
CDK4/6 Inhibition Reprograms Mitochondrial Metabolism in BRAFV600 Melanoma via a p53 Dependent Pathway

 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
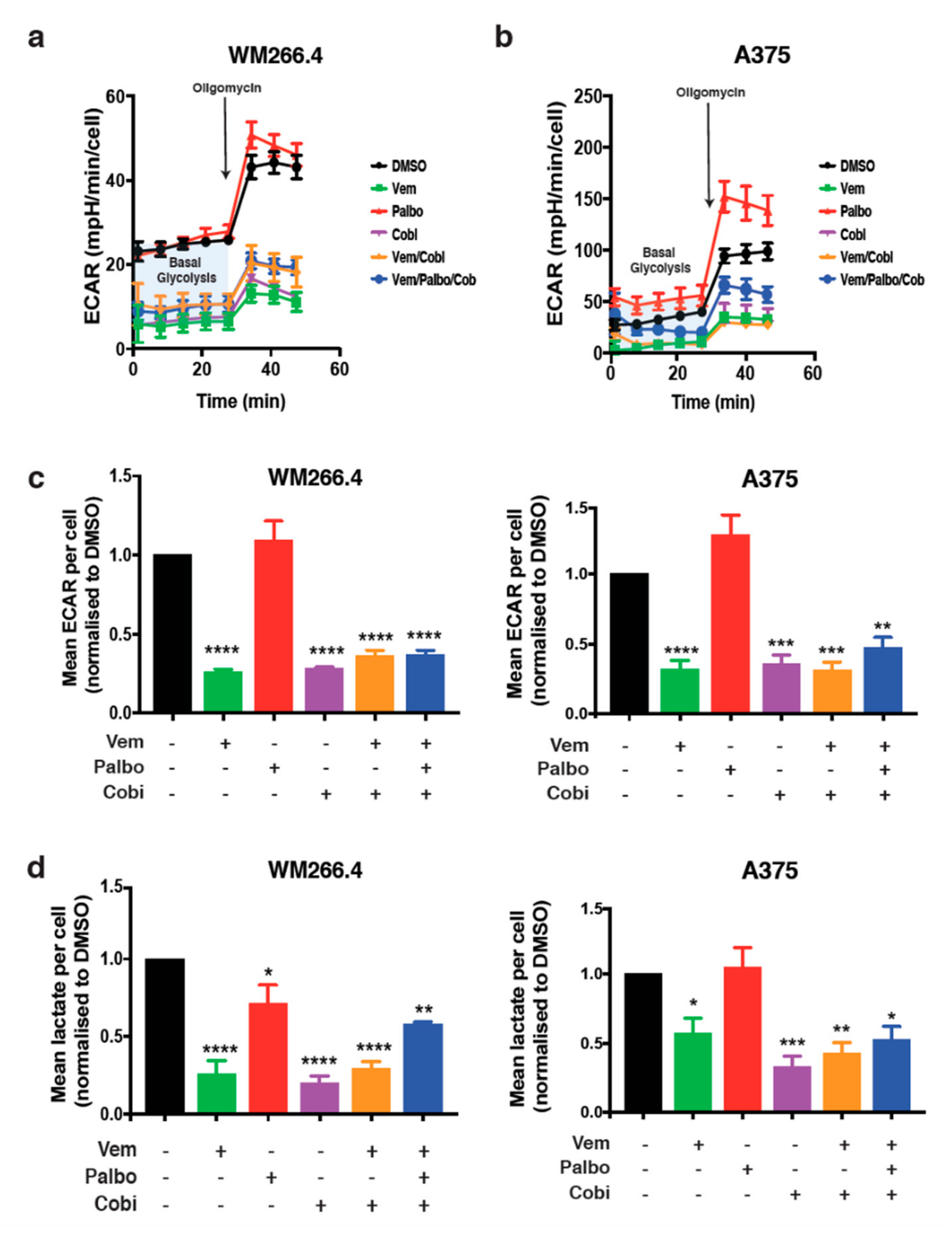
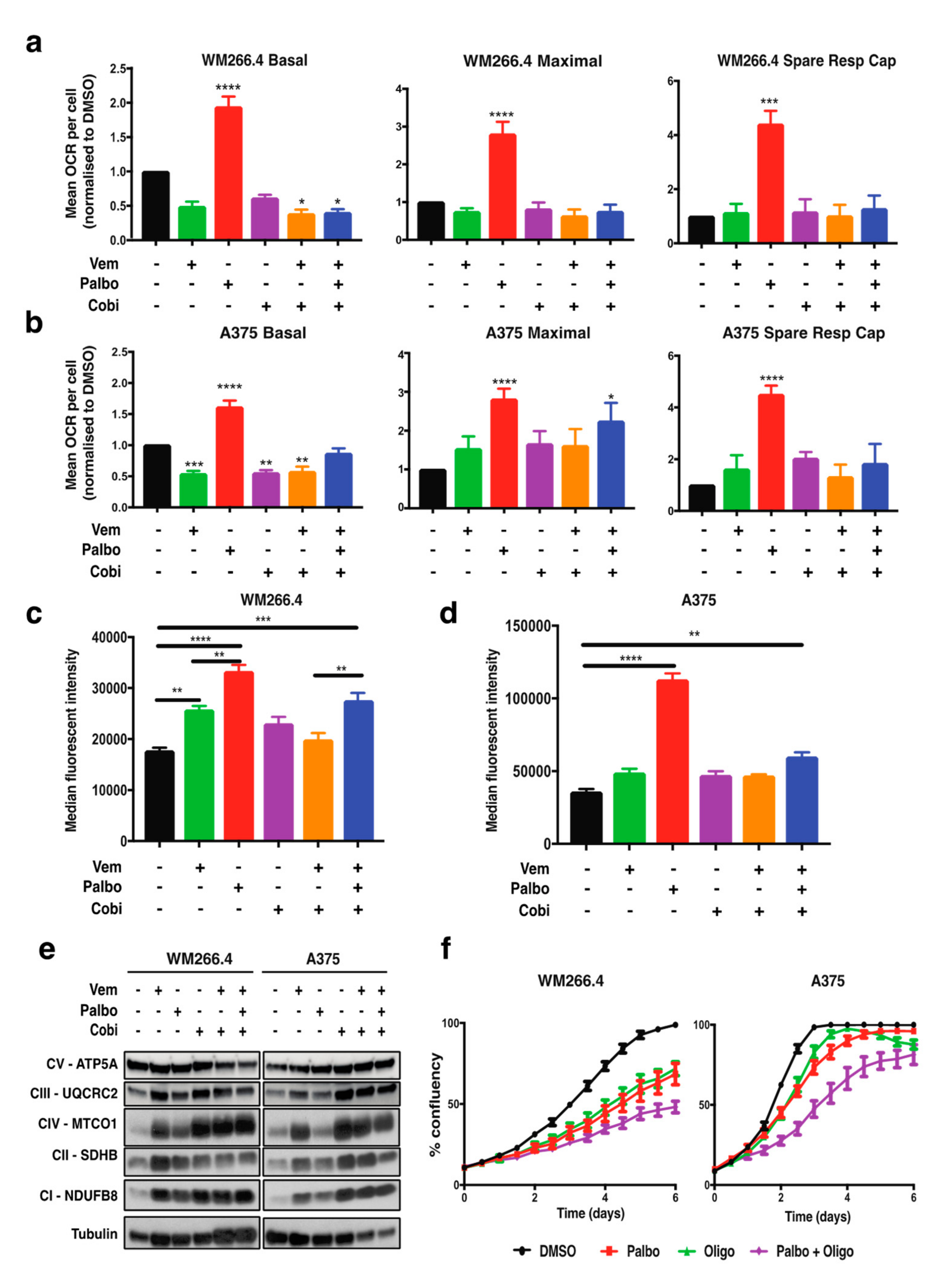
2.1. CDK4/6 Inhibition Increases Mitochondrial Metabolism but Not the Response to BRAF/MEK Inhibition
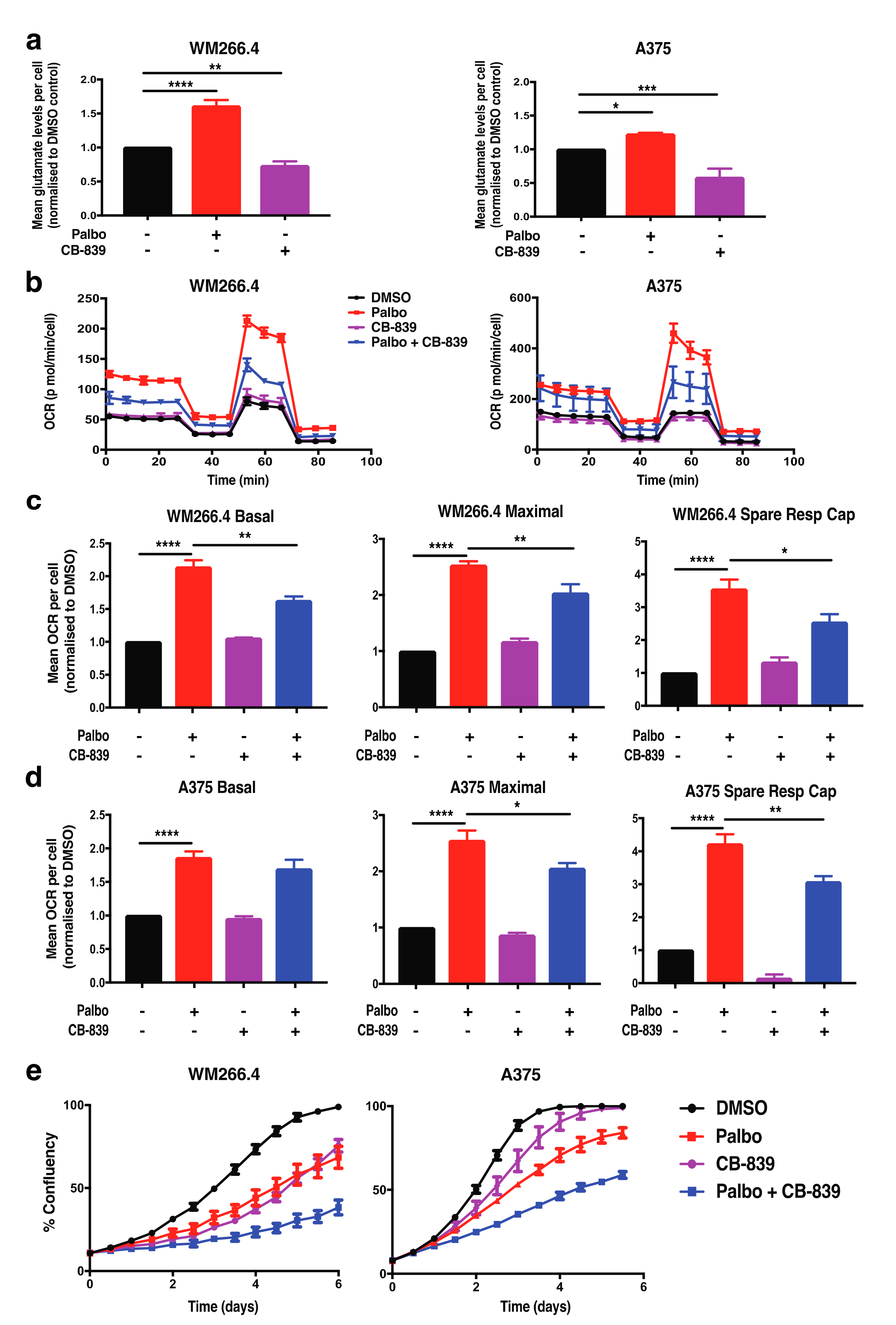
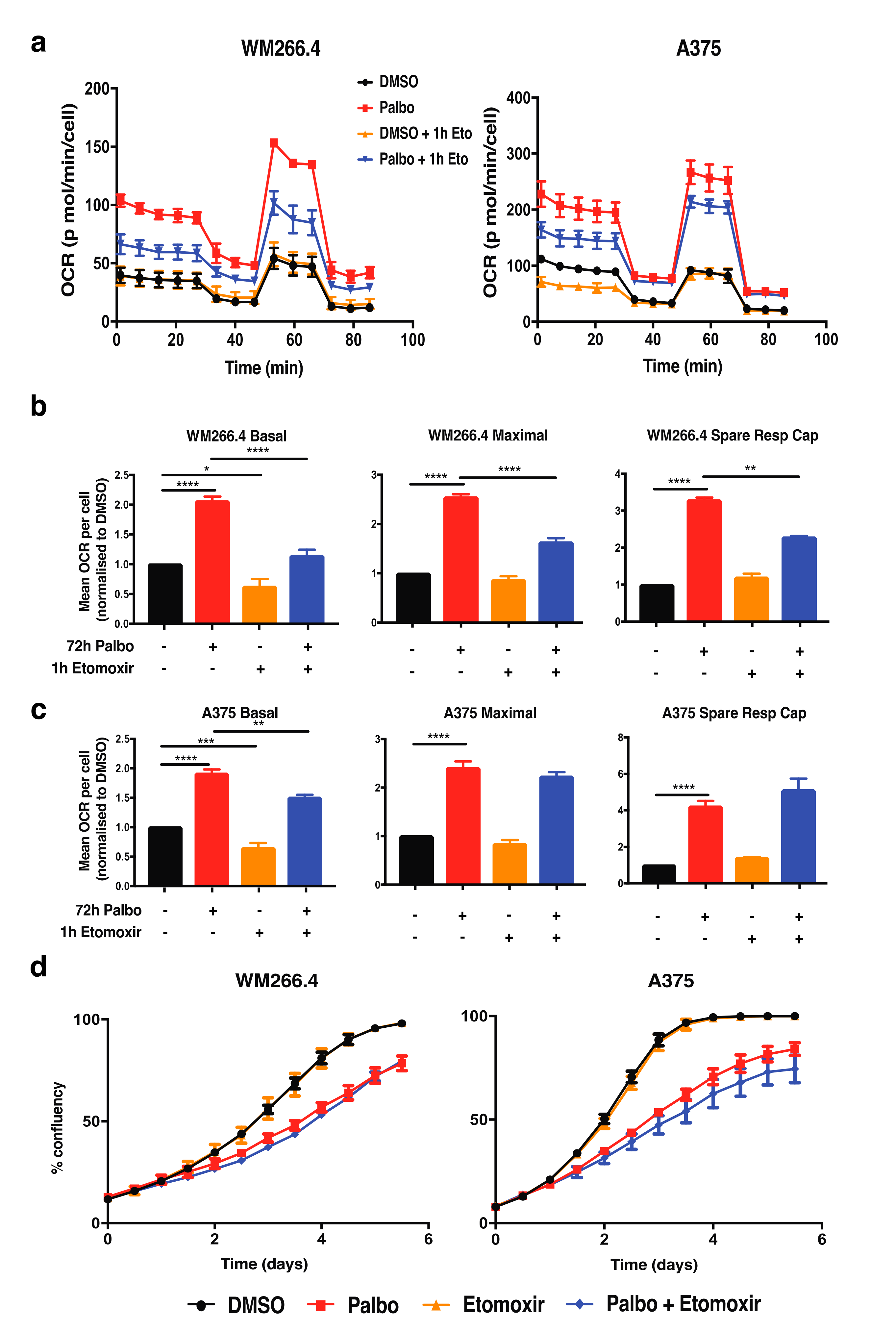
2.2. CDK4/6 Inhibition Alters Glutamine Metabolism and Fatty Acid Oxidation in BRAFV600 Melanoma Cells
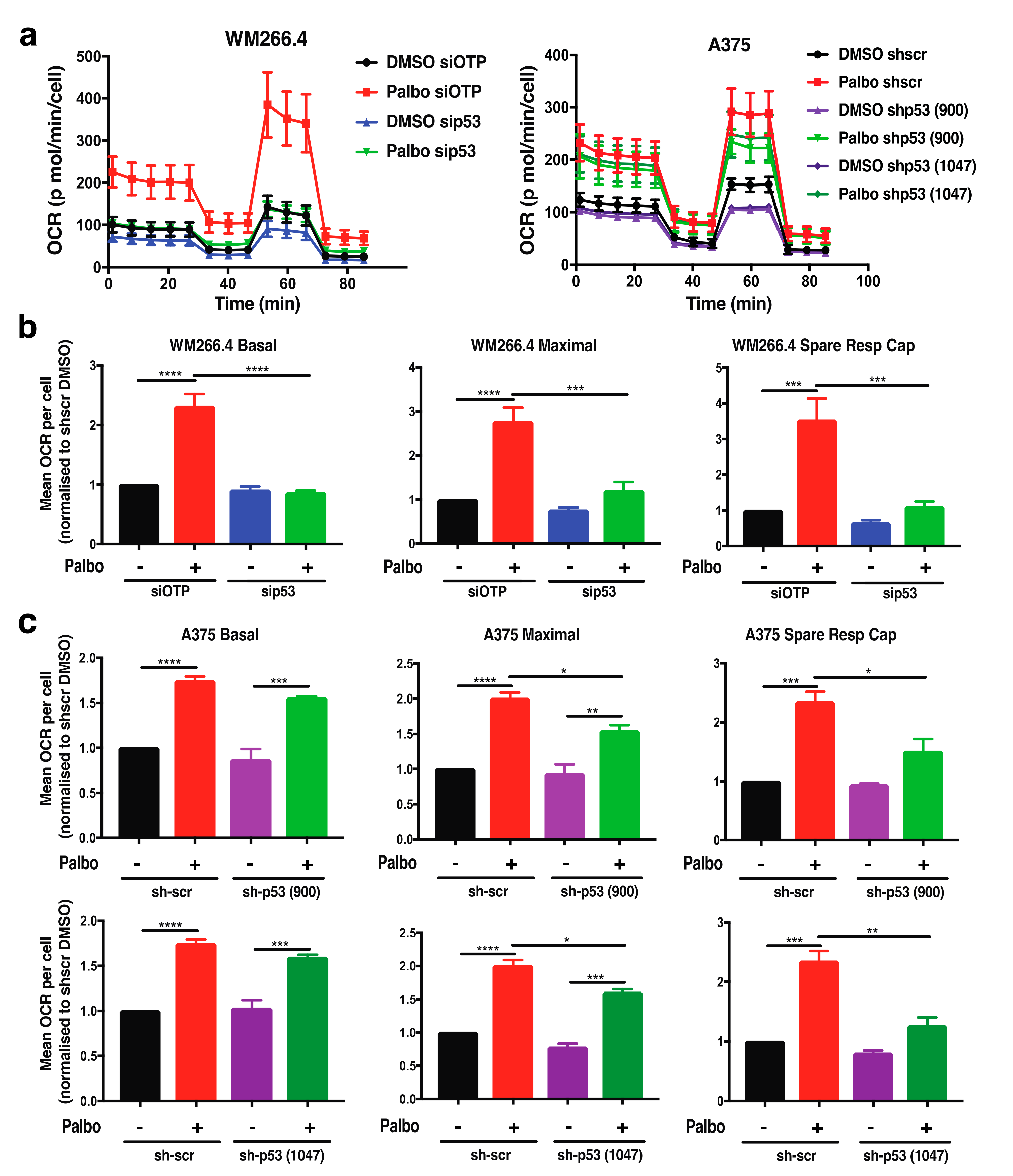
2.3. Adaptive Metabolic Responses to CDK4/6 Inhibition Are Partially Dependent on p53 in BRAFV600 Melanoma Cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Pharmacologic Inhibitors
4.3. Western Immunoblotting
4.4. qRT-PCR
4.5. Lactate Assay
4.6. Extracellular Flux Analysis
4.7. Glutamate Detection Assay
4.8. Mitotracker Assay
4.9. siRNA Mediated Gene Knockdown
4.10. Proliferation and Viability Assays
4.11. Drug Dose–Response Assays
4.12. Gene Set Enrichment Analysis (GSEA)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sebolt-Leopold, J.S.; Herrera, R. Targeting the mitogen-activated protein kinase cascade to treat cancer. Nat. Rev. Cancer 2004, 4, 937–947. [Google Scholar] [CrossRef]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.-P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas, N. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [Green Version]
