Breast Tumor Cell Invasion and Pro-Invasive Activity of Cancer-Associated Fibroblasts Co-Targeted by Novel Urokinase-Derived Decapeptides

 , , , , , and
, , , , , and
Abstract
:
1. Introduction
2. Results
2.1. Peptide Design
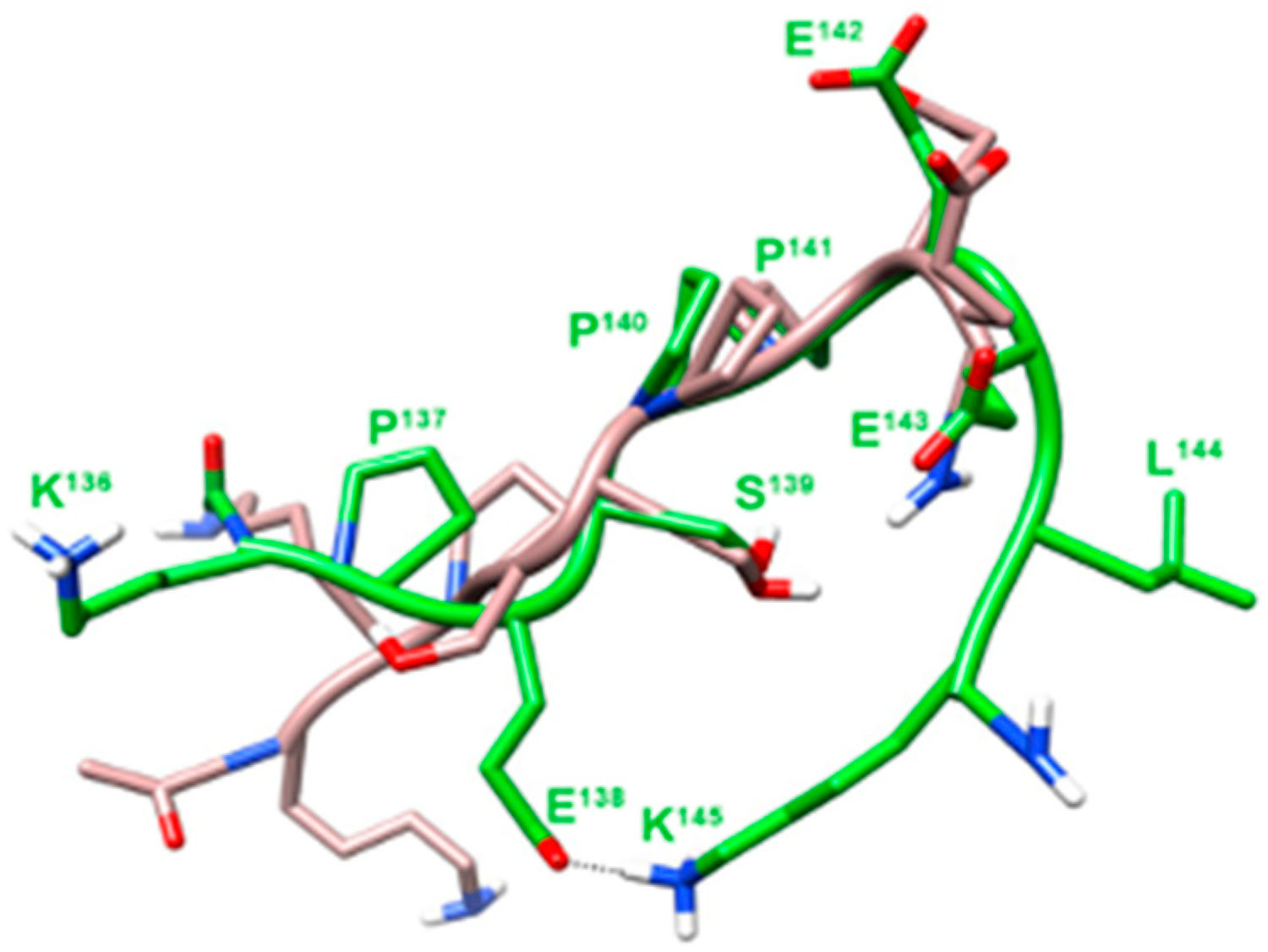
2.2. Conformational Analysis
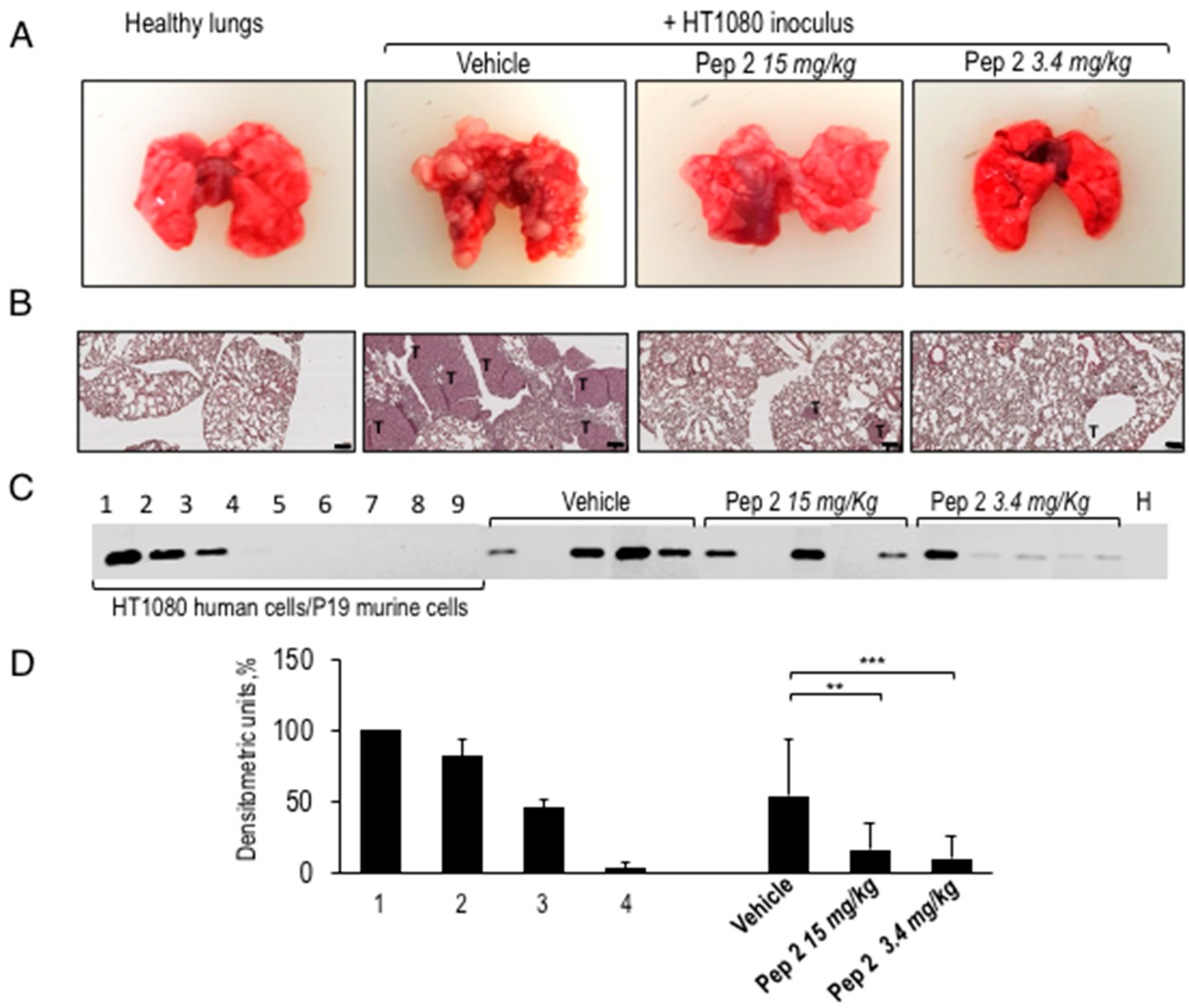
2.3. Inhibition of Malignant Cell Lung Colonization In Vivo by Pep 2
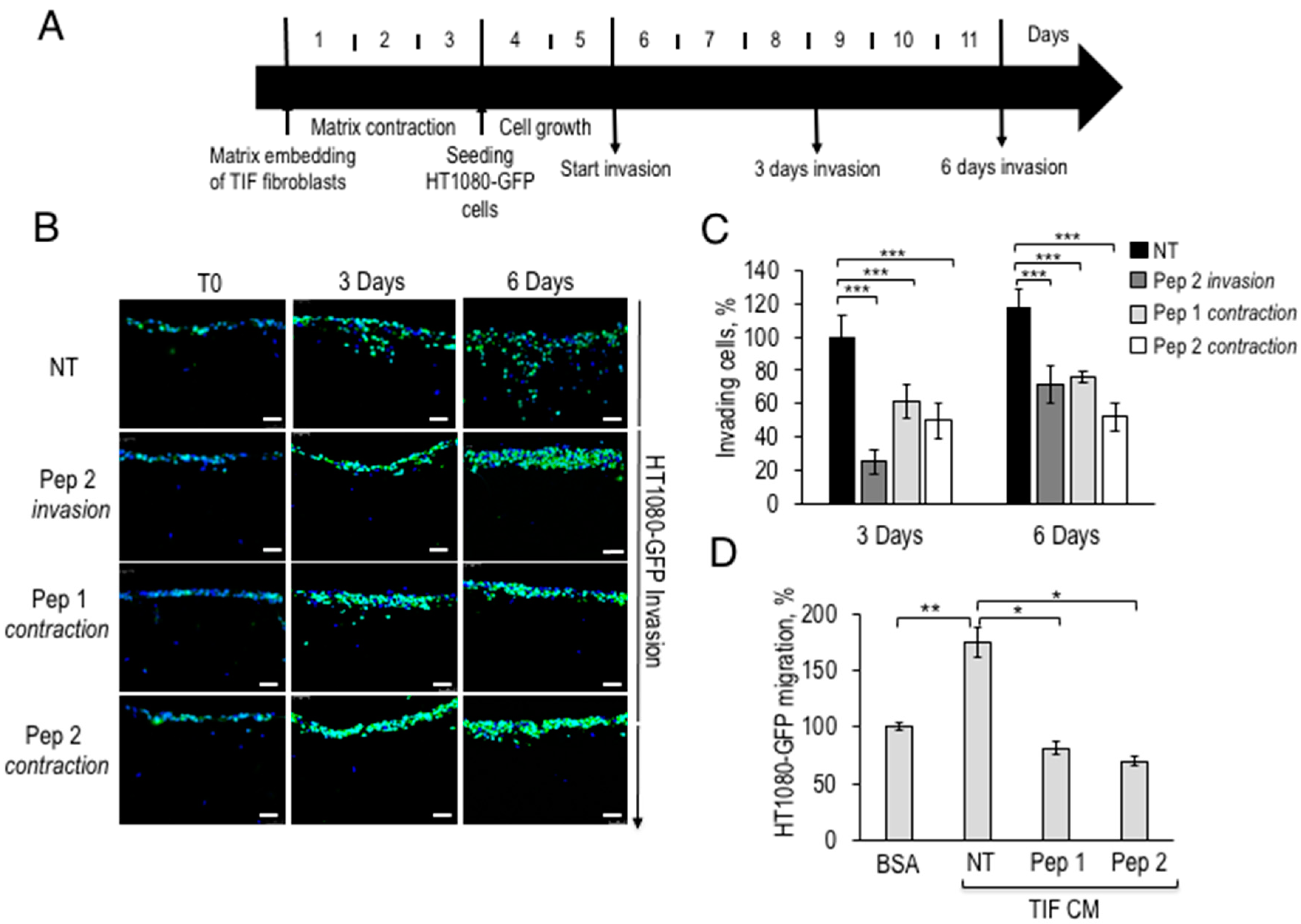
2.4. Inhibition of Fibrosarcoma Invasion in 3D-Organotypic Assay by Pep 1 or Pep 2 Decapeptides
2.5. Inhibition of HT1080 Cell Invasion by αv Integrin Receptor Interaction with Pep 1 or Pep 2
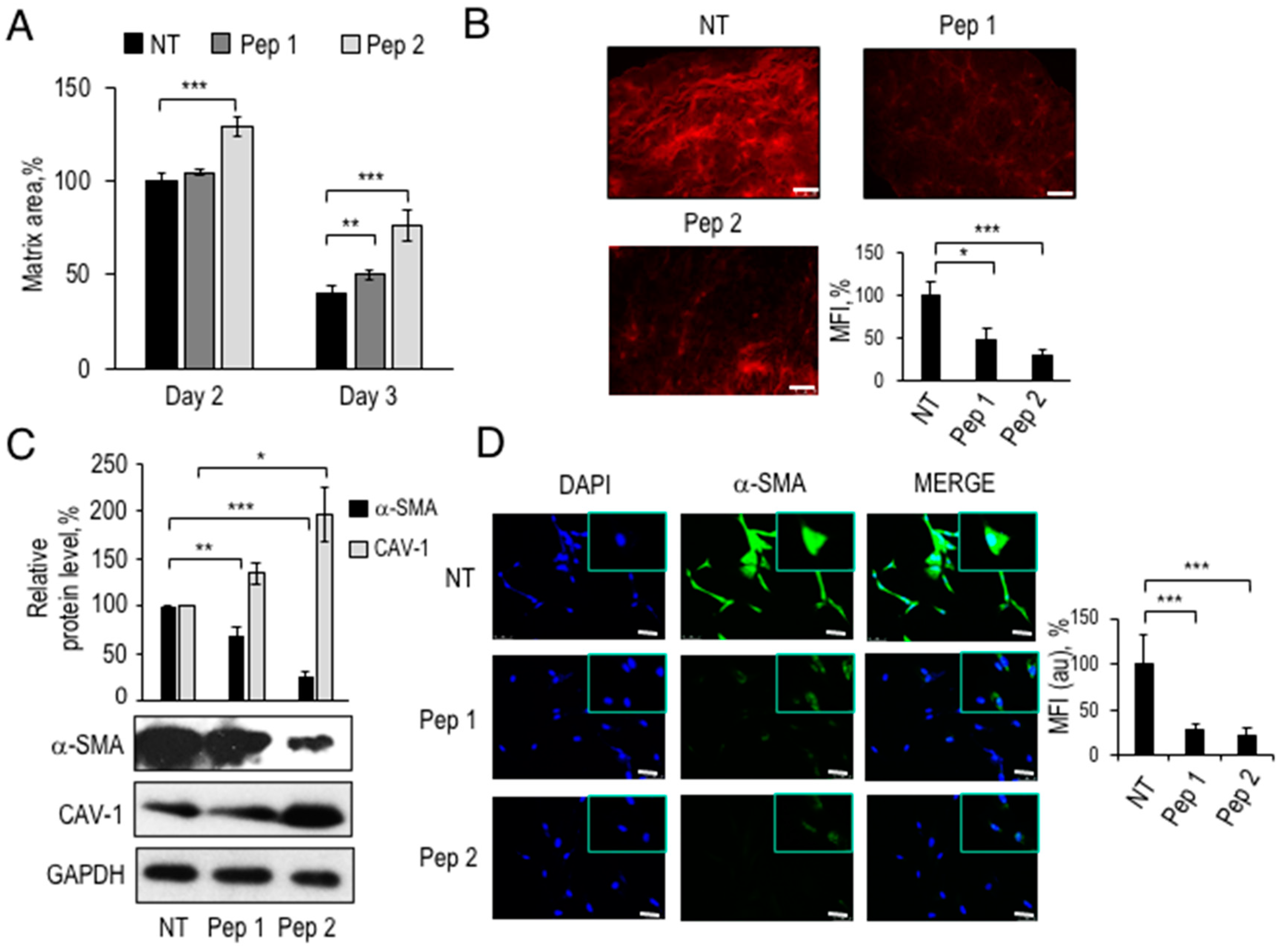
2.6. Partial Reversion of CAF-Like Phenotype in TIF Fibroblasts Exposed to Pep 1 or Pep 2
2.7. Proliferation and Apoptosis of TIF and HT1080-GFP Cells Unaffected by Pep 2
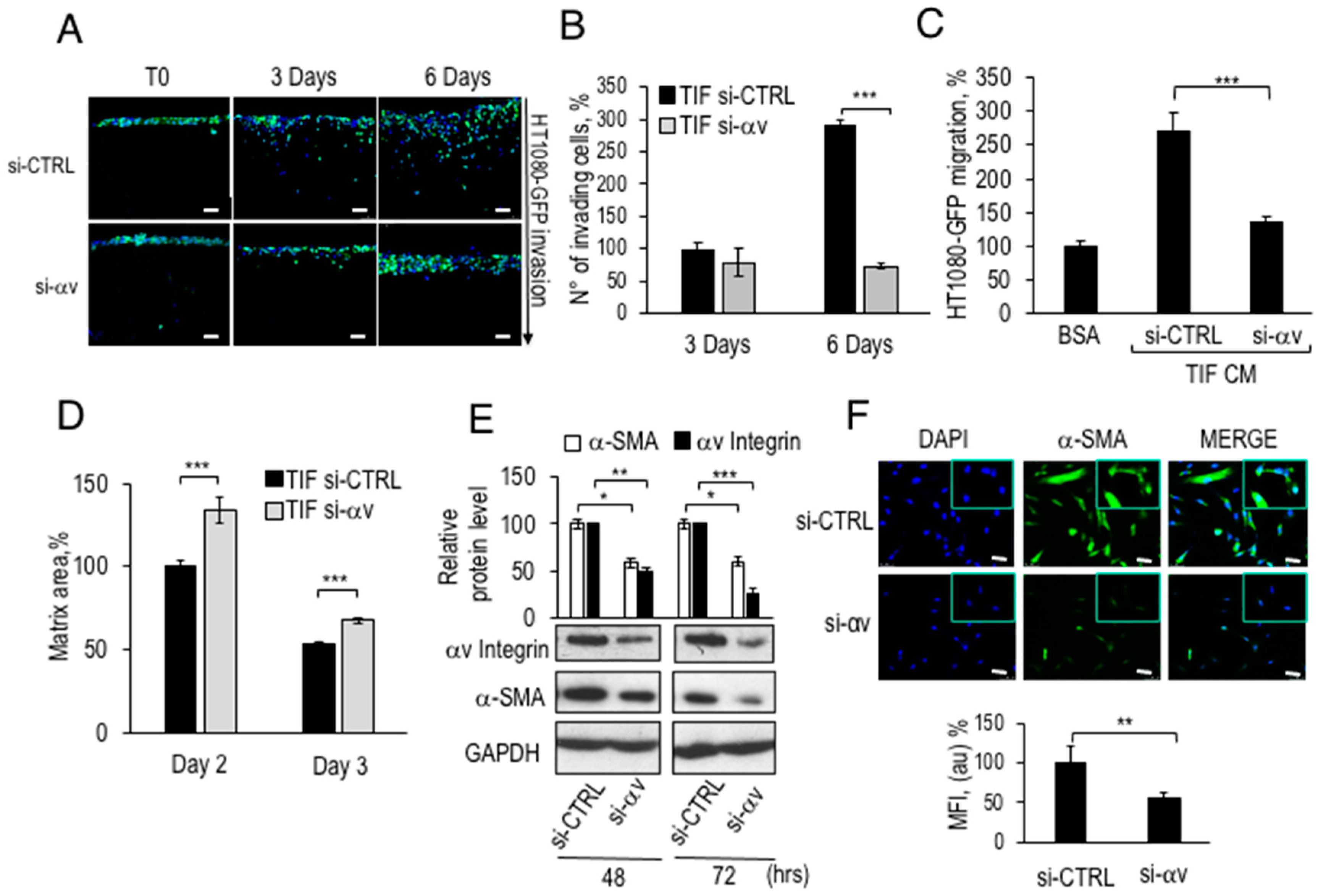
2.8. Inhibition of CAF-Like Phenotype and Pro-Invasive Activity of αv-Silenced TIF Fibroblasts
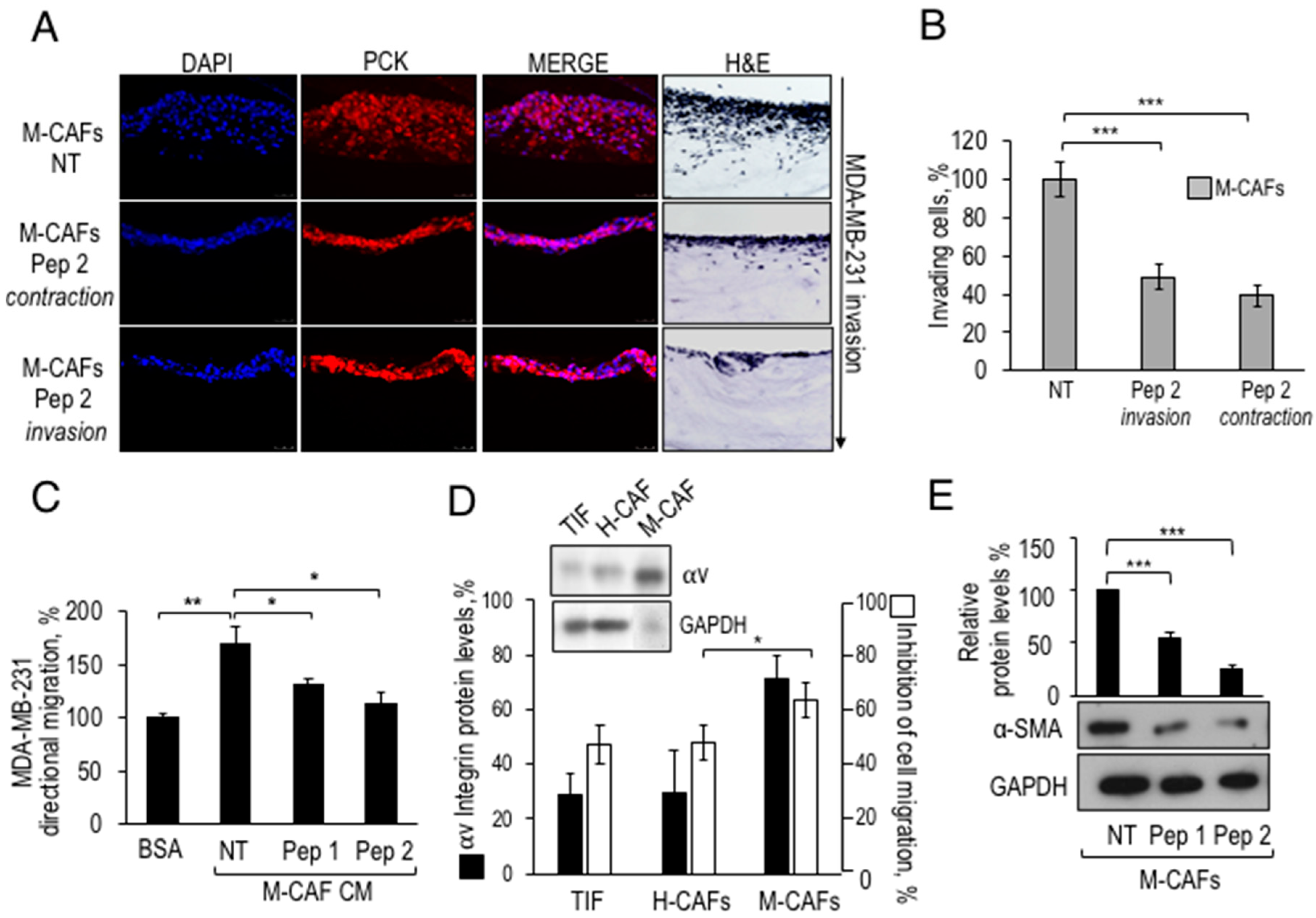
2.9. Exposure to Pep 2 of Primary CAFs Prevents Their Pro-Invasive Activity in a 3D-Assay
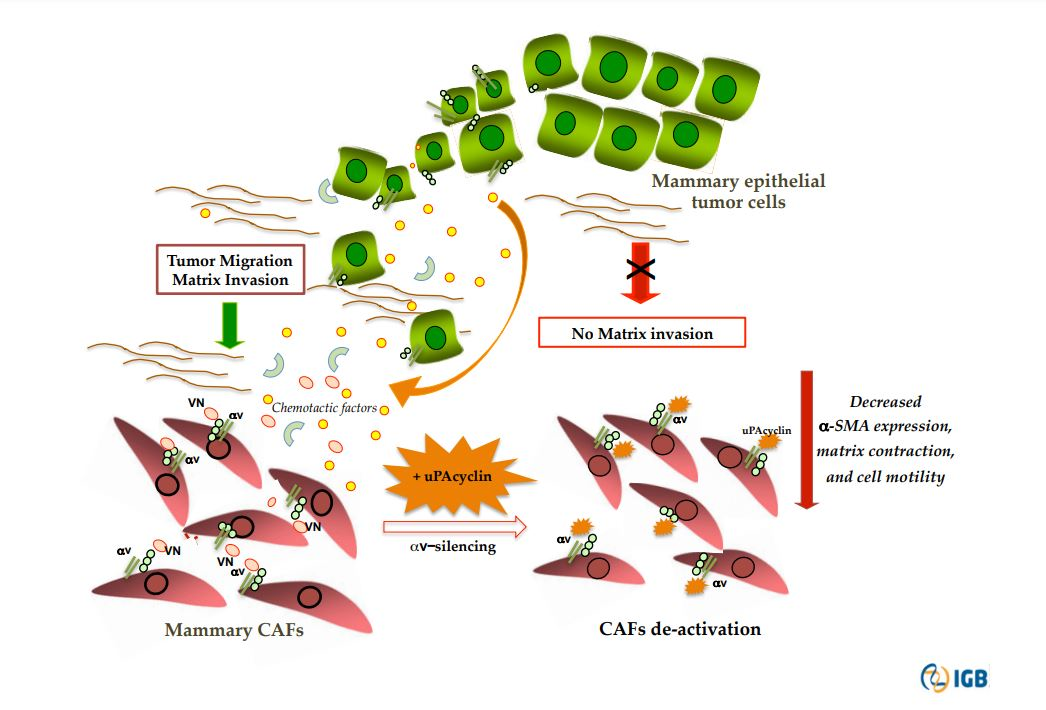
3. Discussion
4. Materials and Methods
4.1. Cell lines and Culture Conditions
4.2. Peptide Synthesis
4.3. Conformational Analysis
4.4. Binding Assay
4.5. Protein Extraction and Western Blot Analysis
- polyclonal anti-α-SMA (Abcam; Ab5694)
- polyclonal anti-Caveolin-1 (Santa Cruz Biotechnology; sc-894)
- monclonal ant-GAPDH (Abcam; Ab9484)
- polyclonal anti-αv (Millipore; Ab1930)
4.6. Apoptosis Assay
4.7. RNA Silencing
4.8. Tail Vein Assay of Lung Metastasis
4.9. Ex Vivo Lung Analysis
4.10. Quantification of Human DNA in Murine Lungs
4.11. Immunofluorescence
4.12. Migration Assays
4.13. Matrix Contraction Assay
4.14. Organotypic Invasion Assay
4.15. Statistical Analyses
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Coussens, L.M. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Bierie, B.; Moses, H.L. Tumour microenvironment: TGFβ: The molecular Jekyll and Hyde of cancer. Nat. Rev. Cancer 2006, 6, 506–520. [Google Scholar] [CrossRef] [PubMed]
