Will Next-Generation Immunotherapy Overcome the Intrinsic Diversity and Low Immunogenicity of Sarcomas to Improve Clinical Benefit?



Abstract
:Simple Summary
Abstract
1. Introduction
2. Immunogenic Landscape of Sarcomas
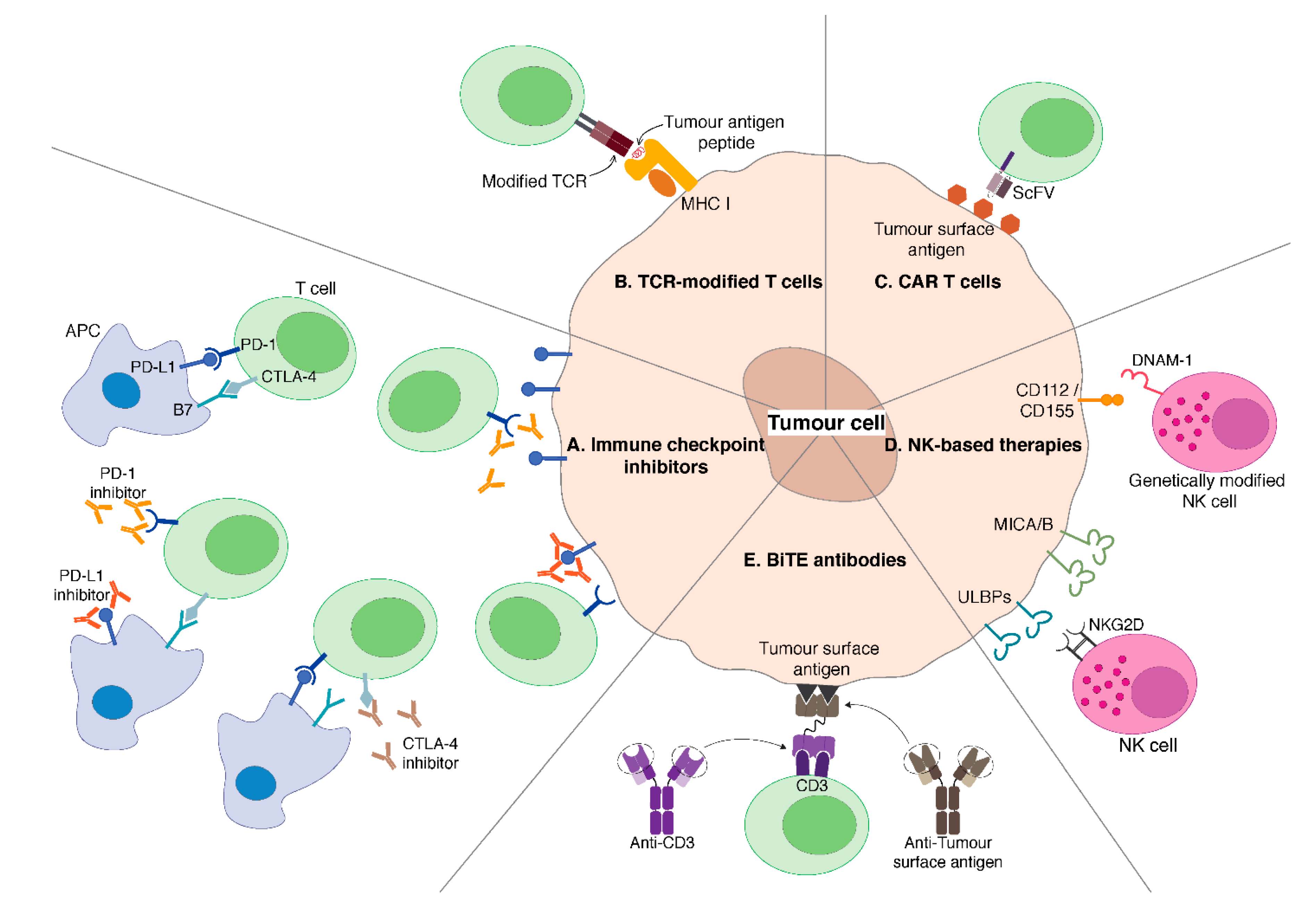
3. Immune Checkpoint Inhibitors
3.1. CTLA-4 Blockade
3.2. PD-1/PD-L1 Blockade
3.3. CTLA-4 + PD-1/PD-L1 Immune Checkpoint Inhibitors
4. Adoptive Transfer of Genetically Modified T Cells
4.1. T Cells Engineered to Express TAA-Specific TCRs
4.2. CAR T Cells
5. NK Cell-Based Therapies
6. Bispecific T Cell Engager (BiTE) Antibodies
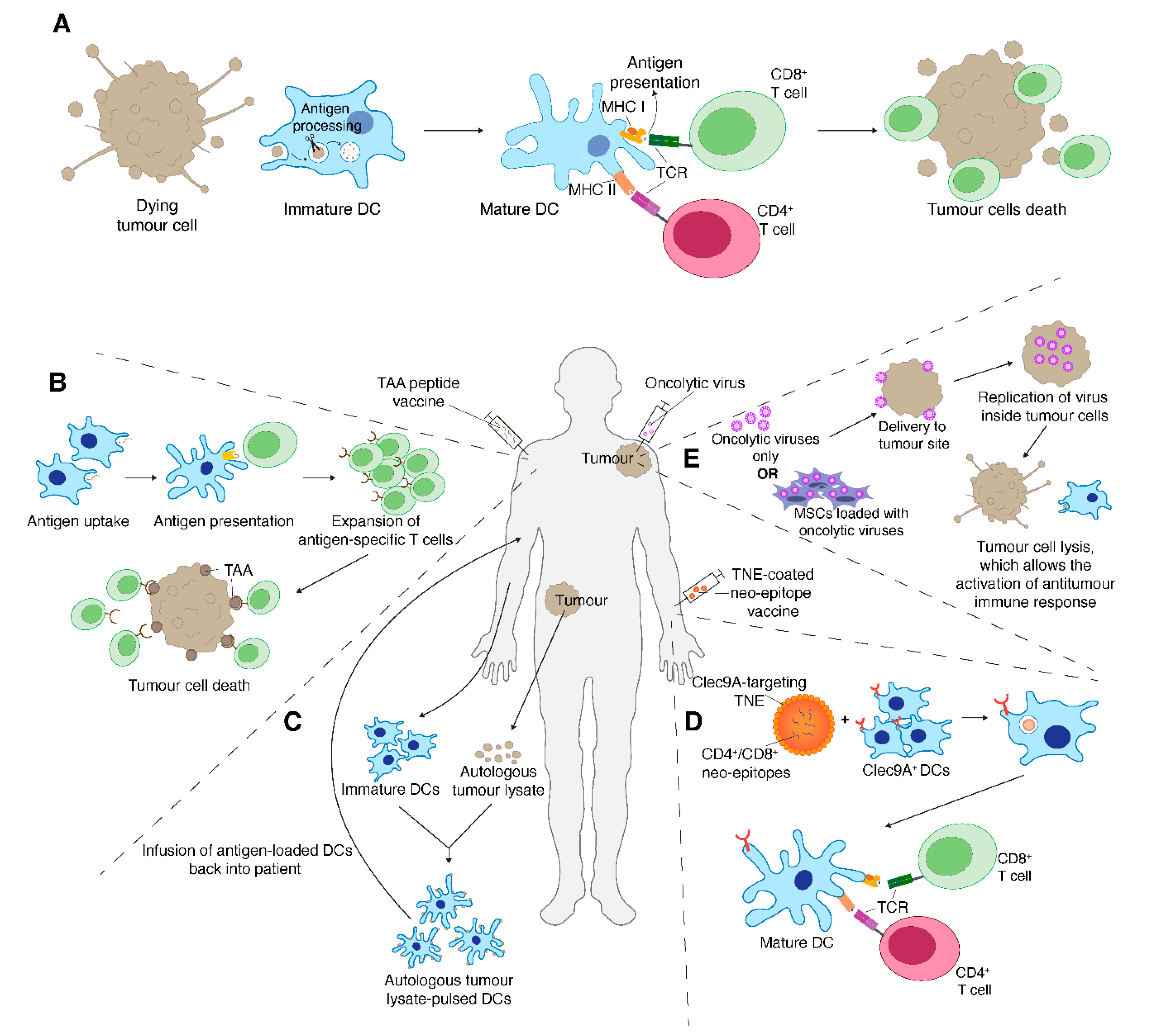
7. Therapeutic Cancer Vaccines
8. Oncolytic Virus Therapy
9. Future Directions
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lahat, G.; Lazar, A.; Lev, D. Sarcoma Epidemiology and Etiology: Potential Environmental and Genetic Factors. Surg. Clin. N. Am. 2008, 88, 451–481. [Google Scholar] [CrossRef] [PubMed]
- Burningham, Z.; Hashibe, M.; Spector, L.G.; Schiffman, J.D. The Epidemiology of Sarcoma. Clin. Sarcoma Res. 2012, 2, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, V.; Denniston, K.A.; Lin, C.J.; Lin, C. A Comparison of Pediatric vs. Adult Patients with the Ewing Sarcoma Family of Tumors. Front. Oncol. 2017, 7, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Zaid, T.; Wang, W.-L.; Somaiah, N.; Lazar, A.J.F. Molecular profiling of sarcomas: New vistas for precision medicine. Virchows Arch. 2017, 471, 243–255. [Google Scholar] [CrossRef]
- Helman, L.J.; Meltzer, P. Mechanisms of sarcoma development. Nat. Rev. Cancer 2003, 3, 685–694. [Google Scholar] [CrossRef]
- Taylor, B.S.; Barretina, J.; Maki, R.G.; Antonescu, C.R.; Singer, S.; Ladanyi, M. Advances in sarcoma genomics and new therapeutic targets. Nat. Rev. Cancer 2011, 11, 541–557. [Google Scholar] [CrossRef] [Green Version]
- Brien, G.L.; Stegmaier, K.; Armstrong, S.A. Targeting chromatin complexes in fusion protein-driven malignancies. Nat. Rev. Cancer 2019, 19, 255–269. [Google Scholar] [CrossRef]
- Tucker, M.A.; D’Angio, G.J.; Boice, J.D.; Strong, L.C.; Li, F.P.; Stovall, M.; Stone, B.J.; Green, D.M.; Lombardi, F.; Newton, W.; et al. Bone Sarcomas Linked to Radiotherapy and Chemotherapy in Children. N. Engl. J. Med. 1987, 317, 588–593. [Google Scholar] [CrossRef]
- Van Geel, A.N.; Pastorino, U.; Jauch, K.W.; Judson, I.R.; Van Coevorden, F.; Buesa, J.M.; Nielsen, O.S.; Boudinet, A.; Tursz, T.; Schmitz, P.I.M. Surgical treatment of lung metastases: The European Organization for Research and Treatment of Cancer-Soft Tissue and Bone Sarcoma Group study of 255 patients. Cancer 1996, 77, 675–682. [Google Scholar] [CrossRef]
- Bielack, S.S.; Kempf-Bielack, B.; Delling, G.; Exner, G.U.; Flege, S.; Helmke, K.; Kotz, R.; Salzer-Kuntschik, M.; Werner, M.; Winkelmann, W.; et al. Prognostic Factors in High-Grade Osteosarcoma of the Extremities or Trunk: An Analysis of 1,702 Patients Treated on Neoadjuvant Cooperative Osteosarcoma Study Group Protocols. J. Clin. Oncol. 2002, 20, 776–790. [Google Scholar] [CrossRef]
- Miwa, S.; Yamamoto, N.; Hayashi, K.; Takeuchi, A.; Igarashi, K.; Tsuchiya, H. Therapeutic Targets for Bone and Soft-Tissue Sarcomas. Int. J. Mol. Sci. 2019, 20, 170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Andrea, F.P. Intrinsic radiation resistance of mesenchymal cancer stem cells and implications for treatment response in a murine sarcoma model. Dan. Med. J. 2012, 59, B4388. [Google Scholar] [PubMed]
- Xu-Monette, Z.Y.; Zhang, M.; Li, J.; Young, K.H. PD-1/PD-L1 Blockade: Have We Found the Key to Unleash the Antitumor Immune Response? Front. Immunol. 2017, 8, 1597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, E.F. The toxins of William B. Coley and the treatment of bone and soft-tissue sarcomas. Iowa Orthop. J. 2006, 26, 154–158. [Google Scholar]
- Hirota, S. Gain-of-Function Mutations of c-kit in Human Gastrointestinal Stromal Tumors. Science 1998, 279, 577–580. [Google Scholar] [CrossRef]
- Hirota, S.; Ohashi, A.; Nishida, T.; Isozaki, K.; Kinoshita, K.; Shinomura, Y.; Kitamura, Y. Gain-of-function mutations of platelet-derived growth factor receptor α gene in gastrointestinal stromal tumors. Gastroenterology 2003, 125, 660–667. [Google Scholar] [CrossRef]
- Lasota, J.; Miettinen, M. Clinical significance of oncogenic KIT and PDGFRA mutations in gastrointestinal stromal tumours. Histopathology 2008, 53, 245–266. [Google Scholar] [CrossRef]
- Reilley, M.J.; Bailey, A.; Subbiah, V.; Janku, F.; Naing, A.; Falchook, G.; Karp, D.; Piha-Paul, S.; Tsimberidou, A.; Fu, S.; et al. Phase I clinical trial of combination imatinib and ipilimumab in patients with advanced malignancies. J. Immunother. Cancer 2017, 5, 35. [Google Scholar] [CrossRef]
- Demetri, G.D.; Von Mehren, M.; Blanke, C.D.; Van den Abbeele, M.D.; Eisenberg, B.; Roberts, P.J.; Heinrich, M.C.; Tuveson, D.A.; Singer, S.; Janicek, M.; et al. Efficacy and Safety of Imatinib Mesylate in Advanced Gastrointestinal Stromal Tumors. N. Engl. J. Med. 2002, 347, 472–480. [Google Scholar] [CrossRef]
- Balachandran, V.P.; Cavnar, M.J.; Zeng, S.; Bamboat, Z.M.; Ocuin, L.M.; Obaid, H.; Sorenson, E.C.; Popow, R.; Ariyan, E.C.; Rossi, F.; et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat. Med. 2011, 17, 1094–1100. [Google Scholar] [CrossRef]
- Gasparotto, D.; Sbaraglia, M.; Rossi, S.; Baldazzi, D.; Brenca, M.; Mondello, A.; Nardi, F.; Racanelli, D.; Cacciatore, M.; Tos, A.P.D.; et al. Tumor genotype, location and malignant potential shape the immunogenicity of primary, untreated Gastrointestinal Stromal Tumors. JCI Insight 2020. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef]
