Activation of Epidermal Growth Factor Receptor Sensitizes Glioblastoma Cells to Hypoxia-Induced Cell Death

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
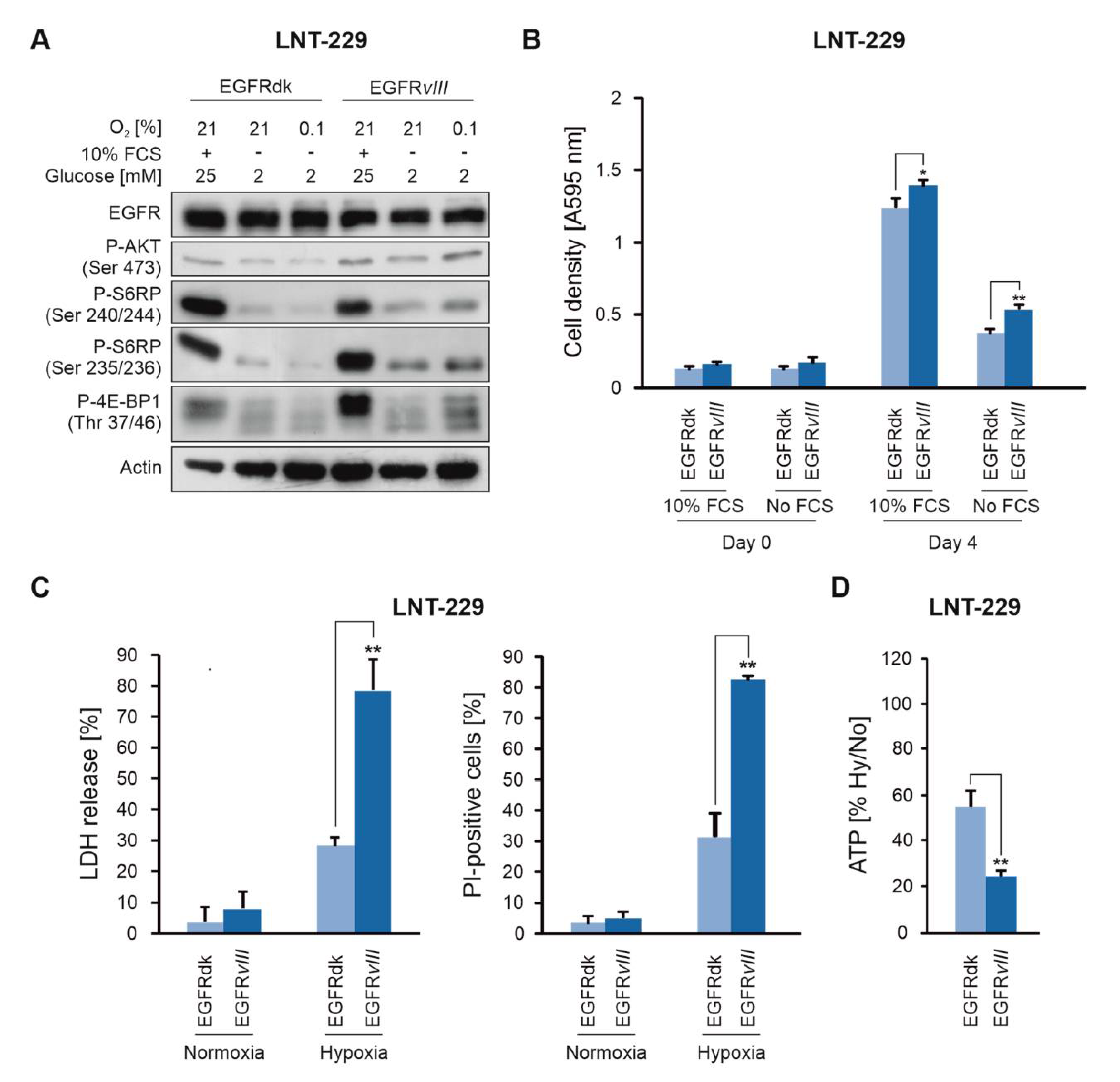
2.1. EGFRvIII Expression Sensitizes Human GB Cells to Hypoxia-Induced Cell Death
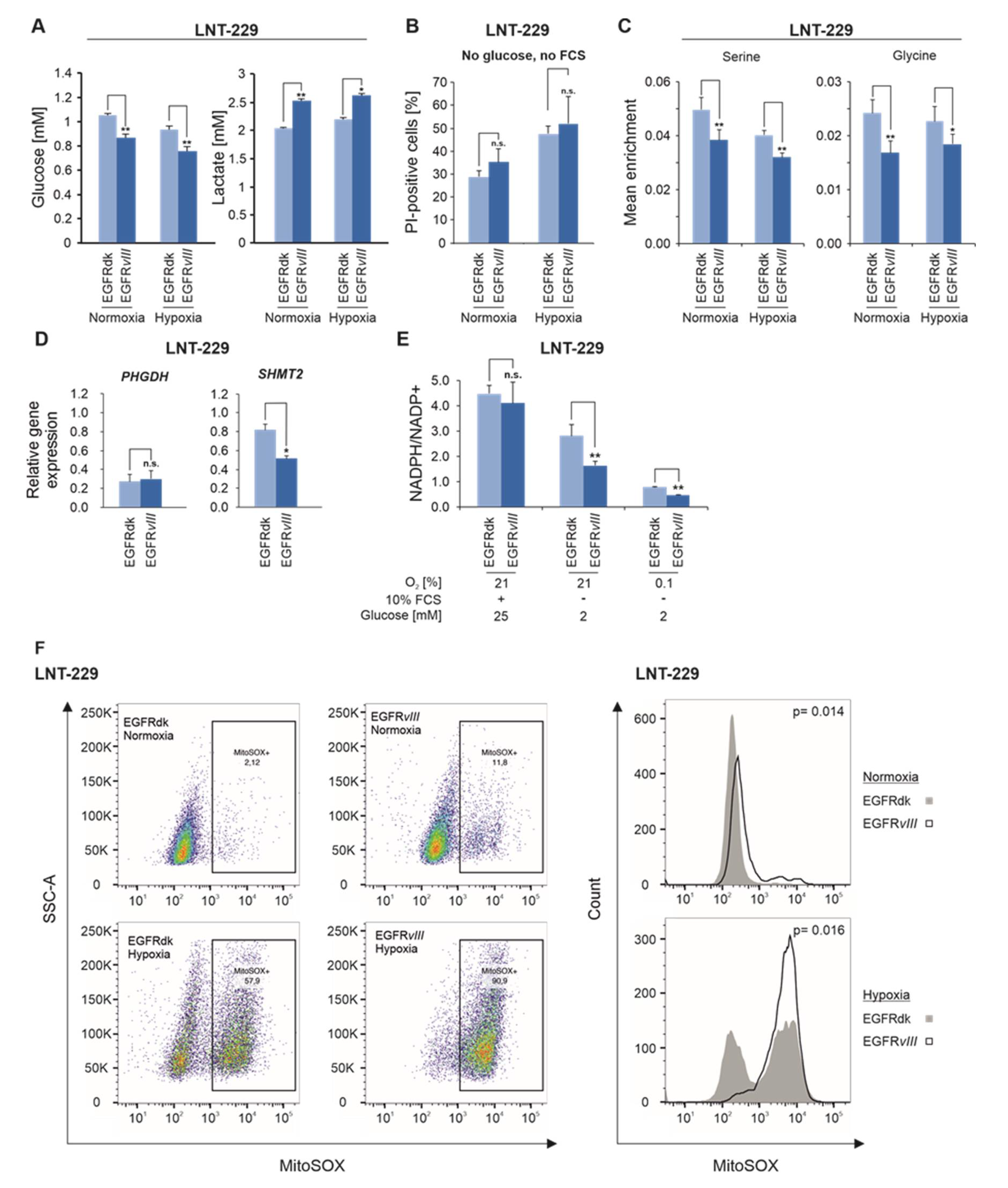
2.2. EGFRvIII Enhances Glucose Consumption and Lactate Production under Hypoxic Conditions and Reduces Flux Into the One-Carbon Metabolism
2.3. EGF Enhances the Sensitivity of GB Cells to Hypoxia-Induced Cell Death
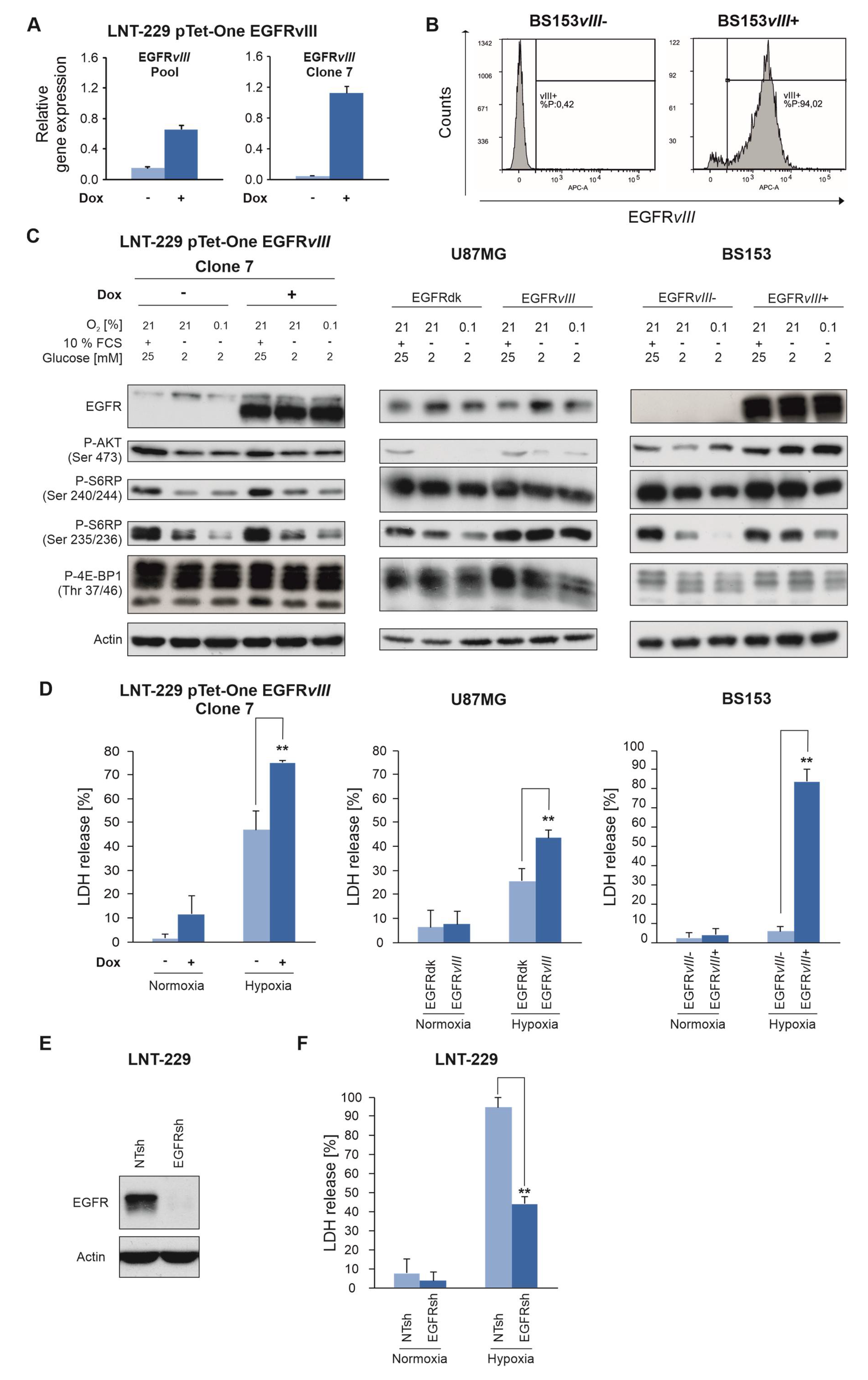
2.4. EGFR Governs Sensitivity of GB Cells To Hypoxia and Nutrient Deprivation
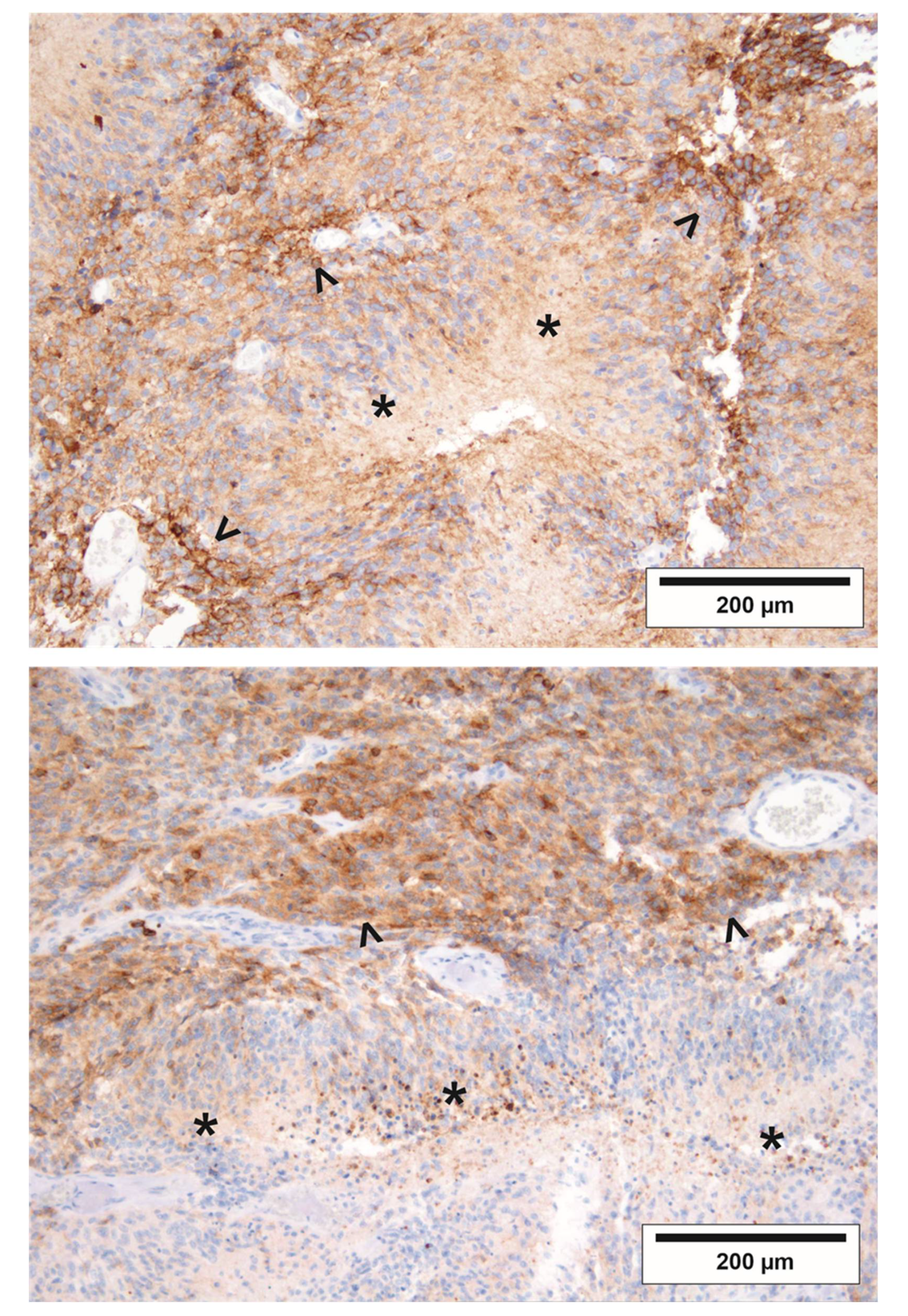
2.5. The Regional Pattern of EGFRvIII in GBs is Compatible with Increased Vulnerability to Starvation Conditions In Vivo
3. Discussion
4. Materials and Methods
4.1. Reagents, Cell Lines and Culture Conditions
4.2. Generation of LNT-229 pTetOne EGFRvIII Cells
4.3. Generation of LNT-229 NTsh and EGFRsh Cells
4.4. Generation of BS153 EGFRvIII- and EGFRvIII+ Cells
4.5. Induction of Hypoxia
4.6. Quantitative Reverse Transcription-PCR (qRT-PCR) Analysis
4.7. Immunoblot Analysis
4.8. Cell Density and Cell Viability Assays
4.9. Measurement of Glucose, Lactate, ATP and Oxygen
4.10. Stable Isotope Tracer Analysis and Quantification of Intracellular Metabolites
4.11. NADPH/NADP+ Measurement
4.12. MitoSOX FACS Analysis
4.13. Immunohistochemistry of Formalin-Fixed, Paraffin-Embedded Tissue