- De Vincenzo, A.; Belli, S.; Franco, P.; Telesca, M.; Iaccarino, I.; Botti, G.; Carriero, M.V.; Ranson, M.; Stoppelli, M.P. Paracrine recruitment and activation of fibroblasts by c-myc expressing breast epithelial cells through the IGFs/IGF-1R axis. Int. J. Cancer 2019, 145, 2827–2839. [Google Scholar] [CrossRef] [PubMed]
- Gaggioli, C.; Hooper, S.; Hidalgo-Carcedo, C.; Grosse, R.; Marshall, J.F.; Harrington, K.; Sahai, E. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat. Cell Biol. 2007, 9, 1392–1400. [Google Scholar] [CrossRef]
- Attieh, Y.; Clark, A.G.; Grass, C.; Richon, S.; Pocard, M.; Mariano, P.; Elkhatib, N.; Betz, T.; Gurchenkov, B.; Vignjevic, D.M. Cancer-associated fibroblasts lead tumor invasion through integrin- β 3-dependent fibronectin assembly. J. Cell Biol. 2017, 16, 3509–3520. [Google Scholar] [CrossRef] [Green Version]
- Raab-Westphal, S.; Marshall, J.F.; Goodman, S.L. Integrins as therapeutic targets: Successes and cancers. Cancers 2017, 9, 110. [Google Scholar] [CrossRef]
- Seguin, L.; Desgrosellier, J.S.; Weis, S.M.; Cheresh, D.A. Integrins and cancer: Regulators of cancer stemness, metastasis, and drug resistance. Trends Cell Biol. 2015, 25, 234–240. [Google Scholar] [CrossRef] [Green Version]
- Vogetseder, A.; Thies, S.; Ingold, B.; Roth, P.; Weller, M.; Schraml, P.; Goodman, S.L.; Moch, H. αv-Integrin isoform expression in primary human tumors and brain metastases. Int. J. Cancer 2013, 133, 2362–2371. [Google Scholar] [CrossRef] [PubMed]
- Alday-Parejo, B.; Stupp, R.; Ruegg, C. Are integrins still practicable targets for Anti-Cancer Therapy? Cancers 2019, 11, 978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carriero, M.V.; Stoppelli, M.P. The Urokinase-type Plasminogen Activator and the Generation of Inhibitors of Urokinase Activity and Signaling. Curr. Pharm. Des. 2011, 17, 1944–1961. [Google Scholar] [CrossRef] [PubMed]