- Larkin, J.; Ascierto, P.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined Vemurafenib and Cobimetinib in BRAF-Mutated Melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved Overall Survival in Melanoma with Combined Dabrafenib and Trametinib. N. Engl. J. Med. 2014, 372, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtin, J.A.; Fridlyand, J.; Kageshita, T.; Patel, H.N.; Busam, K.J.; Kutzner, H.; Cho, K.-H.; Aiba, S.; Bröcker, E.-B.; LeBoit, P.E.; et al. Distinct Sets of Genetic Alterations in Melanoma. N. Engl. J. Med. 2005, 353, 2135–2147. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, K.E.; McArthur, G.A. The Cell-Cycle Regulator CDK4: An Emerging Therapeutic Target in Melanoma. Clin. Cancer Res. 2013, 19, 5320. [Google Scholar] [CrossRef] [Green Version]
- Coleman, M.L.; Marshall, C.J.; Olson, M.F. RAS and RHO GTPases in G1-phase cell-cycle regulation. Nat. Rev. Mol. Cell Biol. 2004, 5, 355. [Google Scholar] [CrossRef]
- VanArsdale, T.; Boshoff, C.; Arndt, K.T.; Abraham, R.T. Molecular Pathways: Targeting the Cyclin D–CDK4/6 Axis for Cancer Treatment. Clin. Cancer Res. 2015, 21, 2905. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, A.; Lee, E.K.; Diehl, J.A. Induction of therapeutic senescence in vemurafenib-resistant melanoma by extended Inhibition of CDK4/6. Cancer Res. 2016, 76, 2990–3002. [Google Scholar] [CrossRef] [Green Version]