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ostrom, Q.T.; Gittleman, H.; Truitt, G.; Boscia, A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011–2015. Neuro-Oncology 2018, 20, iv1–iv86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Taillibert, S.; Kanner, A.A.; Kesari, S.; Steinberg, D.M.; Toms, S.A.; Taylor, L.P.; Lieberman, F.; Silvani, A.; Fink, K.L.; et al. Maintenance Therapy With Tumor-Treating Fields Plus Temozolomide vs Temozolomide Alone for Glioblastoma: A Randomized Clinical Trial. JAMA 2015, 314, 2535–2543. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.K.; Cvrljevic, A.N.; Johns, T.G. The epidermal growth factor receptor variant III (EGFRvIII): Where wild things are altered. FEBS J. 2013, 280, 5350–5370. [Google Scholar] [CrossRef]
- Yoshimoto, K.; Dang, J.; Zhu, S.; Nathanson, D.; Huang, T.; Dumont, R.; Seligson, D.B.; Yong, W.H.; Xiong, Z.; Rao, N.; et al. Development of a real-time RT-PCR assay for detecting EGFRvIII in glioblastoma samples. Clin. Cancer Res. 2008, 14, 488–493. [Google Scholar] [CrossRef] [Green Version]
- Padfield, E.; Ellis, H.P.; Kurian, K.M. Current Therapeutic Advances Targeting EGFR and EGFRvIII in Glioblastoma. Front. Oncol. 2015, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [Green Version]