- Franco, P.; Vocca, I.; Carriero, M.V.; Alfano, D.; Cito, L.; Longamesi-Cattani, I.; Grieco, P.; Ossowski, L.; Stoppelli, M.P. Activation of urokinase receptor by a novel interaction between the connecting peptide region of urokinase and αvβ5 integrin. J. Cell Sci. 2006, 119, 3424–3434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, P.; Carotenuto, A.; Marcozzi, C.; Votta, G.; Sarno, C.; Iaccarino, I.; Brancaccio, D.; De Vincenzo, A.; Novellino, E.; Grieco, P.; et al. Opposite modulation of cell migration by distinct subregions of urokinase connecting peptide. ChemBioChem 2013, 14, 882–889. [Google Scholar] [CrossRef]
- Franco, P.; Iaccarino, C.; Chiaradonna, F.; Brandazza, A.; Iavarone, C.; Mastronicola, M.R.; Nolli, M.L.; Stoppelli, M.P. Phosphorylation of human pro-urokinase on Ser138/303 impairs its receptor-dependent ability to promote myelomonocytic adherence and motility. J. Cell Biol. 1997, 137, 779–791. [Google Scholar] [CrossRef] [Green Version]
- Vocca, I.; Franco, P.; Alfano, D.; Votta, G.; Carriero, M.V.; Estrada, Y.; Caputi, M.; Netti, P.; Ossowski, L.; Stoppelli, M.P. Inhibition of migration and invasion of carcinoma cells by urokinase-derived antagonists of alphavbeta5 integrin activation. Int. J. Cancer 2009, 124, 316–325. [Google Scholar] [CrossRef]
- Baranyi, U.; Winter, B.; Gugerell, A.; Hegedus, B.; Brostjan, C.; Laufer, G.; Messner, B. Primary Human Fibroblasts in Culture Switch to a Myofibroblast-Like Phenotype Independently of TGF Beta. Cells 2019, 8, 721. [Google Scholar] [CrossRef] [Green Version]
- Waisberg, J.; De Souza Viana, L.; Affonso, R.J.; Morini Silva, S.R.; Denadai, M.V.; Margeotto, F.B.; De Souza, C.S.; Matos, D. Overexpression of the ITGAV gene is associated with progression and spread of colorectal cancer. Anticancer Res. 2014, 34, 5599–5607. [Google Scholar]
- Cheuk, I.W.; Siu, M.T.; Ho, J.C.; Shin, V.Y.; Kwong, A. ITGAV targeting as a therapeutic approach for treatment of metastatic breast cancer. Am. J. Cancer Res. 2020, 10, 211–223. [Google Scholar]