- Martin, C.A.; Cullinane, C.; Kirby, L.; Abuhammad, S.; Lelliott, E.J.; Waldeck, K.; Young, R.J.; Brajanovski, N.; Cameron, D.P.; Walker, R.; et al. Palbociclib synergizes with BRAF and MEK inhibitors in treatment naïve melanoma but not after the development of BRAF inhibitor resistance. Int. J. Cancer 2017, 142, 2139–2152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parmenter, T.J.; Kleinschmidt, M.; Kinross, K.M.; Bond, S.T.; Li, J.; Kaadige, M.R.; Rao, A.; Sheppard, K.E.; Hugo, W.; Pupo, G.M.; et al. Response of BRAF-mutant melanoma to BRAF inhibition is mediated by a network of transcriptional regulators of glycolysis. Cancer Discov. 2014, 4, 423–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haq, R.; Shoag, J.; Andreu-Perez, P.; Yokoyama, S.; Edelman, H.; Rowe, G.C.; Frederick, D.T.; Hurley, A.D.; Nellore, A.; Kung, A.L.; et al. Oncogenic BRAF Regulates Oxidative Metabolism via PGC1α and MITF. Cancer Cell 2013, 23, 302–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopal, Y.N.; Rizos, H.; Chen, G.; Deng, W.; Frederick, D.T.; Cooper, Z.A.; Scolyer, R.A.; Pupo, G.; Komurov, K.; Sehgal, V.; et al. Inhibition of mTORC1/2 overcomes resistance to MAPK pathway inhibitors mediated by PGC1alpha and oxidative phosphorylation in melanoma. Cancer Res. 2014, 74, 