- Chinnaiyan, P.; Won, M.; Wen, P.Y.; Rojiani, A.M.; Werner-Wasik, M.; Shih, H.A.; Ashby, L.S.; Michael Yu, H.-H.; Stieber, V.W.; Malone, S.C.; et al. A randomized phase II study of everolimus in combination with chemoradiation in newly diagnosed glioblastoma: Results of NRG Oncology RTOG 0913. Neuro-Oncology 2018, 20, 666–673. [Google Scholar] [CrossRef] [Green Version]
- Ronellenfitsch, M.W.; Steinbach, J.P.; Wick, W. Epidermal growth factor receptor and mammalian target of rapamycin as therapeutic targets in malignant glioma: Current clinical status and perspectives. Target. Oncol. 2010, 5, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef] [Green Version]
- Westphal, M.; Heese, O.; Steinbach, J.P.; Schnell, O.; Schackert, G.; Mehdorn, M.; Schulz, D.; Simon, M.; Schlegel, U.; Senft, C.; et al. A randomised, open label phase III trial with nimotuzumab, an anti-epidermal growth factor receptor monoclonal antibody in the treatment of newly diagnosed adult glioblastoma. Eur. J. Cancer 2015, 51, 522–532. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Gorlia, T.; Bady, P.; Platten, M.; van den Bent, M.J.; Taphoorn, M.J.B.; Steuve, J.; Brandes, A.A.; Hamou, M.-F.; Wick, A.; et al. Phase II Study of Radiotherapy and Temsirolimus versus Radiochemotherapy with Temozolomide in Patients with Newly Diagnosed Glioblastoma without MGMT Promoter Hypermethylation (EORTC 26082). Clin. Cancer Res. 2016, 22, 4797–4806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronellenfitsch, M.W.; Brucker, D.P.; Burger, M.C.; Wolking, S.; Tritschler, F.; Rieger, J.; Wick, W.; Weller, M.; Steinbach, J.P. Antagonism of the mammalian target of rapamycin