Dichotomous Impact of Myc on rRNA Gene Activation and Silencing in B Cell Lymphomagenesis

 , , and
, , and 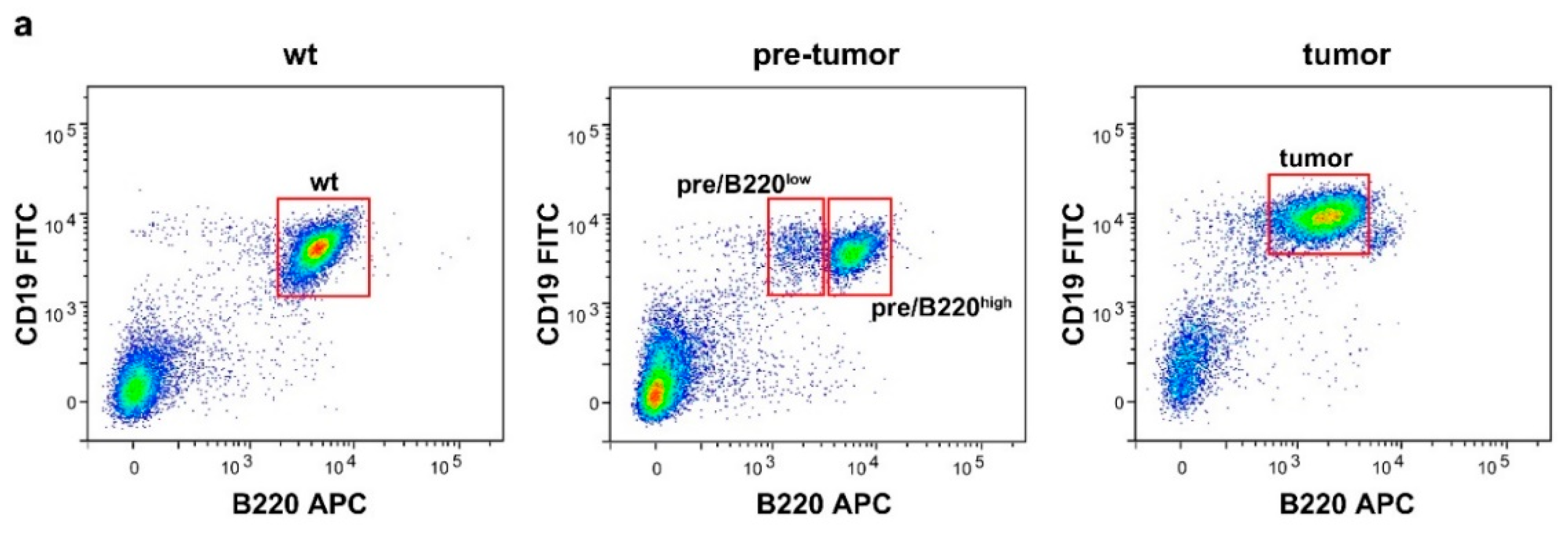{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
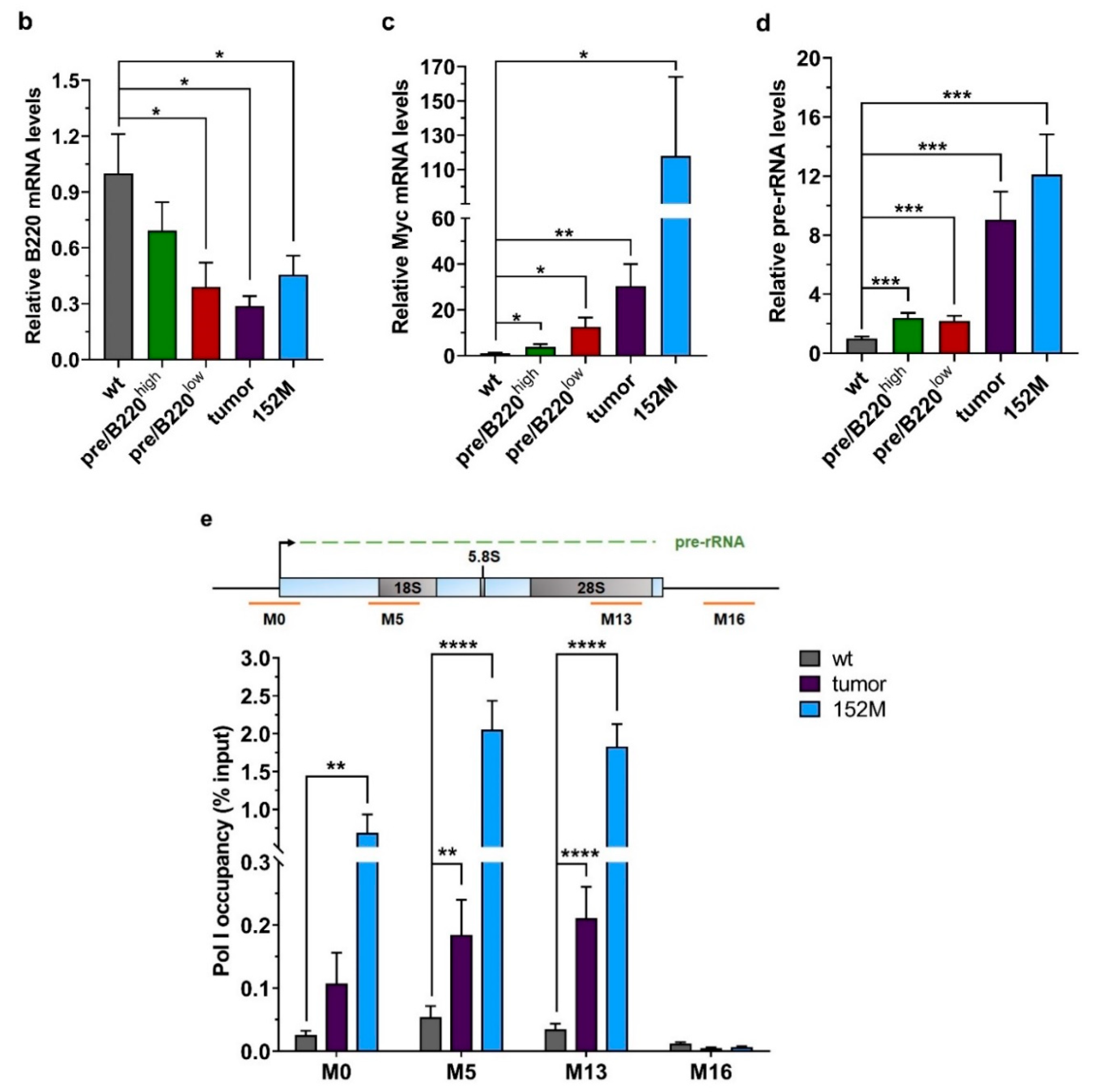
2.1. Drastic Increase of Myc and rRNA Gene Expression upon Lymphoma Progression
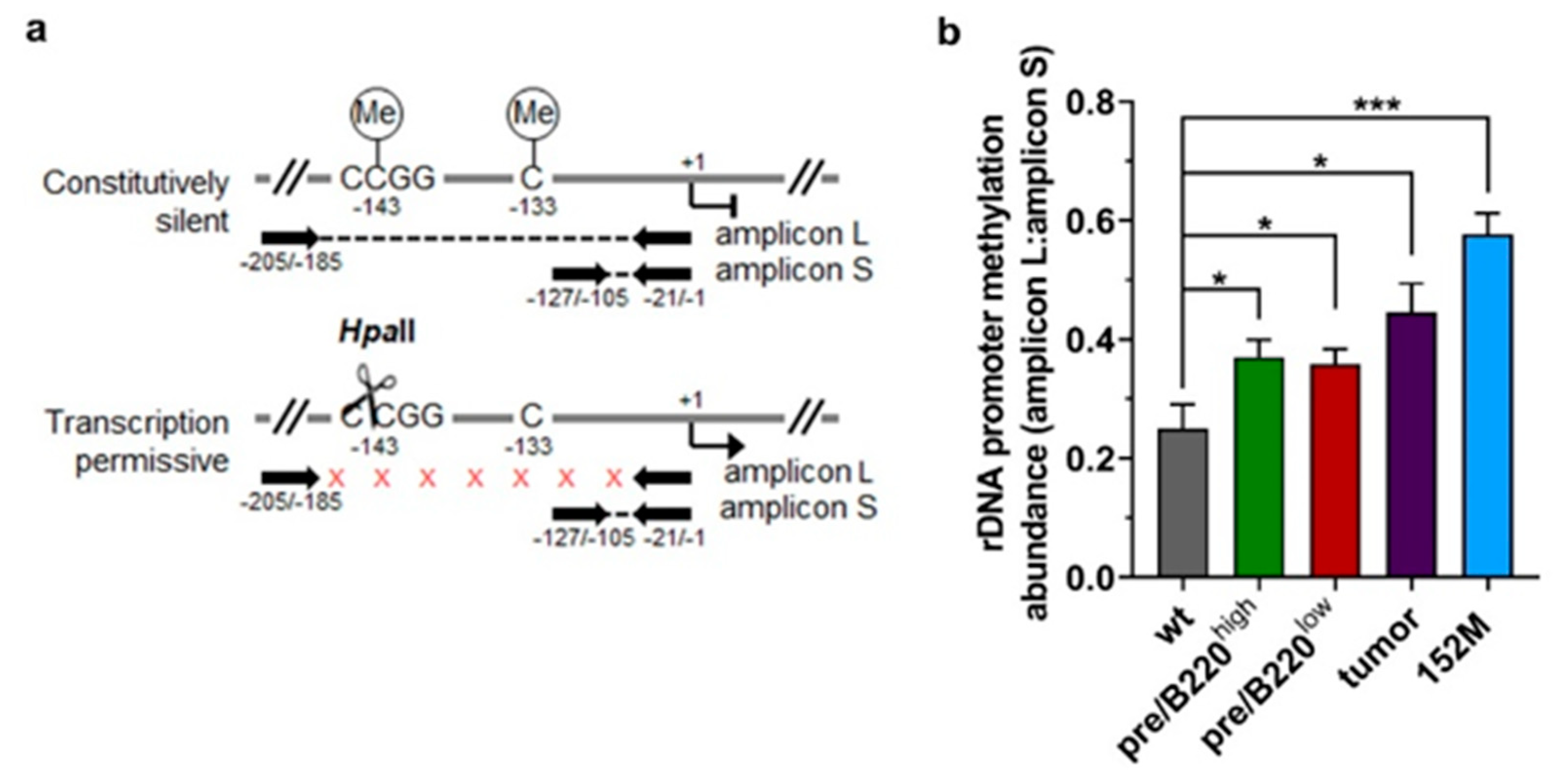
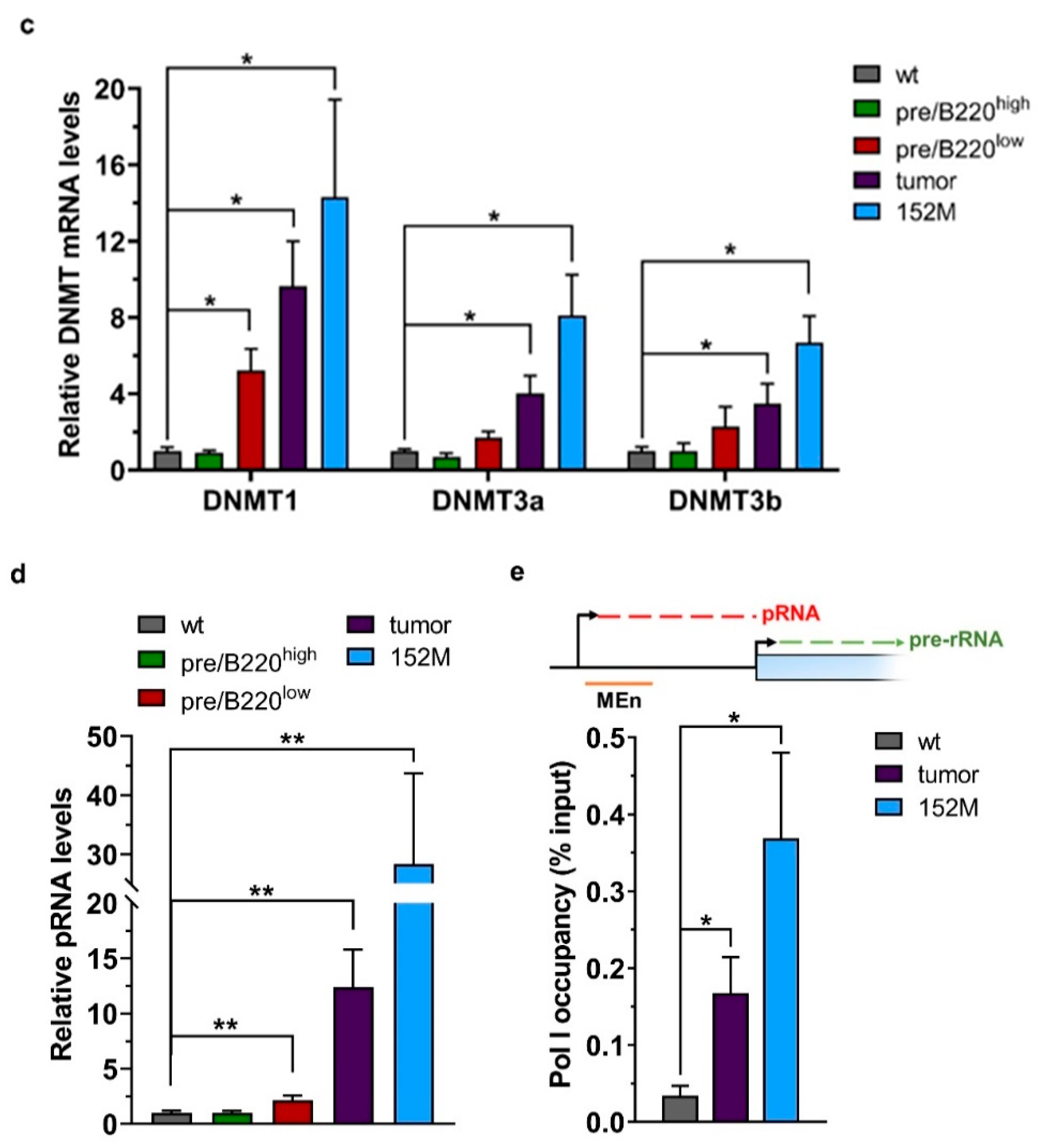
2.2. Hypertranscription of rDNA Coexists with Reinforced Epigenetic Silencing
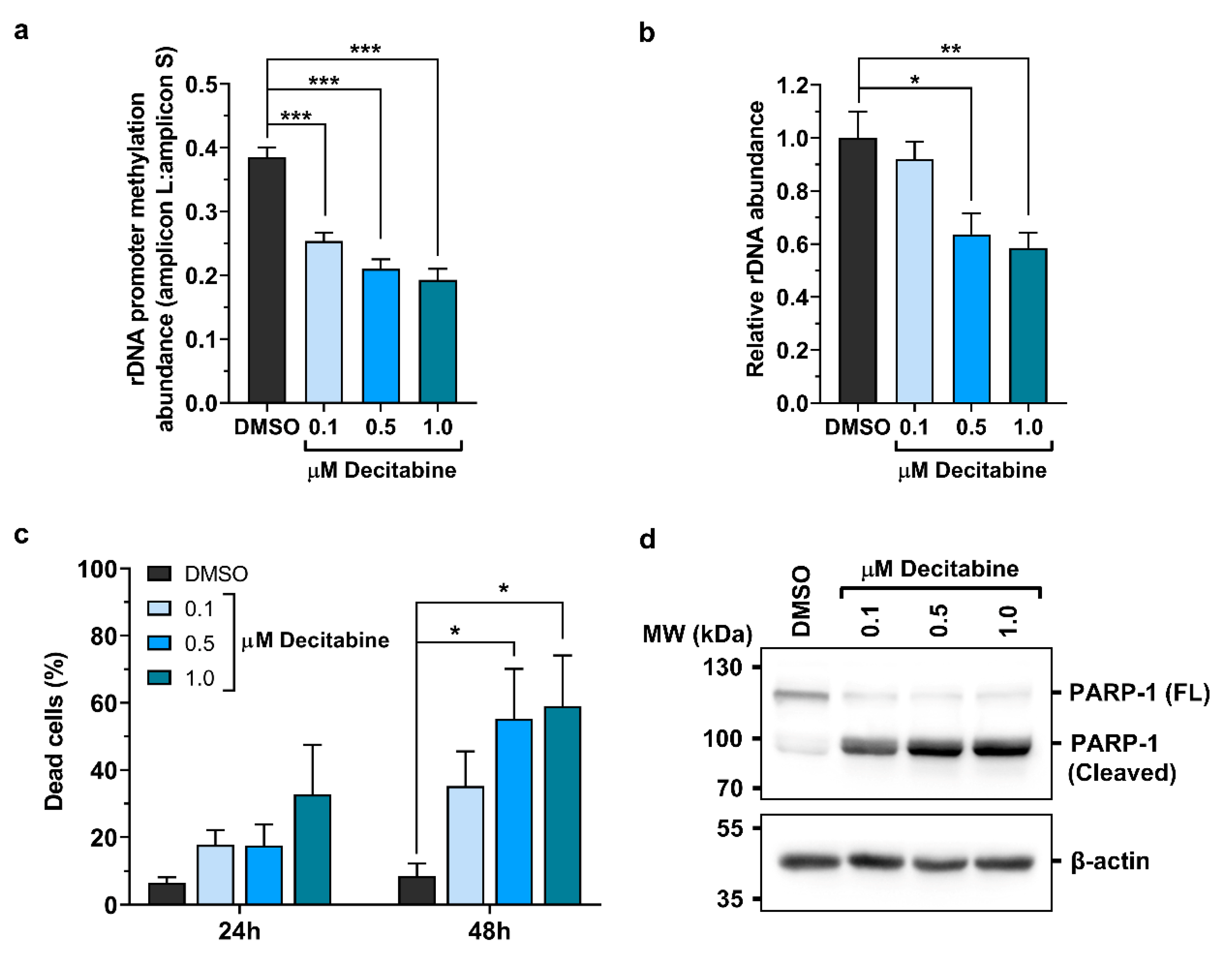
2.3. Promoter Methylation Protects rDNA Stability in Lymphoma Cells
3. Discussion
4. Materials and Methods
4.1. Eμ-Myc Mice and Isolation of B Cells
4.2. Fluorescence-Activated Cell Sorting (FACS)
4.3. Cell Culture and Decitabine Treatment
4.4. Gene Expression Analysis
4.5. Analyses of rDNA Methylation and Abundance
4.6. Chromatin Immunoprecipitation (ChIP)
4.7. Western Blotting
4.8. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Nunney, L.; Muir, B. Peto’s paradox and the hallmarks of cancer: Constructing an evolutionary framework for understanding the incidence of cancer. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20150161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baluapuri, A.; Wolf, E.; Eilers, M. Target gene-independent functions of MYC oncoproteins. Nat. Rev. Mol. Cell Biol. 2020, 21, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Dalla-Favera, R.; Bregni, M.; Erikson, J.; Patterson, D.; Gallo, R.C.; Croce, C.M. Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc. Natl. Acad. Sci. USA 1982, 79, 7824–7827. [Google Scholar] [CrossRef] [Green Version]
- Taub, R.; Kirsch, I.; Morton, C.; Lenoir, G.; Swan, D.; Tronick, S.; Aaronson, S.; Leder, P. Translocation of the c-myc gene into the immunoglobulin heavy chain locus in human Burkitt lymphoma and murine plasmacytoma cells. Proc. Natl. Acad. Sci. USA 1982, 79, 7837–7841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Van