7037–7047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.; Frederick, D.T.; Wu, L.; Wei, Z.; Krepler, C.; Srinivasan, S.; Chae, Y.C.; Xu, X.; Choi, H.; Dimwamwa, E.; et al. Targeting mitochondrial biogenesis to overcome drug resistance to MAPK inhibitors. J. Clin. Investig. 2016, 126, 1834–1856. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Mejia, I.C.; Lagarrigue, S.; Giralt, A.; Martinez-Carreres, L.; Zanou, N.; Denechaud, P.-D.; Castillo-Armengol, J.; Chavey, C.; Orpinell, M.; Delacuisine, B.; et al. CDK4 Phosphorylates AMPKα2 to Inhibit Its Activity and Repress Fatty Acid Oxidation. Mol. Cell 2017, 68, 336–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarrado-Castellarnau, M.; de Atauri, P.; Tarragó-Celada, J.; Perarnau, J.; Yuneva, M.; Thomson, T.M.; Cascante, M. De novo MYC addiction as an adaptive response of cancer cells to CDK4/6 inhibition. Mol. Syst. Biol. 2017, 13. [Google Scholar] [CrossRef]
- Franco, J.; Balaji, U.; Freinkman, E.; Witkiewicz, A.K.; Knudsen, E.S. Metabolic re-programming of pancreatic cancer mediated by CDK4/6 inhibition elicits unique vulnerabilities. Cell Rep. 2016, 14, 979–990. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Dominy, J.E.; Choi, Y.J.; Jurczak, M.; Tolliday, N.; Camporez, J.P.; Chim, H.; Lim, J.-H.; Ruan, H.-B.; Yang, X.; et al. Cyclin D1-Cdk4 controls glucose metabolism independently of cell cycle progression. Nature 2014, 510, 547–551. [Google Scholar] [CrossRef]