- Pardoll, D. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowshanravan, B.; Halliday, N.; Sansom, D.M. CTLA-4: A moving target in immunotherapy. Blood 2018, 131, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Read, S.; Greenwald, R.; Izcue, A.; Robinson, N.; Mandelbrot, D.; Francisco, L.; Sharpe, A.H.; Powrie, F. Blockade of CTLA-4 on CD4+CD25+ regulatory T cells abrogates their function in vivo. J. Immunol. 2006, 177, 4376–4383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [Green Version]
- Contardi, E.; Palmisano, G.L.; Tazzari, P.L.; Martelli, A.M.; Falà, F.; Fabbi, M.; Kato, T.; Lucarelli, E.; Donati, D.; Polito, L.; et al. CTLA-4 is constitutively expressed on tumor cells and can trigger apoptosis upon ligand interaction. Int. J. Cancer 2005, 117, 538–550. [Google Scholar] [CrossRef]
- Hingorani, P.; Maas, M.L.; Gustafson, M.P.; Dickman, P.S.; Adams, R.H.; Watanabe, M.; Eshun, F.; Williams, J.; Seidel, M.J.; Dietz, A.B. Increased CTLA-4+ T cells and an increased ratio of monocytes with loss of class II (CD14+ HLA-DRlo/neg) found in aggressive pediatric sarcoma patients. J. Immunother. Cancer 2015, 3, 35. [Google Scholar] [CrossRef] [Green Version]
- Merchant, M.S.; Wright, M.; Baird, K.; Wexler, L.H.; Rodriguez-Galindo, C.; Bernstein, D.; Delbrook, C.; Lodish, M.B.; Bishop, R.J.; Wolchok, J.D.; et al. Phase I Clinical Trial of Ipilimumab in Pediatric Patients with Advanced Solid Tumors. Clin. Cancer Res. 2016, 22, 1364–1370. [Google Scholar] [CrossRef] [Green Version]
- Maki, R.G.; Jungbluth, A.A.; Gnjatic, S.; Schwartz, G.K.; D’Adamo, D.R.; Keohan, M.L.; Wagner, M.J.; Scheu, K.; Chiu, R.; Ritter, E.; et al. A Pilot Study of Anti-CTLA4 Antibody Ipilimumab in Patients with Synovial Sarcoma. Sarcoma 2013, 2013, 168145. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Shoushtari, A.N.; Keohan, M.L.; Dickson, M.A.; Gounder, M.M.; Chi, P.; Loo, J.K.; Gaffney, L.; Schneider, L.; Patel, Z.; et al. Combined KIT and CTLA-4 Blockade in Patients with Refractory GIST and Other Advanced Sarcomas: A Phase Ib Study of Dasatinib plus Ipilimumab. Clin. Cancer Res. 2017, 23, 2972–2980. [Google Scholar] [CrossRef] [Green Version]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Kim, J.R.; Moon, Y.J.; Kwon, K.S.; Bae, J.S.; Wagle, S.; Kim, K.M.; Park, H.S.; Lee, H.; Moon, W.S.; Chung, M.J.; et al. Tumor infiltrating PD1-positive lymphocytes and the expression of PD-L1 predict poor prognosis of soft tissue sarcomas. PLoS ONE 2013, 8, e82870. [Google Scholar] [CrossRef]
- Kim, C.; Kim, E.K.; Jung, H.; Chon, H.J.; Han, J.W.; Shin, K.H.; Hu, H.; Kim, K.S.; Choi, Y.D.; Kim, S.; et al. Prognostic implications of PD-L1 expression in patients with soft tissue sarcoma. BMC Cancer 2016, 16, 434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vargas, A.C.; Maclean, F.M.; Sioson, L.; Tran, D.; Bonar, F.; Mahar, A.; Cheah, A.L.; Russell, P.; Grimison, P.; Richardson, L.; et al. Prevalence of PD-L1 expression in matched recurrent and/or metastatic sarcoma samples and in a range of selected sarcomas subtypes. PLoS ONE 2020, 15, e0222551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Angelo, S.P.; Shoushtari, A.N.; Agaram, N.P.; Kuk, D.; Qin, L.-X.; Carvajal, R.D.; Dickson, M.A.; Gounder, M.; Keohan, M.L.; Schwartz, G.K.; et al. Prevalence of tumor-infiltrating lymphocytes and PD-L1 expression in the soft tissue sarcoma microenvironment. Hum. Pathol. 2015, 46, 357–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.-A.; Reed, K.; et al. Nivolumab plus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollack, S.M.; He, Q.; Yearley, J.H.; Emerson, R.; Vignali, M.; Zhang, Y.; Redman, M.W.; Baker, K.K.; Cooper, S.; Donahue, B.; et al. T-cell infiltration and clonality correlate with programmed cell death protein 1 and programmed death-ligand 1 expression in patients with soft tissue sarcomas. Cancer 2017, 123, 3291–3304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Issels, R.; Büclein, V.; Kampmann, E.; Knösel, T.; Nössner, E.; Subklewe, M.; Lindner, L. Dissecting the role of tumor-infiltrating lymphocytes (TIL) in patients with high-risk soft-tissue sarcoma (STS) receiving neo-adjuvant chemotherapy (NAC) with regional hyperthermia (RHT). Ann. Oncol. 2016, 27, vi488. [Google Scholar] [CrossRef]
- Park, H.K.; Kim, M.; Sung, M.; Lee, S.E.; Kim, Y.J.; Choi, Y.-L. Status of programmed death-ligand 1 expression in sarcomas. J. Transl. Med. 2018, 16, 303. [Google Scholar] [CrossRef] [PubMed]
- Tawbi, H.; Burgess, M.; Bolejack, V.; Van Tine, B.A.; Schuetze, S.M.; Hu, J.; D’Angelo, S.; Attia, S.; Riedel, R.F.; A Priebat, D.; et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): A multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1493–1501. [Google Scholar] [CrossRef]
- Burgess, M.A.; Bolejack, V.; Schuetze, S.; Van Tine, B.A.; Attia, S.; Riedel, R.F.; Hu, J.S.; Davis, L.E.; Okuno, S.H.; Priebat, D.A.; et al. Clinical activity of pembrolizumab (P) in undifferentiated pleomorphic sarcoma (UPS) and dedifferentiated/pleomorphic liposarcoma (LPS): Final results of SARC028 expansion cohorts. J. Clin. Oncol. 2019, 37, 11015. [Google Scholar] [CrossRef]
- Ben-Ami, E.; Barysauskas, C.M.; Solomon, S.; Tahlil, K.; Malley, R.; Hohos, M.; Polson, K.; Loucks, M.; Severgnini, M.; Patel, T.; et al. Immunotherapy with single agent nivolumab for advanced leiomyosarcoma of the uterus: Results of a phase 2 study. Cancer 2017, 123, 3285–3290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keung, E.Z.; Burgess, M.; Salazar, R.; Parra, E.R.; Rodrigues-Canales, J.; Bolejack, V.; Van Tine, B.A.; Schuetze, S.M.; Attia, S.; Riedel, R.F.; et al. Correlative Analyses of the SARC028 Trial Reveal an Association Between Sarcoma-Associated Immune Infiltrate and Response to Pembrolizumab. Clin. Cancer Res. 2020, 26, 1258–1266. [Google Scholar] [CrossRef]