Calcar, S.; Qu, C.; Cavenee, W.K.; Zhang, M.Q.; Ren, B. A global transcriptional regulatory role for c-Myc in Burkitt’s lymphoma cells. Proc. Natl. Acad. Sci. USA 2003, 100, 8164–8169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeller, K.I.; Zhao, X.; Lee, C.W.; Chiu, K.P.; Yao, F.; Yustein, J.T.; Ooi, H.S.; Orlov, Y.L.; Shahab, A.; Yong, H.C.; et al. Global mapping of c-Myc binding sites and target gene networks in human B cells. Proc. Natl. Acad. Sci. USA 2006, 103, 17834–17839. [Google Scholar] [CrossRef] [Green Version]
- Walz, S.; Lorenzin, F.; Morton, J.; Wiese, K.E.; von Eyss, B.; Herold, S.; Rycak, L.; Dumay-Odelot, H.; Karim, S.; Bartkuhn, M.; et al. Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature 2014, 511, 483–487. [Google Scholar] [CrossRef]
- Arabi, A.; Wu, S.; Ridderstrale, K.; Bierhoff, H.; Shiue, C.; Fatyol, K.; Fahlen, S.; Hydbring, P.; Soderberg, O.; Grummt, I.; et al. c-Myc associates with ribosomal DNA and activates RNA polymerase I transcription. Nat. Cell Biol. 2005, 7, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Grandori, C.; Gomez-Roman, N.; Felton-Edkins, Z.A.; Ngouenet, C.; Galloway, D.A.; Eisenman, R.N.; White, R.J. c-Myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nat. Cell Biol. 2005, 7, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Roman, N.; Grandori, C.; Eisenman, R.N.; White, R.J. Direct activation of RNA polymerase III transcription by c-Myc. Nature 2003, 421, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.J.; White, R.J. MYC regulation of cell growth through control of transcription by RNA polymerases I and III. Cold Spring Harb. Perspect. Med. 2014, 4, a018408. [Google Scholar] [CrossRef]
- Destefanis, F.; Manara, V.; Bellosta, P. Myc as a Regulator of Ribosome Biogenesis and Cell Competition: A Link to Cancer. Int. J. Mol. Sci. 2020, 21, 4037. [Google Scholar] [CrossRef]