selectively mediates metabolic effects of epidermal growth factor receptor inhibition and protects human malignant glioma cells from hypoxia-induced cell death. Brain 2009, 132, 1509–1522. [Google Scholar] [CrossRef] [Green Version]
- Steinbach, J.P.; Klumpp, A.; Wolburg, H.; Weller, M. Inhibition of Epidermal Growth Factor Receptor Signaling Protects Human Malignant Glioma Cells from Hypoxia-Induced Cell Death. Cancer Res. 2004, 64, 1575–1578. [Google Scholar] [CrossRef] [Green Version]
- Thiepold, A.-L.; Lorenz, N.I.; Foltyn, M.; Engel, A.L.; Divé, I.; Urban, H.; Heller, S.; Bruns, I.; Hofmann, U.; Dröse, S.; et al. Mammalian target of rapamycin complex 1 activation sensitizes human glioma cells to hypoxia-induced cell death. Brain 2017, 140, 2623–2638. [Google Scholar] [CrossRef]
- Pandita, A.; Aldape, K.D.; Zadeh, G.; Guha, A.; James, C.D. Contrasting in vivo and in vitro fates of glioblastoma cell subpopulations with amplified EGFR. Genes Chromosomes Cancer 2004, 39, 29–36. [Google Scholar] [CrossRef]
- Struve, N.; Riedel, M.; Schulte, A.; Rieckmann, T.; Grob, T.J.; Gal, A.; Rothkamm, K.; Lamszus, K.; Petersen, C.; Dikomey, E.; et al. EGFRvIII does not affect radiosensitivity with or without gefitinib treatment in glioblastoma cells. Oncotarget 2015, 6, 33867–33877. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Fan, J.; Venneti, S.; Wan, Y.-W.; Pawel, B.R.; Zhang, J.; Finley, L.W.S.; Lu, C.; Lindsten, T.; Cross, J.R.; et al. Serine catabolism regulates mitochondrial redox control during hypoxia. Cancer Discov. 2014, 4, 1406–1417. [Google Scholar] [CrossRef] [Green Version]
