000/0.0001 | rs150259097 | No changes | NA | NP | NP | NP | Class 3 | |
| 5 | MSH6 | c.2092C>G | p.(Gln698Glu) | Class 3 | VUS (5)/+++ | NR/NR | rs63750832 | Unconclusive (3/5) | Benign | r.2092C>G¨; p.Gln698Glu | NP | NP | Class 3 |
| 67 | c.1153_1155del | p.(Arg385del) | Class 3 | VUS (2)/+++ | NR/NR | rs267608043 | No changes | Damaging | r.1153_1155del (NP); p.Arg385del | Non allelic imbalance (NP/1.02±0.09) | 0,98 | Class 4 | |
| 72 | c.1450G>A | p.(Glu484Lys) | Not reported | VUS (1)/+ | NR/NR | _ | No changes | Damaging | NP | NP | NP | Class 3 | |
| 70 & 75 | c.1618_1620del | p.(Leu540del) | Not reported | VUS (2) vs Pathogenic (1)/+ | NR/NR | _ | No changes | Damaging | r.[1618_1620del;1607_3172del]; p.[Leu540del;Ser536_Asp1058delinsAsn] | Destabilization (0.69±0.03/0.65±0.06) | >0,99 | Class 5 | |
| 82 | c.3150_3161dup | p.(Val1051_Ile1054dup) | Not reported | Not reported | NR/NR | _ | No changes | Damaging | r.3150_3161dup; p.Val1051_Ile1054dup | Non allelic imbalance(1.04±0.14/1.16±0.26) | >0,99 | Class 5 | |
| 77 | c.3226C>T | p.(Arg1076Cys) | Class 3 | Pathogenic/Likely pathogenic (6)/+++ | NR/NR | rs63750617 | No changes | Damaging | r.3226C>T*; p.Arg1076Cys | NP | NP | Class 4** | |
| 73 | c.3296T>A | p.(Ile1099Asn) | Not reported | Not reported | NR/NR | _ | No changes | Damaging | NP | NP | NP | Class 3 | |
| 85 | PMS2 | c.1320A>G | p.(Pro440=) | Not reported | VUS (1) vs Bening/Likely bening (5)/+ | NR/0.0001 | rs138697590 | No changes | NA | NP | NP | NP | Class 3 |
| 73 | c.3296T>A | p.(Ile1099Asn) | Not reported | Not reported | NR/NR | _ | No changes | Damaging | NP | NP | NP | Class 3 | |
| 85 | PMS2 | c.1320A>G | p.(Pro440=) | Not reported | VUS (1) vs Benign/likely benign (5)/+ | NR/0.0001 | rs138697590 | No changes | NA | NP | NP | NP | Class 3 |
| MSH6 Variant | Frequency in Controls (ExAC/ESP) | Multifactorial Likelihood Analysis | Final Classification | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Prior Probability of PATHOGENICITY | Prior Used | Case ID | Proband (Yes/No) | Ascertainment | Cancer (Age) | MSI/IHC Status | CRC/EC LR | Tumor Characteristics LR | Bayes | Segrega-tion LR | Odds for Causality | Posterior Odds | Posterior Probability of Pathogenicity | |||
| c.1153_1155del | NR/NR | 0,134 | 0,5 | 67 | Yes | clinic | CRC (53) | MSI-H & MSH6 loss | - | 4,16 | 2,15 | 15,22 | 63.31 | 63.31 | 0,984 | Class 4: Likely pathogenic |
| AF1_III4 | Yes | clinic | EC (59) | MSH6 loss | - | 7,08 | ||||||||||
| AF1_III1 | No | clinic | CRC (59) | MSH6 loss | 4,16 | |||||||||||
| c.1618_1620del | NR/NR | 0,959 | 0,9 | 70 | Yes | clinic | CRC (46) | MSI-H & MSH6 loss | - | 6,54 | 1,85 | 1,85 | 12,07 | 108,62 | 0,991 | Class 5 Pathogenic |
| 70 | No | clinic | CRC (43) | MSI-H & MSH6 loss | 6,54 | |||||||||||
| 75 | Yes | clinic | CRC (45) | MSI-H & MSH6 loss | - | - | ||||||||||
| c.3150_3161dup | NR/NR | 0,961 | 0,9 | 82 | Yes | clinic | OC (47) | MSI-H & PMS2 loss | - | 30,28 | - | 28,75 | 870,61 | 7835,47 | 1,000 | Class 5 Pathogenic |
| AF2_II2 | Yes | clinic | CRC (61) | MSI-H & MSH6 loss | - | 0,99 | ||||||||||
| AF3_III3 | Yes | clinic | CRC (47) | MSH6 loss | - | 29,08 | ||||||||||
| AF3_II2 | No | clinic | EC (56) | MSH6 loss | 1,75 | |||||||||||
| AF3_II2 | No | clinic | CRC (75) | MSI-H & MSH6 loss | 4,16 | |||||||||||
| AF3_II11 | No | clinic | CRC (68) | MSI-H | 4,16 | |||||||||||