- Barna, M.; Pusic, A.; Zollo, O.; Costa, M.; Kondrashov, N.; Rego, E.; Rao, P.H.; Ruggero, D. Suppression of Myc oncogenic activity by ribosomal protein haploinsufficiency. Nature 2008, 456, 971–975. [Google Scholar] [CrossRef]
- Diolaiti, D.; McFerrin, L.; Carroll, P.A.; Eisenman, R.N. Functional interactions among members of the MAX and MLX transcriptional network during oncogenesis. Biochim. Biophys. Acta 2015, 1849, 484–500. [Google Scholar] [CrossRef] [Green Version]
- Mathsyaraja, H.; Freie, B.; Cheng, P.F.; Babaeva, E.; Catchpole, J.T.; Janssens, D.; Henikoff, S.; Eisenman, R.N. Max deletion destabilizes MYC protein and abrogates Eµ-Myc lymphomagenesis. Genes Dev. 2019, 33, 1252–1264. [Google Scholar] [CrossRef] [Green Version]
- Sharifi, S.; Bierhoff, H. Regulation of RNA Polymerase I Transcription in Development, Disease, and Aging. Annu. Rev. Biochem. 2018, 87, 51–73. [Google Scholar] [CrossRef]
- Bersaglieri, C.; Santoro, R. Genome Organization in and around the Nucleolus. Cells 2019, 8, 579. [Google Scholar] [CrossRef] [Green Version]
- Santoro, R.; Li, J.; Grummt, I. The nucleolar remodeling complex NoRC mediates heterochromatin formation and silencing of ribosomal gene transcription. Nat. Genet. 2002, 32, 393–396. [Google Scholar] [CrossRef]
- Santoro, R.; Grummt, I. Molecular mechanisms mediating methylation-dependent silencing of ribosomal gene transcription. Mol. Cell 2001, 8, 719–725. [Google Scholar] [CrossRef]
- McStay, B. Nucleolar organizer regions: Genomic ‘dark matter’ requiring illumination. Genes Dev. 2016, 30, 1598–1610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindstrom, M.S.; Jurada, D.; Bursac, S.; Orsolic, I.; Bartek, J.; Volarevic, S. Nucleolus as an emerging hub in maintenance of genome stability and cancer pathogenesis. Oncogene 2018, 37, 2351–2366. [Google Scholar] [CrossRef] [PubMed]
- Drygin, D.; Rice, W.G.; Grummt, I. The RNA polymerase I transcription machinery: An emerging target for the treatment of cancer. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 131–156. [Google Scholar] [CrossRef] [PubMed]