- Affo, S.; Yu, L.X.; Schwabe, R.F. The role of cancer-associated fibroblasts and fibrosis in liver cancer. Annu. Rev. Pathol. 2017, 12, 153–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, J.; Giancotti, F.G. Integrin signaling in cancer: Mechanotransduction, stemness, epithelial plasticity and therapeutic resistance. Cancer Cell 2019, 35, 347–367. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, S.V.; Pasapera, A.M.; Sabass, B.; Waterman, C.M. Force fluctuations within focal adhesion mediate ECM-rigidity sensing to guide directed cell migration. Cell 2012, 151, 1513–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Persio, C.M.; Van De Water, L. Integrin Regulation of CAF Differentiation and Function. Cancers 2019, 11, 715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signalling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Mazar, A.P.; Lebrun, J.; Rabbani, S.A. An antiangiogenic urokinase-derived pepide combined with tamoxifen decreases tumor growth and metastasis in a syngeneic model of breast cancer. Cancer Res. 2002, 62, 4678–4684. [Google Scholar]
- Ghamande, S.A.; Silverman, M.H.; Huh, W.; Behbakht, K.; Ball, G.; Cuasay, L.; Würtz, S.O.; Brunner, N.; Gold, M.A. A phase 2, randomized, double-blind, placebo-controlled trial of clinical activity and safety of subcutaneous Å6 in women with asymptomatic CA125 progression after first-line chemotherapy of epithelial ovarian cancer. Gynecol. Oncol. 2008, 111, 89–94. [Google Scholar] [CrossRef]
- Gold, M.A.; Brady, W.E.; Lankes, H.A.; Rose, P.G.; Kelley, J.L.; De Geest, K.; Crispens, M.A.; Resnick, K.E.; Howell, S.B. A phase II study of a urokinase-derived peptide (Å6) in the treatment of persistent or recurrent epithelial ovarian, fallopian tube, or primary peritoneal carcinomas: A Gynecologic Oncology Group study. Gynecol. Oncol. 2012, 125, 635–639. [Google Scholar] [CrossRef] [Green Version]
- Baggio, C.; Barile, E.; Di Sorbo, G.; Kipps, T.J.; Pellecchia, M. The cell surface receptor CD44: NMR-based characterization of putative ligands. ChemMedChem. 2016, 11, 1097–1106. [Google Scholar] [CrossRef] [Green Version]