- Fack, F.; Espedal, H.; Keunen, O.; Golebiewska, A.; Obad, N.; Harter, P.N.; Mittelbronn, M.; Bähr, O.; Weyerbrock, A.; Stuhr, L.; et al. Bevacizumab treatment induces metabolic adaptation toward anaerobic metabolism in glioblastomas. Acta Neuropathol. 2015, 129, 115–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapisarda, A.; Hollingshead, M.; Uranchimeg, B.; Bonomi, C.A.; Borgel, S.D.; Carter, J.P.; Gehrs, B.; Raffeld, M.; Kinders, R.J.; Parchment, R.; et al. Increased antitumor activity of bevacizumab in combination with hypoxia inducible factor-1 inhibition. Mol. Cancer Ther. 2009, 8, 1867–1877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinot, O.L.; de La Motte Rouge, T.; Moore, N.; Zeaiter, A.; Das, A.; Phillips, H.; Modrusan, Z.; Cloughesy, T. AVAglio: Phase 3 trial of bevacizumab plus temozolomide and radiotherapy in newly diagnosed glioblastoma multiforme. Adv. Ther. 2011, 28, 334–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronellenfitsch, M.W.; Zeiner, P.S.; Mittelbronn, M.; Urban, H.; Pietsch, T.; Reuter, D.; Senft, C.; Steinbach, J.P.; Westphal, M.; Harter, P.N. Akt and mTORC1 signaling as predictive biomarkers for the EGFR antibody nimotuzumab in glioblastoma. Acta Neuropathol. Commun. 2018, 6, 81. [Google Scholar] [CrossRef]