- Ruggero, D. Revisiting the nucleolus: From marker to dynamic integrator of cancer signaling. Sci. Signal. 2012, 5, pe38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penzo, M.; Montanaro, L.; Trere, D.; Derenzini, M. The Ribosome Biogenesis—Cancer Connection. Cells 2019, 8, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stults, D.M.; Killen, M.W.; Williamson, E.P.; Hourigan, J.S.; Vargas, H.D.; Arnold, S.M.; Moscow, J.A.; Pierce, A.J. Human rRNA gene clusters are recombinational hotspots in cancer. Cancer Res. 2009, 69, 9096–9104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, J.M.; Harris, A.W.; Pinkert, C.A.; Corcoran, L.M.; Alexander, W.S.; Cory, S.; Palmiter, R.D.; Brinster, R.L. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 1985, 318, 533–538. [Google Scholar] [CrossRef]
- Harris, A.W.; Pinkert, C.A.; Crawford, M.; Langdon, W.Y.; Brinster, R.L.; Adams, J.M. The E mu-myc transgenic mouse. A model for high-incidence spontaneous lymphoma and leukemia of early B cells. J. Exp. Med. 1988, 167, 353–371. [Google Scholar] [CrossRef] [Green Version]
- Vecchio, E.; Fiume, G.; Correnti, S.; Romano, S.; Iaccino, E.; Mimmi, S.; Maisano, D.; Nisticò, N.; Quinto, I. Insights about MYC and Apoptosis in B-Lymphomagenesis: An Update from Murine Models. Int. J. Mol. Sci. 2020, 21, 4265. [Google Scholar] [CrossRef] [PubMed]
- Ferrad, M.; Ghazzaui, N.; Issaoui, H.; Cook-Moreau, J.; Denizot, Y. Mouse Models of c-myc Deregulation Driven by IgH Locus Enhancers as Models of B-Cell Lymphomagenesis. Front. Immunol. 2020, 11, 1564. [Google Scholar] [CrossRef] [PubMed]
- Sabo, A.; Kress, T.R.; Pelizzola, M.; de Pretis, S.; Gorski, M.M.; Tesi, A.; Morelli, M.J.; Bora, P.; Doni, M.; Verrecchia, A.; et al. Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature 2014, 511, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Vecchio, E.; Golino, G.; Pisano, A.; Albano, F.; Falcone, C.; Ceglia, S.; Iaccino, E.; Mimmi, S.; Fiume, G.; Giurato, G.; et al. IBTK contributes to B-cell lymphomagenesis in Eµ-myc transgenic mice conferring resistance to apoptosis. Cell Death Dis. 2019, 10, 320. [Google Scholar] [CrossRef] [PubMed]