- Feng, Y.; Shen, J.; Gao, Y.; Liao, Y.; Cote, G.M.; Choy, E.; Chebib, I.; Mankin, H.; Hornicek, F.; Duan, Z. Expression of programmed cell death ligand 1 (PD-L1) and prevalence of tumor-infiltrating lymphocytes (TILs) in chordoma. Oncotarget 2015, 6, 11139–11149. [Google Scholar] [CrossRef] [Green Version]
- Zou, M.-X.; Peng, A.-B.; Lv, G.-H.; Wang, X.-B.; Li, J.; She, X.-L.; Jiang, Y. Expression of programmed death-1 ligand (PD-L1) in tumor-infiltrating lymphocytes is associated with favorable spinal chordoma prognosis. Am. J. Transl. Res. 2016, 8, 3274–3287. [Google Scholar]
- Machado, I.; López-Guerrero, J.A.; Scotlandi, K.; Picci, P.; Llombart-Bosch, A. Immunohistochemical analysis and prognostic significance of PD-L1, PD-1, and CD8+ tumor-infiltrating lymphocytes in Ewing’s sarcoma family of tumors (ESFT). Virchows Arch. 2018, 472, 815–824. [Google Scholar] [CrossRef]
- Raj, S.; Bui, M.; Gonzales, R.; Letson, D.; Antonia, S. Impact of Pdl1 Expression on Clinical Outcomes in Subtypes of Sarcoma. Ann. Oncol. 2014, 25, iv498. [Google Scholar] [CrossRef]
- Zhu, Z.; Jin, Z.; Zhang, M.; Tang, Y.; Yang, G.; Yuan, X.; Yao, J.; Sun, D. Prognostic value of programmed death-ligand 1 in sarcoma: A meta-analysis. Oncotarget 2017, 8, 59570–59580. [Google Scholar] [CrossRef] [Green Version]
- Lussier, D.M.; O’Neill, L.; Nieves, L.M.; McAfee, M.S.; Holechek, S.A.; Collins, A.W.; Dickman, P.; Jacobsen, J.; Hingorani, P.; Blattman, J.N. Enhanced T-Cell Immunity to Osteosarcoma Through Antibody Blockade of PD-1/PD-L1 Interactions. J. Immunother. 2015, 38, 96–106. [Google Scholar] [CrossRef]
- Koirala, P.; Roth, M.E.; Gill, J.; Piperdi, S.; Chinai, J.M.; Geller, D.S.; Hoang, B.H.; Park, A.; Fremed, M.A.; Zang, X.; et al. Immune infiltration and PD-L1 expression in the tumor microenvironment are prognostic in osteosarcoma. Sci. Rep. 2016, 6, 30093. [Google Scholar] [CrossRef] [PubMed]
- Fritzsching, B.; Fellenberg, J.; Moskovszky, L.; Sápi, Z.; Krenacs, T.; Machado, I.; Poeschl, J.; Lehner, B.; Szendrõi, M.; Bosch, A.L.; et al. CD8+/FOXP3+-ratio in osteosarcoma microenvironment separates survivors from non-survivors: A multicenter validated retrospective study. OncoImmunology 2015, 4, e990800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Erp, A.; Versleijen-Jonkers, Y.M.H.; Hillebrandt-Roeffen, M.H.; Van Houdt, L.; Gorris, M.A.; Van Dam, L.S.; Mentzel, T.; Weidema, M.; Savci-Heijink, C.D.; Desar, I.M.; et al. Expression and clinical association of programmed cell death-1, programmed death-ligand-1 and CD8+ lymphocytes in primary sarcomas is subtype dependent. Oncotarget 2017, 8, 71371–71384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wedekind, M.F.; Wagner, L.M.; Cripe, T.P. Immunotherapy for osteosarcoma: Where do we go from here? Pediatr. Blood Cancer 2018, 65, e27227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, K.; Okamoto, M.; Sasaki, J.; Kuroda, C.; Ishida, H.; Ueda, K.; Okano, S.; Ideta, H.; Kamanaka, T.; Sobajima, A.; et al. Clinical outcome of osteosarcoma and its correlation with programmed death-ligand 1 and T cell activation markers. OncoTargets Ther. 2019, 12, 2513–2518. [Google Scholar] [CrossRef] [Green Version]
- Zheng, B.; Ren, T.; Huang, Y.; Sun, K.; Wang, S.; Bao, X.; Liu, K.; Guo, W. PD-1 axis expression in musculoskeletal tumors and antitumor effect of nivolumab in osteosarcoma model of humanized mouse. J. Hematol. Oncol. 2018, 11, 16. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-C.; Beird, H.C.; Livingston, J.A.; Advani, S.; Mitra, A.; Cao, S.; Reuben, A.; Ingram, D.; Wang, W.-L.; Ju, Z.; et al. Immuno-genomic landscape of osteosarcoma. Nat. Commun. 2020, 11, 1008. [Google Scholar] [CrossRef] [Green Version]
- Lussier, D.M.; Johnson, J.L.; Hingorani, P.; Blattman, J.N. Combination immunotherapy with α-CTLA-4 and α-PD-L1 antibody blockade prevents immune escape and leads to complete control of metastatic osteosarcoma. J. Immunother. Cancer 2015, 3, 21. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Mahoney, M.R.; Van Tine, B.A.; Atkins, J.; Milhem, M.M.; Jahagirdar, B.N.; Antonescu, C.R.; Horvath, E.; Tap, W.D.; Schwartz, G.K.; et al. Nivolumab with or without ipilimumab treatment for metastatic sarcoma (Alliance A091401): Two open-label, non-comparative, randomised, phase 2 trials. Lancet Oncol. 2018, 19, 416–426. [Google Scholar] [CrossRef]
- Juretić, E.; Spagnoli, G.C.; Schultz-Thater, E.; Sarcevic, B. Cancer/testis tumour-associated antigens: Immunohistochemical detection with monoclonal antibodies. Lancet Oncol. 2003, 4, 104–109. [Google Scholar] [CrossRef]
- Smith, S.M.; Iwenofu, O.H. NY-ESO-1: A promising cancer testis antigen for sarcoma immunotherapy and diagnosis. Chin. Clin. Oncol. 2018, 7, 44. [Google Scholar] [CrossRef] [PubMed]
- Jungbluth, A.A.; Antonescu, C.R.; Busam, K.J.; Iversen, K.; Kolb, D.; Coplan, K.; Chen, Y.T.; Stockert, E.; Ladanyi, M.; Old, L.J. Monophasic and biphasic synovial sarcomas abundantly express cancer/testis antigen NY-ESO-1 but not MAGE-A1 or CT7. Int. J. Cancer 2001, 94, 252–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robbins, P.F.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; Dudley, M.E.; Wunderlich, J.R.; Nahvi, A.V.; Helman, L.J.; Mackall, C.L.; et al. Tumor Regression in Patients with Metastatic Synovial Cell Sarcoma and Melanoma Using Genetically Engineered Lymphocytes Reactive With NY-ESO-1. J. Clin. Oncol. 2011, 29, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.F.; Kassim, S.H.; Tran, T.L.N.; Crystal, J.S.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Dudley, M.E.; Wunderlich, J.R.; Sherry, R.M.; et al. A Pilot Trial Using Lymphocytes Genetically Engineered with an NY-ESO-1-Reactive T-cell Receptor: Long-term Follow-up and Correlates with Response. Clin. Cancer Res. 2015, 21, 1019–1027. [Google Scholar] [CrossRef] [Green Version]