- Maurer, G.D.; Trischler, I.; Adams, B.; Tabatabai, G.; Roger, W.W.; Stupp, R.; Weller, M. Cilengitide modulates attachment and viability of human glioma cells, but not sensitivity to irradiation or temozolomide in vitro. Neuro Oncol. 2009, 11, 747–756. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Hegi, M.E.; Gorlia, T.; Erridge, S.C.; Perry, J.; Hong, Y.K.; Aldape, K.D.; Lhermitte, B.; Pietsch, T.; Grujicic, D.; et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 1008–1100. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Dai, J.; Zao, Z.; Shelley, G.; Keller, E.T. Abituzumab targeting of av-class integrins inhibits prostate cancer progression. Mol. Cancer Res. 2017, 15, 875–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasinska, L.; Mackiewicz, J. Integrins as a new target for cancer treatment. Anticancer Agents Med. Chem. 2019, 19, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, A.R.; Hart, I.R.; Watson, A.R.; Welti, J.C.; Silva, R.G.; Robinson, S.D.; Violante, G.D.; Gourlaouen, M.; Salih, M.; Jones, M.C.; et al. Stimulation of tumor growth and angiogenesis by low concentrations of RGD-mimetic integrin inhibitors. Nat. Med. 2009, 15, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Yeung, T.L.; Leung, C.S.; Li, F.; Wong, S.T.; Mok, S.C. Targeting Stromal-Cancer Cell Crosstalk Networks in Ovarian Cancer Treatment. Biomolecules 2016, 6, 3. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.S.K.; Sng, M.K.; Teo, Z.Q.; Chong, H.C.; Twang, J.S.; Tan, N.S. Targeting nuclear receptors in cancer-associated fibroblasts as concurrent therapy to inhibit development of chemoresistant tumors. Oncogene 2018, 37, 160–173. [Google Scholar] [CrossRef] [Green Version]
- Jiang, G.M.; Xu, W.; Du, J.; Zhang, K.S.; Zhang, Q.G.; Wang, X.W.; Liu, Z.G.; Liu, S.Q.; Xie, W.Y.; Liu, H.F.; et al. The application of the fibroblast activation protein α-targeted immunotherapy strategy. Oncotarget 2016, 7, 33472–33482. [Google Scholar] [CrossRef] [Green Version]
- Tabai, K.; Le, A.; Weaver, V.M.; Werb, Z. Targeting the cancer-associated fibroblasts as a treatment in triple-negative breast cancer. Oncotarget 2016, 7, 82889–82901. [Google Scholar] [CrossRef] [Green Version]