- Harding, J.J.; Telli, M.L.; Munster, P.N.; Le, M.H.; Molineaux, C.; Bennett, M.K.; Mittra, E.; Burris, H.A.; Clark, A.S.; Dunphy, M.; et al. Safety and tolerability of increasing doses of CB-839, a first-in-class, orally administered small molecule inhibitor of glutaminase, in solid tumors. J. Clin. Oncol. 2015, 33, 2512. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. US National Library of Medicine: CB-839. Available online: https://clinicaltrials.gov/ct2/results?cond=&term=cb-839&cntry=&state=&city=&dist= (accessed on 10 November 2020).
- Qu, Q.; Zeng, F.; Liu, X.; Wang, Q.J.; Deng, F. Fatty acid oxidation and carnitine palmitoyltransferase I: Emerging therapeutic targets in cancer. Cell Death Dis. 2016, 7, e2226. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Puzio-Kuter, A.M. The Control of the Metabolic Switch in Cancers by Oncogenes and Tumor Suppressor Genes. Science 2010, 330, 1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- AbuHammad, S.; Cullinane, C.; Martin, C.; Bacolas, Z.; Ward, T.; Chen, H.; Slater, A.; Ardley, K.; Kirby, L.; Chan, K.T.; et al. Regulation of PRMT5-MDM4 axis is critical in the response to CDK4/6 inhibitors in melanoma. Proc. Natl. Acad. Sci. USA 2019, 116, 17990–18000. [Google Scholar] [CrossRef] [PubMed]