- D’Angelo, S.P.; Melchiori, L.; Merchant, M.S.; Bernstein, D.; Glod, J.; Kaplan, R.N.; Grupp, S.; Tap, W.D.; Chagin, K.; Binder, G.K.; et al. Antitumor Activity Associated with Prolonged Persistence of Adoptively Transferred NY-ESO-1 c259T Cells in Synovial Sarcoma. Cancer Discov. 2018, 8, 944–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eleftheriadou, I.; Brett, S.; Domogala, A.; Patasic, L.; Kijewska, M.; Soor, K.; Georgouli, M.; Dopierala, J.; Fisher, P.; Jing, J.; et al. NY-ESO-1 and LAGE1A: An emerging target for cell therapies in solid tumours. Ann. Oncol. 2019, 30, v503. [Google Scholar] [CrossRef]
- Ramachandran, I.; Lowther, D.E.; Dryer-Minnerly, R.; Wang, R.; Fayngerts, S.; Nunez, D.; Betts, G.; Bath, N.; Tipping, A.J.; Melchiori, L.; et al. Systemic and local immunity following adoptive transfer of NY-ESO-1 SPEAR T cells in synovial sarcoma. J. Immunother. Cancer 2019, 7, 276. [Google Scholar] [CrossRef]
- Van Tine, B.; Butler, M.; Araujo, D.; Johnson, M.; Clarke, J.; Liebner, D.; Odunsi, K.; Olszanski, A.; Basu, S.; Brophy, F.; et al. ADP-A2M4 (MAGE-A4) in patients with synovial sarcoma. Ann. Oncol. 2019, 30, v684–v685. [Google Scholar] [CrossRef]
- Hong, D.S.; Van Tine, B.A.; Olszanski, A.J.; Johnson, M.L.; Liebner, D.A.; Trivedi, T.; Lin, Q.; Elefant, E.; Dryer-Minnerly, R.; Navenot, J.-M.; et al. Phase I dose escalation and expansion trial to assess the safety and efficacy of ADP-A2M4 SPEAR T cells in advanced solid tumors. J. Clin. Oncol. 2020, 38, 102. [Google Scholar] [CrossRef]
- Suzuki, M.; Curran, K.J.; Cheung, N.K. Chimeric antigen receptors and bispecific antibodies to retarget T cells in pediatric oncology. Pediatr. Blood Cancer 2015, 62, 1326–1336. [Google Scholar] [CrossRef] [Green Version]
- Kowolik, C.M.; Topp, M.S.; Gonzalez, S.; Pfeiffer, T.; Olivares, S.; Gonzalez, N.; Smith, D.D.; Forman, S.J.; Jensen, M.C.; Cooper, L.J. CD28 Costimulation Provided through a CD19-Specific Chimeric Antigen Receptor Enhances In vivo Persistence and Antitumor Efficacy of Adoptively Transferred T Cells. Cancer Res. 2006, 66, 10995–11004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadelain, M.; Brentjens, R.; Rivière, I. The promise and potential pitfalls of chimeric antigen receptors. Curr. Opin. Immunol. 2009, 21, 215–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, C.U.; Savoldo, B.; Dotti, G.; Pule, M.; Yvon, E.; Myers, G.D.; Rossig, C.; Russell, H.V.; Diouf, O.; Liu, E.; et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011, 118, 6050–6056. [Google Scholar] [CrossRef] [PubMed]
- Pule, M.A.; Savoldo, B.; Myers, G.D.; Rossig, C.; Russell, H.V.; Dotti, G.; Huls, M.H.; Liu, E.; Gee, A.P.; Mei, Z.; et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: Persistence and antitumor activity in individuals with neuroblastoma. Nat. Med. 2008, 14, 1264–1270. [Google Scholar] [CrossRef] [PubMed]
- Long, A.H.; Highfill, S.L.; Cui, Y.; Smith, J.P.; Walker, A.J.; Ramakrishna, S.; El-Etriby, R.; Galli, S.; Tsokos, M.G.; Orentas, R.J.; et al. Reduction of MDSCs with All-trans Retinoic Acid Improves CAR Therapy Efficacy for Sarcomas. Cancer Immunol. Res. 2016, 4, 869–880. [Google Scholar] [CrossRef] [Green Version]
- Chulanetra, M.; Morchang, A.; Sayour, E.; Eldjerou, L.; Milner, R.; Lagmay, J.; Cascio, M.; Stover, B.; Slayton, W.; Chaicumpa, W.; et al. GD2 chimeric antigen receptor modified T cells in synergy with sub-toxic level of doxorubicin targeting osteosarcomas. Am. J. Cancer Res. 2020, 10, 674–687. [Google Scholar]
- Ebb, D.H.; Meyers, P.; Grier, H.; Bernstein, M.; Gorlick, R.; Lipshultz, S.E.; Krailo, M.; Devidas, M.; Barkauskas, D.A.; Siegal, G.P.; et al. Phase II Trial of Trastuzumab in Combination with Cytotoxic Chemotherapy for Treatment of Metastatic Osteosarcoma with Human Epidermal Growth Factor Receptor 2 Overexpression: A Report from the Children’s Oncology Group. J. Clin. Oncol. 2012, 30, 2545–2551. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, N.; Salsman, V.S.; Yvon, E.; Louis, C.U.; Perlaky, L.; Wels, W.S.; Dishop, M.K.; Kleinerman, E.E.; Pule, M.; Rooney, C.M.; et al. Immunotherapy for Osteosarcoma: Genetic Modification of T cells Overcomes Low Levels of Tumor Antigen Expression. Mol. Ther. 2009, 17, 1779–1787. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.S.; Hegde, M.; Robertson, C.; Ghazi, A.; Gerken, C.; Liu, E.; Dakhova, O.; Ashoori, A.; Corder, A.; et al. Human Epidermal Growth Factor Receptor 2 (HER2) –Specific Chimeric Antigen Receptor–Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J. Clin. Oncol. 2015, 33, 1688–1696. [Google Scholar] [CrossRef]
- Hegde, M.; Derenzo, C.; Zhang, H.; Mata, M.; Gerken, C.; Shree, A.; Yi, Z.; Brawley, V.; Dakhova, O.; Wu, M.-F.; et al. Expansion of HER2-CAR T cells after lymphodepletion and clinical responses in patients with advanced sarcoma. J. Clin. Oncol. 2017, 35, 10508. [Google Scholar] [CrossRef]