- Conway, J.R.W.; Vennin, C.; Cazet, A.S.; Hermann, D.; Murphy, K.J.; Warren, S.C.; Wullkopf, L.; Boulghourjian, A.; Zaratzian, A.; Da Silva, A.M.; et al. Three-dimensional organotypic matrices from alternative collagen sources as pre-clinical models for cell bioloy. Sci. Rep. 2017, 7, 16887. [Google Scholar] [CrossRef]
- Merlino, F.; Billard, E.; Yousif, A.M.; Di Maro, S.; Brancaccio, D.; Abate, L.; Carotenuto, A.; Bellavita, R.; d’Emmanuele di Villa Bianca, R.; Santicioli, P.; et al. Functional selectivity revealed by N-methylation scanning of human urotensin II and related peptides. J. Med. Chem. 2019, 62, 1455–1467. [Google Scholar] [CrossRef]
- Merlino, F.; Yousif, A.M.; Billard, É; Dufour-Gallant, J.; Turcotte, S.; Grieco, P.; Chatenet, D.; Lubell, W.D. Urotensin II(4−11) azasulfuryl peptides: Synthesis and biological activity. J. Med. Chem. 2016, 59, 4740–4752. [Google Scholar] [CrossRef] [PubMed]
- Merlino, F.; Zhou, Y.; Cai, M.; Carotenuto, A.; Yousif, A.M.; Brancaccio, D.; Di Maro, S.; Zappavigna, S.; Limatola, A.; Novellino, E.; et al. Development of macrocyclic peptidomimetics containing constrained α,α-dialkylated amino acids with potent and selective activity at human melanocortin receptors. J. Med. Chem. 2018, 61, 4263–4269. [Google Scholar] [CrossRef] [PubMed]
- Jullian, M.; Hernandez, A.; Maurras, A.; Puget, K.; Amblard, M.; Martinez, J.; Subra, G. N-terminus FITC labeling of peptides on solid support: The truth behind the spacer. Tetrahedron Lett. 2009, 50, 260–263. [Google Scholar] [CrossRef]
- Hwang, T.L.; Shaka, A.J. Water suppression that works. Excitation sculpting using arbitrary wave-forms and pulsed-field gradients. J. Magn. Res. 1995, 112, 275–279. [Google Scholar] [CrossRef]
- Piantini, U.; Sorensen, O.W.; Ernst, R.R. Multiple quantum filters for elucidating NMR coupling network. J. Am. Chem. Soc. 1982, 104, 6800–6801. [Google Scholar] [CrossRef]
- Marion, D.; Wüthrich, K. Application of phase sensitive two-dimensional correlated spectroscopy (COSY) for measurements of 1H-1H spin-spin coupling constants in proteins. Biochem. Biophys. Res. Commun. 1983, 113, 967–997. [Google Scholar] [CrossRef]