- Terzian, T.; Torchia, E.C.; Dai, D.; Robinson, S.E.; Murao, K.; Stiegmann, R.A.; Gonzalez, V.; Boyle, G.M.; Powell, M.B.; Pollock, P.M.; et al. p53 prevents progression of nevi to melanoma predominantly through cell cycle regulation. Pigment Cell Melanoma Res. 2010, 23, 781–794. [Google Scholar] [CrossRef]
- Maddocks, O.D.; Vousden, K.H. Metabolic regulation by p53. J. Mol. Med. 2011, 89, 237–245. [Google Scholar] [CrossRef] [Green Version]
- Berkers, C.R.; Maddocks, O.D.; Cheung, E.C.; Mor, I.; Vousden, K.H. Metabolic regulation by p53 family members. Cell Metab. 2013, 18, 617–633. [Google Scholar] [CrossRef] [Green Version]
- Cristofanilli, M.; Turner, N.C.; Bondarenko, I.; Ro, J.; Im, S.-A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): Final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2016, 17, 425–439. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.S.; Crown, J.P.; Lang, I.; Boer, K.; Bondarenko, I.M.; Kulyk, S.O.; Ettl, J.; Patel, R.; Pinter, T.; Schmidt, M.; et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): A randomised phase 2 study. Lancet Oncol. 2015, 16, 25–35. [Google Scholar] [CrossRef]
- Sherr, C.J.; Beach, D.; Shapiro, G.I. Targeting cdk4 and cdk6: From discovery to therapy. Cancer Discov. 2016, 6, 353–367. [Google Scholar] [CrossRef] [Green Version]
- ClinicalTrials.gov. US National Library of Medicine: CDK4/6 Inhibitor. Available online: https://clinicaltrials.gov/ct2/results?cond=&term=CDK4%2F6+inhibitor&cntry=&state=&city=&dist= (accessed on 10 November 2020).