- Hegde, M.; Joseph, S.K.; Pashankar, F.; DeRenzo, C.; Sanber, K.; Navai, S.; Byrd, T.T.; Hicks, J.; Xu, M.L.; Gerken, C.; et al. Tumor response and endogenous immune reactivity after administration of HER2 CAR T cells in a child with metastatic rhabdomyosarcoma. Nat. Commun. 2020, 11, 3549. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Park, H.; Greene, J.; Pao, J.; Mulvey, E.; Zhou, S.X.; Albert, C.M.; Moy, F.; Sachdev, D.; Yee, D.; et al. IGF1R- and ROR1-Specific CAR T Cells as a Potential Therapy for High Risk Sarcomas. PLoS ONE 2015, 10, e0133152. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Yu, L.; Cooper, L.J.; Hollomon, M.; Huls, H.; Kleinerman, E.S. Genetically modified T cells targeting interleukin-11 receptor alpha-chain kill human osteosarcoma cells and induce the regression of established osteosarcoma lung metastases. Cancer Res. 2012, 72, 271–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majzner, R.G.; Theruvath, J.L.; Nellan, A.; Heitzeneder, S.; Cui, Y.; Mount, C.W.; Rietberg, S.P.; Linde, M.H.; Xu, P.; Rota, C.; et al. CAR T Cells Targeting B7-H3, a Pan-Cancer Antigen, Demonstrate Potent Preclinical Activity Against Pediatric Solid Tumors and Brain Tumors. Clin. Cancer Res. 2019, 25, 2560–2574. [Google Scholar] [CrossRef]
- Bonifant, C.L.; Jackson, H.J.; Brentjens, R.J.; Curran, K.J. Toxicity and management in CAR T-cell therapy. Mol. Ther. Oncolytics 2016, 3, 16011. [Google Scholar] [CrossRef]
- Tsukahara, T.; Kawaguchi, S.; Torigoe, T.; Asanuma, H.; Nakazawa, E.; Shimozawa, K.; Nabeta, Y.; Kimura, S.; Kaya, M.; Nagoya, S.; et al. Prognostic significance of HLA class I expression in osteosarcoma defined by anti-pan HLA class I monoclonal antibody, EMR8-5. Cancer Sci. 2006, 97, 1374–1380. [Google Scholar] [CrossRef]
- Berghuis, D.; De Hooge, A.S.; Santos, S.J.; Horst, D.; Wiertz, E.J.; Van Eggermond, M.C.; Elsen, P.J.V.D.; Taminiau, A.H.; Ottaviano, L.; Schaefer, K.-L.; et al. Reduced human leukocyte antigen expression in advanced-stage Ewing sarcoma: Implications for immune recognition. J. Pathol. 2009, 218, 222–231. [Google Scholar] [CrossRef]
- Kiessling, R.; Klein, E.; Wigzell, H. “Natural” killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. Eur. J. Immunol. 1975, 5, 112–117. [Google Scholar] [CrossRef]
- Markiewicz, K.; Zeman, K.; Kozar, A.; Golebiowska-Wawrzyniak, M.; Wozniak, W. Evaluation of selected parameters of cellular immunity in children with osteosarcoma at diagnosis. Med. Wieku Rozw. 2012, 16, 212–221. [Google Scholar]
- Buddingh, E.P.; Ruslan, S.E.; Berghuis, D.; Gelderblom, H.; Anninga, J.K.; Hogendoorn, P.C.; Egeler, R.M.; Schilham, M.W.; Lankester, A.C. Intact interferon signaling in peripheral blood leukocytes of high-grade osteosarcoma patients. Cancer Immunol. Immunother. 2012, 61, 941–947. [Google Scholar] [CrossRef] [Green Version]
- Buddingh, E.P.; Schilham, M.W.; Ruslan, S.E.; Berghuis, D.; Szuhai, K.; Suurmond, J.; Taminiau, A.H.; Gelderblom, H.; Egeler, R.M.; Serra, M.; et al. Chemotherapy-resistant osteosarcoma is highly susceptible to IL-15-activated allogeneic and autologous NK cells. Cancer Immunol. Immunother. 2011, 60, 575–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, C.; Eslin, D.; Levy, A.; Roberson, J.; Giusti, V.; Sutphin, R. Prognostic significance of early lymphocyte recovery in pediatric osteosarcoma. Pediatr. Blood Cancer 2010, 55, 1096–1102. [Google Scholar] [CrossRef] [PubMed]
- Luksch, R.; Perotti, D.; Cefalo, G.; Gambacorti Passerini, C.; Massimino, M.; Spreafico, F.; Casanova, M.; Ferrari, A.; Terenziani, M.; Polastri, D.; et al. Immunomodulation in a treatment program including pre- and post-operative interleukin-2 and chemotherapy for childhood osteosarcoma. Tumori 2003, 89, 263–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Zhang, W.; Xu, P. NK cell and macrophages confer prognosis and reflect immune status in osteosarcoma. J. Cell Biochem. 2018, 120, 8792–8797. [Google Scholar] [CrossRef] [PubMed]
- Delgado, D.; Webster, D.E.; DeSantes, K.B.; Durkin, E.T.; Shaaban, A.F. KIR receptor-ligand incompatibility predicts killing of osteosarcoma cell lines by allogeneic NK cells. Pediatr. Blood Cancer 2010, 55, 1300–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariani, E.; Tarozzi, A.; Meneghetti, A.; Cattini, L.; Facchini, A. Human osteosarcoma cell susceptibility to natural killer cell lysis depends on CD54 and increases after TNF alpha incubation. FEBS Lett. 1997, 406, 83–88. [Google Scholar] [CrossRef] [Green Version]
- Meneghetti, A.; Mariani, E.; Santi, S.; Riccio, M.; Cattini, L.; Paoletti, S.; Facchini, A. NK binding capacity and lytic activity depend on the expression of ICAM-1 on target bone tumours. Int. J. Oncol. 1999, 15, 909–914. [Google Scholar] [CrossRef] [PubMed]
- Zamai, L.; Zauli, G.; Bavelloni, A.; Marmiroli, S.; Cataldi, A.; Weber, G.; Vitale, M. Tiazofurin induces a down-modulation of ICAM-1 expression on K562 target cells impairing NK adhesion and killing. Cell Immunol. 1995, 164, 100–104. [Google Scholar] [CrossRef]
- Cho, D.; Shook, D.R.; Shimasaki, N.; Chang, Y.-H.; Fujisaki, H.; Campana, D. Cytotoxicity of Activated Natural Killer Cells against Pediatric Solid Tumors. Clin. Cancer Res. 2010, 16, 3901–3909. [Google Scholar] [CrossRef] [Green Version]
- Leung, W.H.; Vong, Q.P.; Lin, W.; Janke, L.; Chen, T.; Leung, W. Modulation of NKG2D ligand expression and metastasis in tumors by spironolactone via RXRgamma activation. J. Exp. Med. 2013, 210, 2675–2692. [Google Scholar] [CrossRef]
- Fernandez, L.; Valentin, J.; Zalacain, M.; Leung, W.; Patino-Garcia, A.; Perez-Martinez, A. Activated and expanded natural killer cells target osteosarcoma tumor initiating cells in an NKG2D-NKG2DL dependent manner. Cancer Lett. 2015, 368, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Sayitoglu, E.C.; Georgoudaki, A.M.; Chrobok, M.; Ozkazanc, D.; Josey, B.J.; Arif, M.; Kusser, K.; Hartman, M.; Chinn, T.M.; Potens, R.; et al. Boosting Natural Killer Cell-Mediated Targeting of Sarcoma Through DNAM-1 and NKG2D. Front. Immunol. 2020, 11, 40. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Cheng, M.; Guo, H.; Chen, Y.; Huse, M.; Cheung, N.-K.V. Retargeting T cells to GD2 pentasaccharide on human tumors using Bispecific humanized antibody. Cancer Immunol. Res. 2014, 3, 266–277. [Google Scholar] [CrossRef] [Green Version]