- Uemura, M.; Zheng, Q.; Koh, C.M.; Nelson, W.G.; Yegnasubramanian, S.; De Marzo, A.M. Overexpression of ribosomal RNA in prostate cancer is common but not linked to rDNA promoter hypomethylation. Oncogene 2012, 31, 1254–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Wang, Y.; Lv, Q.; Zhang, J.; Wang, Q.; Gao, F.; Hou, H.; Zhang, H.; Zhang, W.; Li, L. Overexpression of Ribosomal RNA in the Development of Human Cervical Cancer Is Associated with rDNA Promoter Hypomethylation. PLoS ONE 2016, 11, e0163340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McStay, B.; Grummt, I. The epigenetics of rRNA genes: From molecular to chromosome biology. Annu. Rev. Cell Dev. Biol. 2008, 24, 131–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; D’Alessio, A.C.; Ito, S.; Xia, K.; Wang, Z.; Cui, K.; Zhao, K.; Sun, Y.E.; Zhang, Y. Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature 2011, 473, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Poole, C.J.; Zheng, W.; Lodh, A.; Yevtodiyenko, A.; Liefwalker, D.; Li, H.; Felsher, D.W.; van Riggelen, J. DNMT3B overexpression contributes to aberrant DNA methylation and MYC-driven tumor maintenance in T-ALL and Burkitt’s lymphoma. Oncotarget 2017, 8, 76898–76920. [Google Scholar] [CrossRef] [Green Version]
- Lefebure, M.; Tothill, R.W.; Kruse, E.; Hawkins, E.D.; Shortt, J.; Matthews, G.M.; Gregory, G.P.; Martin, B.P.; Kelly, M.J.; Todorovski, I.; et al. Genomic characterisation of Eµ-Myc mouse lymphomas identifies Bcor as a Myc co-operative tumour-suppressor gene. Nat. Commun. 2017, 8, 14581. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Issa, J.J.; Kropf, P. DNA Hypomethylating Drugs in Cancer Therapy. Cold Spring Harb. Perspect. Med. 2017, 7, a026948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaitanya, G.V.; Steven, A.J.; Babu, P.P. PARP-1 cleavage fragments: Signatures of cell-death proteases in neurodegeneration. Cell Commun. Signal. 2010, 8, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. p53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef]