- Braunschweiler, L.; Ernst, R.R. Coherence transfer by isotropic mixing: Application to proton correlation spectroscopy. J. Magn. Reson. 1983, 53, 521–528. [Google Scholar] [CrossRef]
- Jenner, J.; Meyer, B.H.; Bachman, P.; Ernst, R.R. Investigation of exchange processes by two-dimensional NMR spectroscopy. J. Chem. Phys. 1979, 71, 4546–4553. [Google Scholar] [CrossRef]
- States, D.J.; Haberkorn, R.A.; Ruben, D.J. A Two-dimensional nuclear Overhauser experiment with pure absorption phase in four quadrants. J. Magn. Reson. 1982, 48, 286–292. [Google Scholar] [CrossRef]
- Bartels, C.; Xia, T.; Billeter, M.; Guentert, P.; Wüthrich, K. The program XEASY for computer-supported NMR spectral analysis of biological macromolecules. J. Biomol. NMR 1995, 6, 1–10. [Google Scholar] [CrossRef]
- Wüthrich, K. NMR of Proteins and Nucleic Acids; John Wiley & Sons: New York, NY, USA, 1986; ISBN 978-0-471-82893-8. [Google Scholar]
- Güntert, P.; Buchner, L. Combined automated NOE assignment and structure calculation with CYANA. J. Biomol. NMR 2015, 62, 453–471. [Google Scholar] [CrossRef] [PubMed]
- Maple, J.; Dinur, U.; Hagler, A.T. Derivation of force fields for molecular mechanics and dynamics from ab initio energy surface. Proc. Natl. Acad. Sci. USA 1988, 85, 5350–5354. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoppelli, M.P.; Tacchetti, C.; Cubellis, M.V.; Corti, A.; Hearing, V.J.; Cassani, G.; Appella, E.; Blaso, F. Autocrine saturation of pro-urokinase receptors on human A431 cells. Cell 1986, 45, 675–684. [Google Scholar] [CrossRef]
- Minopoli, M.; Botti, G.; Gigantino, V.; Ragone, C.; Sarno, S.; Motti, M.L.; Scognamoglio, G.; Greggi, S.; Scaffa, C.; Roca, M.S.; et al. Targeting the Formyl peptide receptor type 1 to prevent the adhesion of ovarian cancer cells onto mesothelium and subsequent invasion. J. Exp. Clin. Cancer Res. 2019, 38, 459–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcaraz, J.; Carrasco, J.L.; Millares, L.; Luis, I.C.; Fernández-Porras, F.J.; Martínez-Romero, A.; Diaz-Valdivia, N.; De