- Mitsis, D.; Francescutti, V.; Skitzki, J. Current Immunotherapies for Sarcoma: Clinical Trials and Rationale. Sarcoma 2016, 2016, 9757219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pender, A.; Jones, R.L.; Pollack, S. Optimising Cancer Vaccine Design in Sarcoma. Cancers 2018, 11, 1. [Google Scholar] [CrossRef] [Green Version]
- Pollack, S.M.; Loggers, E.T.; Rodler, E.T.; Yee, C.; Jones, R.L. Immune-based therapies for sarcoma. Sarcoma 2011, 2011, 438940. [Google Scholar] [CrossRef] [Green Version]
- Wilky, B.A.; Goldberg, J.M. Immunotherapy in sarcoma: A new frontier. Discov. Med. 2014, 17, 201–206. [Google Scholar]
- Jager, E.; Gnjatic, S.; Nagata, Y.; Stockert, E.; Jager, D.; Karbach, J.; Neumann, A.; Rieckenberg, J.; Chen, Y.T.; Ritter, G.; et al. Induction of primary NY-ESO-1 immunity: CD8+ T lymphocyte and antibody responses in peptide-vaccinated patients with NY-ESO-1+ cancers. Proc. Natl. Acad. Sci. USA 2000, 97, 12198–12203. [Google Scholar] [CrossRef] [Green Version]
- Davis, I.D.; Chen, W.; Jackson, H.; Parente, P.; Shackleton, M.; Hopkins, W.; Chen, Q.; Dimopoulos, N.; Luke, T.; Murphy, R.; et al. Recombinant NY-ESO-1 protein with ISCOMATRIX adjuvant induces broad integrated antibody and CD4(+) and CD8(+) T cell responses in humans. Proc. Natl. Acad. Sci. USA 2004, 101, 10697–10702. [Google Scholar] [CrossRef] [Green Version]
- Carvajal, R.D.; Agulnik, M.; Ryan, C.W.; Milhem, M.; George, S.; Jones, R.L.; Chmielowski, B.; Van Tine, B.A.; Tawbi, H.A.-H.; Elias, A.D.; et al. Trivalent ganglioside vaccine and immunologic adjuvant versus adjuvant alone in metastatic sarcoma patients rendered disease-free by surgery: A randomized phase 2 trial. J. Clin. Oncol. 2014, 32, 10520. [Google Scholar] [CrossRef]
- Clark, J.; Rocques, P.J.; Crew, A.J.; Gill, S.; Shipley, J.; Chan, A.M.; Gusterson, B.A.; Cooper, C.S. Identification of novel genes, SYT and SSX, involved in the t(X;18)(p11.2;q11.2) translocation found in human synovial sarcoma. Nat. Genet. 1994, 7, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Ida, K.; Kawaguchi, S.; Sato, Y.; Tsukahara, T.; Nabeta, Y.; Sahara, H.; Ikeda, H.; Torigoe, T.; Ichimiya, S.; Kamiguchi, K.; et al. Crisscross CTL Induction by SYT-SSX Junction Peptide and Its HLA-A*2402 Anchor Substitute. J. Immunol. 2004, 173, 1436–1443. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Nabeta, Y.; Tsukahara, T.; Hirohashi, Y.; Syunsui, R.; Maeda, A.; Sahara, H.; Ikeda, H.; Torigoe, T.; Ichimiya, S.; et al. Detection and Induction of CTLs Specific for SYT-SSX-Derived Peptides in HLA-A24+ Patients with Synovial Sarcoma. J. Immunol. 2002, 169, 1611–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawaguchi, S.; Tsukahara, T.; Ida, K.; Kimura, S.; Murase, M.; Kano, M.; Emori, M.; Nagoya, S.; Kaya, M.; Torigoe, T.; et al. SYT-SSX breakpoint peptide vaccines in patients with synovial sarcoma: A study from the Japanese Musculoskeletal Oncology Group. Cancer Sci. 2012, 103, 1625–1630. [Google Scholar] [CrossRef]
- Fields, R.C.; Shimizu, K.; Mule, J.J. Murine dendritic cells pulsed with whole tumor lysates mediate potent antitumor immune responses in vitro and in vivo. Proc. Natl. Acad. Sci. USA 1998, 95, 9482–9487. [Google Scholar] [CrossRef] [Green Version]
- He, Y.T.; Zhang, Q.M.; Kou, Q.C.; Tang, B. In vitro generation of cytotoxic T lymphocyte response using dendritic cell immunotherapy in osteosarcoma. Oncol. Lett. 2016, 12, 1101–1106. [Google Scholar] [CrossRef] [Green Version]
- Miwa, S.; Nishida, H.; Tanzawa, Y.; Takeuchi, A.; Hayashi, K.; Yamamoto, N.; Mizukoshi, E.; Nakamoto, Y.; Kaneko, S.; Tsuchiya, H. Phase 1/2 study of immunotherapy with dendritic cells pulsed with autologous tumor lysate in patients with refractory bone and soft tissue sarcoma. Cancer 2017, 123, 1576–1584. [Google Scholar] [CrossRef] [Green Version]
- Geiger, J.D.; Hutchinson, R.J.; Hohenkirk, L.F.; McKenna, E.A.; Yanik, G.A.; Levine, J.E.; Chang, A.E.; Braun, T.M.; Mule, J.J. Vaccination of pediatric solid tumor patients with tumor lysate-pulsed dendritic cells can expand specific T cells and mediate tumor regression. Cancer Res. 2001, 61, 8513–8519. [Google Scholar]
- Himoudi, N.; Wallace, R.; Parsley, K.L.; Gilmour, K.; Barrie, A.-U.; Howe, K.; Dong, R.; Sebire, N.J.; Michalski, A.; Thrasher, A.J.; et al. Lack of T-cell responses following autologous tumour lysate pulsed dendritic cell vaccination, in patients with relapsed osteosarcoma. Clin. Transl. Oncol. 2012, 14, 271–279. [Google Scholar] [CrossRef]
- Merchant, M.S.; Bernstein, D.; Amoako, M.; Baird, K.; Fleisher, T.A.; Morre, M.; Steinberg, S.M.; Sabatino, M.; Stroncek, D.F.; Venkatasan, A.M.; et al. Adjuvant Immunotherapy to Improve Outcome in High-Risk Pediatric Sarcomas. Clin. Cancer Res. 2016, 22, 3182–3191. [Google Scholar] [CrossRef] [Green Version]
- Dhodapkar, M.V.; Sznol, M.; Zhao, B.; Wang, D.; Carvajal, R.D.; Keohan, M.L.; Chuang, E.; Sanborn, R.E.; Lutzky, J.; Powderly, J.; et al. Induction of antigen-specific immunity with a vaccine targeting NY-ESO-1 to the dendritic cell receptor DEC-205. Sci. Transl. Med. 2014, 6, 232ra251. [Google Scholar] [CrossRef] [PubMed]
- Zeng, B.; Middelberg, A.P.; Gemiarto, A.; MacDonald, K.; Baxter, A.G.; Talekar, M.; Moi, D.; Tullett, K.M.; Caminschi, I.; Lahoud, M.H.; et al. Self-adjuvanting nanoemulsion targeting dendritic cell receptor Clec9A enables antigen-specific immunotherapy. J. Clin. Investig. 2018, 128, 1971–1984. [Google Scholar] [CrossRef] [PubMed]
- Masterman, K.A.; Haigh, O.L.; Tullett, K.M.; Leal-Rojas, I.M.; Walpole, C.; Pearson, F.E.; Cebon, J.; Schmidt, C.; O’Brien, L.; Rosendahl, N.; et al. Human CLEC9A antibodies deliver NY-ESO-1 antigen to CD141(+) dendritic cells to activate naive and memory NY-ESO-1-specific CD8(+) T cells. J. Immunother. Cancer 2020, 8, e000691. [Google Scholar] [CrossRef] [PubMed]