| Case ID | Variant Calling | RefSNP (rs) | MAF | In Silico predictions | ClinVar Classification | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Splicing | Protein Function | |||||||||||
| Gene | cDNA Change | Predicted Protein Change | ExAC/ESP | SIFT (score) | Mutation Taster (p-value) | Polyphen2/HumDiv (score) | Polyphen2/HumVar (score) | Provean | ||||
| 10 | EPCAM (^) | c.811G>T | p.(Val271Phe) | NR/NR | No changes | D (0) | D (1) | PrD (1.000) | PrD (0.989) | NP | Not reported | |
| 29 | POLD1 | c.2275G>A | p.(Val759Ile) | rs145473716 | 0.002/0.001 | No changes | D (0) | D (1) | PrD (1.000) | PrD (0.988) | NP | VUS (1) vs Benign/Likely benign (6)/+ |
| 30 | APC | c.7936C>G | p.(Gln2646Glu) | NR/NR | No changes | D (0.02) | D (1) | PsD (0.688) | B (0.182) | NP | VUS (1)/+ | |
| 39 | FAN1 | c.149T>G | p.(Met50Arg) | rs148404807 | 0.002/0.002 | No changes | T (0.08) | D (1) | PrD (0.991) | PsD (0.690) | NP | Not reported |
| 55 | PMS1 | c.497A>C | p.(Lys166Thr) | NR/NR | No changes | D (0) | D (1) | PsD (0.757) | PsD (0.599) | NP | Not reported | |
| 58 | EXO1 | c.2212-1G>A | p.Val738_Lys743del | rs4150000 | 0.0019/0.0028 | Loss of ASS | NA | NA | NA | NA | NA | Lhotaa et al., 2016: r.2212_2229del; p.Val738_Lys743del |
| 59 | APC | c.1966C>G | p.(Leu656Val) | rs577466163 | NR/NR | Gain of DSS | D (0) | D (1) | PrD(0.999) | PrD (0.998) | NP | VUS (1)/+ |
| 62 and 74 | MSH3 | c.2732T>G | p.(Leu911Trp) | rs41545019 | 0.002/0.004 | No changes | D (0) | D (0.999) | PrD (1.000) | PrD (0.978) | NP | Not reported |
| 65 | MUTYH | c.1437_1439del | p.Glu480del | rs587778541 | NR/0.000 | No changes | NA | NA | NA | NA | D (−7.78) | Pathogenic (9)/** |
| 74 | MSH3 | c.685T>C | p.(Tyr229His) | NR/NR | No changes | D (0.01) | D (0.999) | PrD (1.000) | PrD (0.973) | NP | Not reported | |
| 81 | BUB1 | c.2473C>T | p.(Pro825Ser) | rs748392521 | NR/NR | No changes | D (0) | D(1) | PrD (1.000) | PrD (0.997) | NP | Not reported |
| 85 | MSH3 | c.3072G>C | p.(Gln1024His) | rs147640909 | 0.000/0.000 | Loss of DSS/Inconclusive at ASS | T (0.39) | P (0.996) | B (0.007) | B (0.013) | NP | Not reported |
| 96 | APC | c.7514G>A | p.(Arg2505Gln) | rs147549623 | 0.001/0.001 | No changes | D (0.04) | D (1) | PrD (1.000) | PrD (0.961) | NP | Benign/Likely benign (8)/++ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dámaso, E.; González-Acosta, M.; Vargas-Parra, G.; Navarro, M.; Balmaña, J.; Ramon y Cajal, T.; Tuset, N.; Thompson, B.A.; Marín, F.; Fernández, A.; et al. Comprehensive Constitutional Genetic and Epigenetic Characterization of Lynch-Like Individuals. Cancers 2020, 12, 1799. https://doi.org/10.3390/cancers12071799
Dámaso E, González-Acosta M, Vargas-Parra G, Navarro M, Balmaña J, Ramon y Cajal T, Tuset N, Thompson BA, Marín F, Fernández A, et al. Comprehensive Constitutional Genetic and Epigenetic Characterization of Lynch-Like Individuals. Cancers. 2020; 12(7):1799. https://doi.org/10.3390/cancers12071799
Chicago/Turabian StyleDámaso, Estela, Maribel González-Acosta, Gardenia Vargas-Parra, Matilde Navarro, Judith Balmaña, Teresa Ramon y Cajal, Noemí Tuset, Bryony A. Thompson, Fátima Marín, Anna Fernández, and et al. 2020. "Comprehensive Constitutional Genetic and Epigenetic Characterization of Lynch-Like Individuals" Cancers 12, no. 7: 1799. https://doi.org/10.3390/cancers12071799