- Harter, P.N.; Jennewein, L.; Baumgarten, P.; Ilina, E.; Burger, M.C.; Thiepold, A.-L.; Tichy, J.; Zörnig, M.; Senft, C.; Steinbach, J.P.; et al. Immunohistochemical Assessment of Phosphorylated mTORC1-Pathway Proteins in Human Brain Tumors. PLoS ONE 2015, 10, e0127123. [Google Scholar] [CrossRef] [Green Version]
- Steinbach, J.P.; Supra, P.; Huang, H.-J.S.; Cavenee, W.K.; Weller, M. CD95-mediated apoptosis of human glioma cells: Modulation by epidermal growth factor receptor activity. Brain Pathol. 2002, 12, 12–20. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luger, A.-L.; Lorenz, N.I.; Urban, H.; Divé, I.; Engel, A.L.; Strassheimer, F.; Dettmer, K.; Zeiner, P.S.; Shaid, S.; Struve, N.; et al. Activation of Epidermal Growth Factor Receptor Sensitizes Glioblastoma Cells to Hypoxia-Induced Cell Death. Cancers 2020, 12, 2144. https://doi.org/10.3390/cancers12082144
Luger A-L, Lorenz NI, Urban H, Divé I, Engel AL, Strassheimer F, Dettmer K, Zeiner PS, Shaid S, Struve N, et al. Activation of Epidermal Growth Factor Receptor Sensitizes Glioblastoma Cells to Hypoxia-Induced Cell Death. Cancers. 2020; 12(8):2144. https://doi.org/10.3390/cancers12082144
Chicago/Turabian StyleLuger, Anna-Luisa, Nadja I. Lorenz, Hans Urban, Iris Divé, Anna L. Engel, Florian Strassheimer, Katja Dettmer, Pia S. Zeiner, Shabnam Shaid, Nina Struve, and et al. 2020. "Activation of Epidermal Growth Factor Receptor Sensitizes Glioblastoma Cells to Hypoxia-Induced Cell Death" Cancers 12, no. 8: 2144. https://doi.org/10.3390/cancers12082144