- Kawano, M.; Itonaga, I.; Iwasaki, T.; Tsumura, H. Enhancement of antitumor immunity by combining anti-cytotoxic T lymphocyte antigen-4 antibodies and cryotreated tumor lysate-pulsed dendritic cells in murine osteosarcoma. Oncol. Rep. 2013, 29, 1001–1006. [Google Scholar] [CrossRef] [Green Version]
- Chawla, S.; Van Tine, B.A.; Pollack, S.; Ganjoo, K.; Elias, A.; Riedel, R.F.; Attia, S.; Choy, E.; Okuno, S.; Agulnik, M.; et al. A phase 2 study of CMB305 and atezolizumab in NY-ESO-1+ soft tissue sarcoma: Interim analysis of immunogenicity, tumor control and survival. Ann. Oncol. 2017, 28, v523. [Google Scholar] [CrossRef]
- Chawla, S.P.; Tine, B.A.V.; Pollack, S.; Ganjoo, K.N.; Elias, A.D.; Riedel, R.F.; Attia, S.; Choy, E.; Okuno, S.H.; Agulnik, M.; et al. A phase II randomized study of CMB305 and atezolizumab versus atezolizumab in NY-ESO-1+ soft tissue sarcoma: Analysis of immunogenicity, tumor control, and patient survival. J. Clin. Oncol. 2019, 37, 11011. [Google Scholar] [CrossRef]
- Lettieri, C.K.; Hingorani, P.; Kolb, E.A. Progress of oncolytic viruses in sarcomas. Expert Rev. Anticancer. Ther. 2012, 12, 229–242. [Google Scholar] [CrossRef]
- Abdelbary, H.; Brown, C.W.; Werier, J.; Bell, J. Using targeted virotherapy to treat a resistant Ewing sarcoma model: From the bedside to the bench and back. Sci. World J. 2014, 2014, 171439. [Google Scholar] [CrossRef]
- Morton, C.L.; Houghton, P.J.; Kolb, E.A.; Gorlick, R.; Reynolds, C.P.; Kang, M.H.; Maris, J.M.; Keir, S.T.; Wu, J.; Smith, M.A. Initial testing of the replication competent Seneca Valley virus (NTX-010) by the pediatric preclinical testing program. Pediatr. Blood Cancer 2010, 55, 295–303. [Google Scholar] [CrossRef] [Green Version]
- Paglino, J.C.; van den Pol, A.N. Vesicular stomatitis virus has extensive oncolytic activity against human sarcomas: Rare resistance is overcome by blocking interferon pathways. J. Virol. 2011, 85, 9346–9358. [Google Scholar] [CrossRef] [Green Version]
- Takakuwa, H.; Goshima, F.; Nozawa, N.; Yoshikawa, T.; Kimata, H.; Nakao, A.; Nawa, A.; Kurata, T.; Sata, T.; Nishiyama, Y. Oncolytic viral therapy using a spontaneously generated herpes simplex virus type 1 variant for disseminated peritoneal tumor in immunocompetent mice. Arch. Virol. 2003, 148, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.M.; Antonescu, C.R.; Bowler, T.; Munhoz, R.; Chi, P.; Dickson, M.A.; Gounder, M.M.; Keohan, M.L.; Movva, S.; Dholakia, R.; et al. Objective Response Rate Among Patients with Locally Advanced or Metastatic Sarcoma Treated with Talimogene Laherparepvec in Combination With Pembrolizumab: A Phase 2 Clinical Trial. JAMA Oncol. 2020, 6, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Hadrys, A.; Sochanik, A.; McFadden, G.; Jazowiecka-Rakus, J. Mesenchymal stem cells as carriers for systemic delivery of oncolytic viruses. Eur. J. Pharmacol. 2020, 874, 172991. [Google Scholar] [CrossRef] [PubMed]
- Mahasa, K.J.; de Pillis, L.; Ouifki, R.; Eladdadi, A.; Maini, P.; Yoon, A.R.; Yun, C.O. Mesenchymal stem cells used as carrier cells of oncolytic adenovirus results in enhanced oncolytic virotherapy. Sci. Rep. 2020, 10, 425. [Google Scholar] [CrossRef]
- Cersosimo, F.; Lonardi, S.; Bernardini, G.; Telfer, B.; Mandelli, G.E.; Santucci, A.; Vermi, W.; Giurisato, E. Tumor-Associated Macrophages in Osteosarcoma: From Mechanisms to Therapy. Int. J. Mol. Sci. 2020, 21, 5207. [Google Scholar] [CrossRef]
- Martinez, M.; Moon, E.K. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef] [Green Version]
- Highfill, S.L.; Cui, Y.; Giles, A.J.; Smith, J.P.; Zhang, H.; Morse, E.; Kaplan, R.N.; Mackall, C.L. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci. Transl. Med. 2014, 6, 237ra267. [Google Scholar] [CrossRef]
- Deng, J.; Zeng, W.; Kong, W.; Shi, Y.; Mou, X. The Study of Sarcoma Microenvironment Heterogeneity Associated with Prognosis Based on an Immunogenomic Landscape Analysis. Front. Bioeng. Biotechnol. 2020, 8, 1003. [Google Scholar] [CrossRef]
- Petitprez, F.; de Reynies, A.; Keung, E.Z.; Chen, T.W.; Sun, C.M.; Calderaro, J.; Jeng, Y.M.; Hsiao, L.P.; Lacroix, L.; Bougouin, A.; et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature 2020, 577, 556–560. [Google Scholar] [CrossRef]
- Dufresne, A.; Lesluyes, T.; Menetrier-Caux, C.; Brahmi, M.; Darbo, E.; Toulmonde, M.; Italiano, A.; Mir, O.; Le Cesne, A.; Le Guellec, S.; et al. Specific immune landscapes and immune checkpoint expressions in histotypes and molecular subtypes of sarcoma. Oncoimmunology 2020, 9, 1792036. [Google Scholar] [CrossRef]
- Menetrier-Caux, C.; Montmain, G.; Dieu, M.C.; Bain, C.; Favrot, M.C.; Caux, C.; Blay, J.Y. Inhibition of the differentiation of dendritic cells from CD34(+) progenitors by tumor cells: Role of interleukin-6 and macrophage colony-stimulating factor. Blood 1998, 92, 4778–4791. [Google Scholar] [CrossRef] [PubMed]
- Sorbye, S.W.; Kilvaer, T.; Valkov, A.; Donnem, T.; Smeland, E.; Al-Shibli, K.; Bremnes, R.M.; Busund, L.T. Prognostic impact of lymphocytes in soft tissue sarcomas. PLoS ONE 2011, 6, e14611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, K.; Li, J.; Zhang, J.; Wang, L.; Zhang, Q.; Ge, J.; Guo, Y.; Wang, B.; Huang, Y.; Yang, T.; et al. SDF-1/CXCR4 axis facilitates myeloid-derived suppressor cells accumulation in osteosarcoma microenvironment and blunts the response to anti-PD-1 therapy. Int. Immunopharmacol. 2019, 75, 105818. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Trial ID | Phase | Treatment | Sarcoma Type | Status |
|---|---|---|---|---|