- Ho, T.; Tan, B.X.; Lane, D. How the Other Half Lives: What p53 Does When It Is Not Being a Transcription Factor. Int. J. Mol. Sci. 2019, 21, 13. [Google Scholar] [CrossRef] [Green Version]
- Eischen, C.M.; Weber, J.D.; Roussel, M.F.; Sherr, C.J.; Cleveland, J.L. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev. 1999, 13, 2658–2669. [Google Scholar] [CrossRef] [Green Version]
- Van Riggelen, J.; Yetil, A.; Felsher, D.W. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat. Rev. Cancer 2010, 10, 301–309. [Google Scholar] [CrossRef]
- Bywater, M.J.; Poortinga, G.; Sanij, E.; Hein, N.; Peck, A.; Cullinane, C.; Wall, M.; Cluse, L.; Drygin, D.; Anderes, K.; et al. Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer-specific activation of p53. Cancer Cell 2012, 22, 51–65. [Google Scholar] [CrossRef] [Green Version]
- Brenner, C.; Deplus, R.; Didelot, C.; Loriot, A.; Vire, E.; De Smet, C.; Gutierrez, A.; Danovi, D.; Bernard, D.; Boon, T.; et al. Myc represses transcription through recruitment of DNA methyltransferase corepressor. EMBO J. 2005, 24, 336–346. [Google Scholar] [CrossRef]
- Hervouet, E.; Vallette, F.M.; Cartron, P.F. Dnmt3/transcription factor interactions as crucial players in targeted DNA methylation. Epigenetics 2009, 4, 487–499. [Google Scholar] [CrossRef] [Green Version]
- Santoro, R.; Schmitz, K.M.; Sandoval, J.; Grummt, I. Intergenic transcripts originating from a subclass of ribosomal DNA repeats silence ribosomal RNA genes in trans. EMBO Rep. 2010, 11, 52–58. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.Y.; Loven, J.; Rahl, P.B.; Paranal, R.M.; Burge, C.B.; Bradner, J.E.; Lee, T.I.; Young, R.A. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 2012, 151, 56–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diesch, J.; Bywater, M.J.; Sanij, E.; Cameron, D.P.; Schierding, W.; Brajanovski, N.; Son, J.; Sornkom, J.; Hein, N.; Evers, M.; et al. Changes in long-range rDNA-genomic interactions associate with altered RNA polymerase II gene programs during malignant transformation. Commun. Biol. 2019, 2. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Lemos, B. Ribosomal DNA copy number amplification and loss in human cancers is linked to tumor genetic context, nucleolus activity, and proliferation. PLoS Genet. 2017, 13, e1006994. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Li, H.; Perry, J.M.; Singh, V.P.; Unruh, J.; Yu, Z.; Zakari, M.; McDowell, W.; Li, L.; Gerton, J.L. Ribosomal DNA copy number loss and sequence variation in cancer. PLoS Genet. 2017, 13, e1006771. [Google Scholar] [CrossRef] [PubMed]
- Gagnon-Kugler, T.; Langlois, F.; Stefanovsky, V.; Lessard, F.; Moss, T. Loss of human ribosomal gene CpG methylation enhances cryptic RNA polymerase II transcription and disrupts ribosomal RNA processing. Mol. Cell 2009, 35, 414–425. [Google Scholar] [CrossRef]
- Hoglund, A.; Nilsson, L.M.; Forshell, L.P.; Maclean, K.H.; Nilsson, J.A. Myc sensitizes p53-deficient cancer cells to the DNA-damaging effects of the DNA methyltransferase inhibitor decitabine. Blood 2009, 113, 4281–4288. [Google Scholar] [CrossRef] [Green Version]
- Khot, A.; Brajanovski, N.; Cameron, D.P.; Hein, N.; Maclachlan, K.H.; Sanij, E.; Lim, J.; Soong, J.; Link, E.; Blombery, P.; et al. First-in-Human RNA Polymerase I Transcription Inhibitor CX-5461 in Patients with Advanced Hematologic Cancers: Results of a Phase I Dose-Escalation Study. Cancer Discov. 2019, 9, 1036–1049. [Google Scholar] [CrossRef] [Green Version]
- Drouin, L.; McGrath, S.; Vidler, L.R.; Chaikuad, A.; Monteiro, O.; Tallant, C.; Philpott, M.; Rogers, C.; Fedorov, O.; Liu, M.; et al. Structure enabled design of BAZ2-ICR, a chemical probe targeting the bromodomains of BAZ2A and BAZ2B. J. Med. Chem. 2015, 58, 2553–2559. [Google Scholar] [CrossRef]