Cos, J.S.; Rami-Porta, R.; Seijo, L. Stromal markers of activated tumor associated fibroblasts predict poor survival and are associated with necrosis in non-small cell lung cancer. Lung Cancer 2019, 135, 151–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timpson, P.; Mcghee, E.J.; Erami, Z.; Nobis, M.; Quinn, J.A.; Edward, M.; Anderson, K.I. Organotypic collagen I assay: A malleable platform to assess cell behaviour in a 3-dimensional context. J. Vis. Exp. 2011, e3089. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cmpd | Sequence * |
|---|---|
| CPp | Ac-K135KPSSPPEELKFQCGQKTLRPRFK158-NH2 |
| [138E]uPA-(135–158) | Ac-K135KPESPPEELKFQCGQKTLRPRFK158-NH2 |
| uPA-(135–143) | Ac-K135KPSSPPEE143-NH2 |
| Å6 | Ac-K136PSSPPEE143-NH2 |
| Pep 1 | Ac-K136PESPPEELK145-NH2 |
| Pep 2 (uPAcyclin) | Ac-K136P[ESPPEELK145]-NH2 |
| Scrambled Pep 2 | Ac-PS[EELKPEPK]-NH2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belli, S.; Franco, P.; Iommelli, F.; De Vincenzo, A.; Brancaccio, D.; Telesca, M.; Merlino, F.; Novellino, E.; Ranson, M.; Del Vecchio, S.; et al. Breast Tumor Cell Invasion and Pro-Invasive Activity of Cancer-Associated Fibroblasts Co-Targeted by Novel Urokinase-Derived Decapeptides. Cancers 2020, 12, 2404. https://doi.org/10.3390/cancers12092404
Belli S, Franco P, Iommelli F, De Vincenzo A, Brancaccio D, Telesca M, Merlino F, Novellino E, Ranson M, Del Vecchio S, et al. Breast Tumor Cell Invasion and Pro-Invasive Activity of Cancer-Associated Fibroblasts Co-Targeted by Novel Urokinase-Derived Decapeptides. Cancers. 2020; 12(9):2404. https://doi.org/10.3390/cancers12092404
Chicago/Turabian StyleBelli, Stefania, Paola Franco, Francesca Iommelli, Anna De Vincenzo, Diego Brancaccio, Marialucia Telesca, Francesco Merlino, Ettore Novellino, Marie Ranson, Silvana Del Vecchio, and et al. 2020. "Breast Tumor Cell Invasion and Pro-Invasive Activity of Cancer-Associated Fibroblasts Co-Targeted by Novel Urokinase-Derived Decapeptides" Cancers 12, no. 9: 2404. https://doi.org/10.3390/cancers12092404