| NCT03074318 | I/II | Avelumab and Trabectedin | Advanced liposarcoma and leiomyosarcoma | Active, not recruiting |
| NCT03006848 | II | Avelumab | Recurrent or progressive osteosarcoma | Active, not recruiting |
| NCT02834013 | II | Niovlumab and ipilimumab | Advanced angiosarcoma | Recruiting |
| NCT03474640 | I | Toripalimab | Advanced soft tissue sarcoma and chondrosarcoma | Recruiting |
| NCT04140526 | I | ONC-392 with/without pembrolizumab | Advanced soft tissue sarcoma | Recruiting |
| NCT02304458 | I/II | Nivolumab with/without ipilimumab | Recurrent/refractory sarcoma: Ewing sarcoma, osteosarcoma, rhabdomyosarcoma | Active, not recruiting |
| NCT04095208 | II | Nivolumab with/without relatlimab | Advanced or metastatic soft tissue sarcoma | Recruiting |
| NCT03899805 | II | Eribulin and pembrolizumab | Refractory liposarcoma, leiomyosarcoma, undifferentiated pleomorphic sarcoma | Recruiting |
| NCT03141684 | II | Atezolizumab | Advanced alveolar soft part sarcoma | Recruiting |
| NCT03338959 | I/II | Pembrolizumab with radiation therapy | Intermediate or high-grade soft tissue sarcoma | Recruiting |
| NCT04458922 | II | Atezolizumab | Newly diagnosed/unresectable/metastatic chondrosarcoma, clear cell sarcoma | Recruiting |
| NCT03307616 | II | Neoadjuvant nivolumab, nivolumab and ipilimumab, nivolumab and radiation therapy, nivolumab and ipilimumab and radiation therapy | Recurrent or resectable undifferentiated pleomorphic sarcoma or dedifferentiated liposarcoma | Active, not recruiting |
| NCT02500797 | II | Nivolumab with/without ipilimumab | Metastatic/unresectable bone sarcoma, liposarcoma, undifferentiated pleomorphic sarcoma | Active, not recruiting |
| NCT03463408 | Early I | Neoadjuvant nivolumab and ipilimumab and radiation therapy | Resectable soft tissue sarcoma | Recruiting |
| NCT03116529 | I/II | Neoadjuvant durvalumab and tremelimumab and radiation therapy | High risk soft tissue sarcoma | Recruiting |
| NCT02815995 | II | Durvalumab and tremelimumab | Advanced/metastatic sarcoma | Active, not recruiting |
| NCT03138161 | I/II | Trabectedin and ipilimumab and nivolumab | Advanced/metastatic soft tissue sarcoma | Recruiting |
| NCT02992743 | II | Autologous NY-ESO-1 genetically modified T cells (NY-ESO-1c259T) | Advanced myxoid/round cell liposarcoma | Recruiting |
| NCT04044768 | II | Autologous ADP-A2M4 genetically modified T cells | Advanced synovial sarcoma or myxoid/ round cell liposarcoma | Recruiting |
| NCT03450122 | I | Autologous NY-ESO-1 genetically modified T cells and chemotherapy and aldesleukin with/without immunostimulatory agents: CMB305 and/or antigen-specific vaccine (ID-LV305) | Advanced or recurrent synovial sarcoma, myxoid liposarcoma, NY-ESO-1 positive sarcoma | Recruiting |
| NCT02650986 | I/II | Autologous NY-ESO-1 genetically modified T cells with/without decitabine | Advanced/metastatic/unresectable synovial sarcoma | Recruiting |
| NCT03250325 | I/II | Autologous NY-ESO-1 genetically modified T cells | Unresectable NY-ESO-1 positive synovial sarcoma | Active, not recruiting |
| NCT04556669 | I | Autologous CD22 CAR genetically modified T cells or TILs with scFv fragment of anti-PD-L1 monoclonal antibody | Sarcoma | Recruiting |
| NCT03635632 | I | Autologous GD2 CAR genetically modified T cells | Relapsed GD2 positive osteosarcoma, Ewing sarcoma, rhabdomyosarcoma | Recruiting |
| NCT03635632 | I/II | Autologous sarcoma specific (CD133, GD2, Muc1, CD117 or other marker) CAR genetically modified T cells | Advanced/ recurrent sarcoma | Recruiting |
| NCT03638206 | I/II | Autologous NY-ESO-1 CAR genetically modified T cells | Synovial sarcoma | Recruiting |
| NCT00902044 | I | Autologous HER2-CD28 CAR genetically modified T cells | Refractory HER2 positive sarcoma, metastatic HER2 positive osteosarcoma | Active, not recruiting |
| NCT04483778 | I | Autologous B7H3 CAR or bispecific B7H3 and CD19 CAR genetically modified T cells | Osteosarcoma, Ewing sarcoma, rhabdomyosarcoma, synovial sarcoma, clear cell sarcoma, soft tissue sarcoma | Recruiting |
| NCT04433221 | I/II | Autologous sarcoma specific (GD2, HER2, PSMA, CD276 or other marker) CAR genetically modified T cells | Advanced/recurrent sarcoma | Recruiting |
| NCT02100891 | II | HLA-haploidentical haematopoietic cell transplantation and donor NK cell infusion | Advanced/recurrent Ewing sarcoma, rhabdomyosarcoma, osteosarcoma | Active, not recruiting |
| NCT02409576 | I/II | Expanded and activated allogenic NK cells | Advanced/metastatic/relapsed Ewing sarcoma, rhabdomyosarcoma | Recruiting |
| NCT03860207 | I/II | Humanised 3F8 bispecific antibody (Hu3F8-BsAb) | Relapsed/refractory GD2 positive osteosarcoma | Recruiting |
| NCT01803152 | I | Autologous tumour lysate with dendritic cell vaccine with/without myeloid derived suppressor cells inhibition | Relapsed sarcoma | Active, not recruiting |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chew, H.Y.; Chan, V.; Simpson, F.; Dolcetti, R. Will Next-Generation Immunotherapy Overcome the Intrinsic Diversity and Low Immunogenicity of Sarcomas to Improve Clinical Benefit? Cancers 2020, 12, 3392. https://doi.org/10.3390/cancers12113392
Chew HY, Chan V, Simpson F, Dolcetti R. Will Next-Generation Immunotherapy Overcome the Intrinsic Diversity and Low Immunogenicity of Sarcomas to Improve Clinical Benefit? Cancers. 2020; 12(11):3392. https://doi.org/10.3390/cancers12113392
Chicago/Turabian StyleChew, Hui Yi, Victor Chan, Fiona Simpson, and Riccardo Dolcetti. 2020. "Will Next-Generation Immunotherapy Overcome the Intrinsic Diversity and Low Immunogenicity of Sarcomas to Improve Clinical Benefit?" Cancers 12, no. 11: 3392. https://doi.org/10.3390/cancers12113392