- Anders, L.; Ke, N.; Hydbring, P.; Choi, Y.J.; Widlund, H.R.; Chick, J.M.; Zhai, H.; Vidal, M.; Gygi, S.P.; Braun, P.; et al. A Systematic Screen for CDK4/6 Substrates Links FOXM1 Phosphorylation to Senescence Suppression in Cancer Cells. Cancer Cell 2011, 20, 620–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonelli, M.; La Monica, S.; Fumarola, C.; Alfieri, R. Multiple effects of CDK4/6 inhibition in cancer: From cell cycle arrest to immunomodulation. Biochem. Pharmacol. 2019, 170, 113676. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.E.; Kovatcheva, M.; Davis, L.E.; Tap, W.D.; Koff, A. CDK4/6 Inhibitors: The Mechanism of Action May Not Be as Simple as Once Thought. Cancer Cell 2018, 34, 9–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwong, L.N.; Costello, J.C.; Liu, H.; Jiang, S.; Helms, T.L.; Langsdorf, A.E.; Jakubosky, D.; Genovese, G.; Muller, F.L.; Jeong, J.H.; et al. Oncogenic NRAS signaling differentially regulates survival and proliferation in melanoma. Nat. Med. 2012, 18, 1503–1510. [Google Scholar] [CrossRef] [Green Version]
- Teh, J.L.F.; Purwin, T.J.; Greenawalt, E.J.; Chervoneva, I.; Goldberg, A.; Davies, M.A.; Aplin, A.E. An In Vivo Reporter to Quantitatively and Temporally Analyze the Effects of CDK4/6 Inhibitor-Based Therapies in Melanoma. Cancer Res. 2016, 76, 5455–5466. [Google Scholar] [CrossRef] [Green Version]
- Yadav, V.; Chen, S.-H.; Yue, Y.G.; Buchanan, S.; Beckmann, R.P.; Peng, S.-B. Co-targeting BRAF and cyclin dependent kinases 4/6 for BRAF mutant cancers. Pharmacol. Ther. 2015, 149, 139–149. [Google Scholar] [CrossRef]
- Vazquez, F.; Lim, J.-H.; Chim, H.; Bhalla, K.; Girnun, G.; Pierce, K.; Clish, C.B.; Granter, S.R.; Widlund, H.R.; Spiegelman, B.M.; et al. PGC1α Expression Defines a Subset of Human Melanoma Tumors with Increased Mitochondrial Capacity and Resistance to Oxidative Stress. Cancer Cell 2013, 23, 287–301. [Google Scholar] [CrossRef] [Green Version]
- Warth, B.; Raffeiner, P.; Granados, A.; Huan, T.; Fang, M.; Forsberg, E.M.; Benton, H.P.; Goetz, L.; Johnson, C.H.; Siuzdak, G. Metabolomics Reveals that Dietary Xenoestrogens Alter Cellular Metabolism Induced by Palbociclib/Letrozole Combination Cancer Therapy. Cell Chem. Biol. 2018, 25, 291–300.e293. [Google Scholar] [CrossRef] [Green Version]
- Cretella, D.; Ravelli, A.; Fumarola, C.; La Monica, S.; Digiacomo, G.; Cavazzoni, A.; Alfieri, R.; Biondi, A.; Generali, D.; Bonelli, M.; et al. The anti-tumor efficacy of CDK4/6 inhibition is enhanced by the combination with PI3K/AKT/mTOR inhibitors through impairment of glucose metabolism in TNBC cells. J. Exp. Clin. Cancer Res. 2018, 37, 72. [Google Scholar] [CrossRef]
- Bonelli, M.; Terenziani, R.; Zoppi, S.; Fumarola, C.; La Monica, S.; Cretella, D.; Alfieri, R.; Cavazzoni, A.; Digiacomo, G.; Galetti, M.; et al. Dual Inhibition of CDK4/6 and PI3K/AKT/mTOR Signaling Impairs Energy Metabolism in MPM Cancer Cells. Int. J. Mol. Sci. 2020, 21, 5165. [Google Scholar] [CrossRef]