- Chen, P.; Chaikuad, A.; Bamborough, P.; Bantscheff, M.; Bountra, C.; Chung, C.W.; Fedorov, O.; Grandi, P.; Jung, D.; Lesniak, R.; et al. Discovery and Characterization of GSK2801, a Selective Chemical Probe for the Bromodomains BAZ2A and BAZ2B. J. Med. Chem. 2016, 59, 1410–1424. [Google Scholar] [CrossRef]
- Guetg, C.; Lienemann, P.; Sirri, V.; Grummt, I.; Hernandez-Verdun, D.; Hottiger, M.O.; Fussenegger, M.; Santoro, R. The NoRC complex mediates the heterochromatin formation and stability of silent rRNA genes and centromeric repeats. EMBO J. 2010, 29, 2135–2146. [Google Scholar] [CrossRef]
- Bierhoff, H.; Dammert, M.A.; Brocks, D.; Dambacher, S.; Schotta, G.; Grummt, I. Quiescence-induced LncRNAs trigger H4K20 trimethylation and transcriptional silencing. Mol. Cell 2014, 54, 675–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seither, P.; Zatsepina, O.; Hoffmann, M.; Grummt, I. Constitutive and strong association of PAF53 with RNA polymerase I. Chromosoma 1997, 106, 216–225. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joshi, G.; Eberhardt, A.O.; Lange, L.; Winkler, R.; Hoffmann, S.; Kosan, C.; Bierhoff, H. Dichotomous Impact of Myc on rRNA Gene Activation and Silencing in B Cell Lymphomagenesis. Cancers 2020, 12, 3009. https://doi.org/10.3390/cancers12103009
Joshi G, Eberhardt AO, Lange L, Winkler R, Hoffmann S, Kosan C, Bierhoff H. Dichotomous Impact of Myc on rRNA Gene Activation and Silencing in B Cell Lymphomagenesis. Cancers. 2020; 12(10):3009. https://doi.org/10.3390/cancers12103009
Chicago/Turabian StyleJoshi, Gaurav, Alexander Otto Eberhardt, Lisa Lange, René Winkler, Steve Hoffmann, Christian Kosan, and Holger Bierhoff. 2020. "Dichotomous Impact of Myc on rRNA Gene Activation and Silencing in B Cell Lymphomagenesis" Cancers 12, no. 10: 3009. https://doi.org/10.3390/cancers12103009