- Vousden, K.H.; Lu, X. Live or let die: The cell’s response to p53. Nat. Rev. Cancer 2002, 2, 594–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, T.Q.; Lowman, X.H.; Reid, M.A.; Mendez-Dorantes, C.; Pan, M.; Yang, Y.; Kong, M. Tumor-associated mutant p53 promotes cancer cell survival upon glutamine deprivation through p21 induction. Oncogene 2017, 36, 1991–2001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowman, X.H.; Hanse, E.A.; Yang, Y.; Ishak Gabra, M.B.; Tran, T.Q.; Li, H.; Kong, M. p53 Promotes Cancer Cell Adaptation to Glutamine Deprivation by Upregulating Slc7a3 to Increase Arginine Uptake. Cell Rep. 2019, 26, 3051–3060.e3054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishak Gabra, M.B.; Yang, Y.; Lowman, X.H.; Reid, M.A.; Tran, T.Q.; Kong, M. IKKβ activates p53 to promote cancer cell adaptation to glutamine deprivation. Oncogenesis 2018, 7, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Treatment | Proliferation + Viability Assays | Metabolism Assays |
|---|---|---|
| PLX (Plx4032, Vemurafenib) | 300 nM | μM |
| Palbo (PD-0332991, Palbociclib) | μM | μM |
| Cobimetinib | 1 nM | 10 nM |
| PLX/Cobimetinib | 300 nM/1 nM | μM/10 nM |
| PLX/Palbo/Cobimetinib | 300 nM/1 nM/1 μM | 1 μM, 1 μM, 10 nM (respectively) |
| Oligomycin | μM | μM |
| CB-839 | 200 nM | 200 nM |
| Etomoxir | μM | μM |
| Gene | Direction | Sequence |
|---|---|---|
| c-Myc | Forward | GGACGACGAGACCTTCATCAA |
| Reverse | CCAGCTTCTCTGAGACGAGCTT | |
| GLUT1 | Forward | TCTCTGTGGGCCTTTTCGTT |
| Reverse | CAGTTTCGAGAAGCCCATGAG | |
| GLUT3 | Forward | GGTGGAAGTACGTTATTGTTGACTTATT |
| Reverse | GTTTGGCTAAAGGGTCTGAGATGT | |
| HK2 | Forward | AAGGCAATAGGGCCTTAAAGTAGAG |
| Reverse | TTCGAGGCTGCAGTGAGCTA | |
| HIF1α | Forward | TTTACCATGCCCCAGATTCAG |
| Reverse | GGTGAACTTTGTCTAGTGCTTCCA | |
| MITF | Forward | CCGTCTCTCACTGGATTGGT |
| Reverse | TACTTGGTGGGGTTTTCGAG | |
| NONO | Forward | CATCAAGGAGGCTCGTGAGAAG |
| Reverse | TGGTTGTGCAGCTCTTCCATCC | |
| PGC1a | Forward | CTGCTAGCAAGTTTGCCTCA |
| Reverse | AGTGGTGCAGTGACCAATCA | |
| TFAM | Forward | TACCGAGGTGGTTTTCATCTG |
| Reverse | AACGCTGGGCAATTCTTCTA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santiappillai, N.T.; Abuhammad, S.; Slater, A.; Kirby, L.; McArthur, G.A.; Sheppard, K.E.; Smith, L.K. CDK4/6 Inhibition Reprograms Mitochondrial Metabolism in BRAFV600 Melanoma via a p53 Dependent Pathway. Cancers 2021, 13, 524. https://doi.org/10.3390/cancers13030524
Santiappillai NT, Abuhammad S, Slater A, Kirby L, McArthur GA, Sheppard KE, Smith LK. CDK4/6 Inhibition Reprograms Mitochondrial Metabolism in BRAFV600 Melanoma via a p53 Dependent Pathway. Cancers. 2021; 13(3):524. https://doi.org/10.3390/cancers13030524
Chicago/Turabian StyleSantiappillai, Nancy T., Shatha Abuhammad, Alison Slater, Laura Kirby, Grant A. McArthur, Karen E. Sheppard, and Lorey K. Smith. 2021. "CDK4/6 Inhibition Reprograms Mitochondrial Metabolism in BRAFV600 Melanoma via a p53 Dependent Pathway" Cancers 13, no. 3: 524. https://doi.org/10.3390/cancers13030524