
Type 2 Innate Lymphoid Cells Protect against Colorectal Cancer Progression and Predict Improved Patient Survival

 ,
,  , , , and
, , , and
Abstract
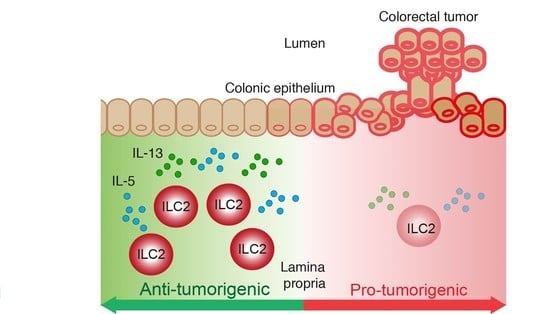
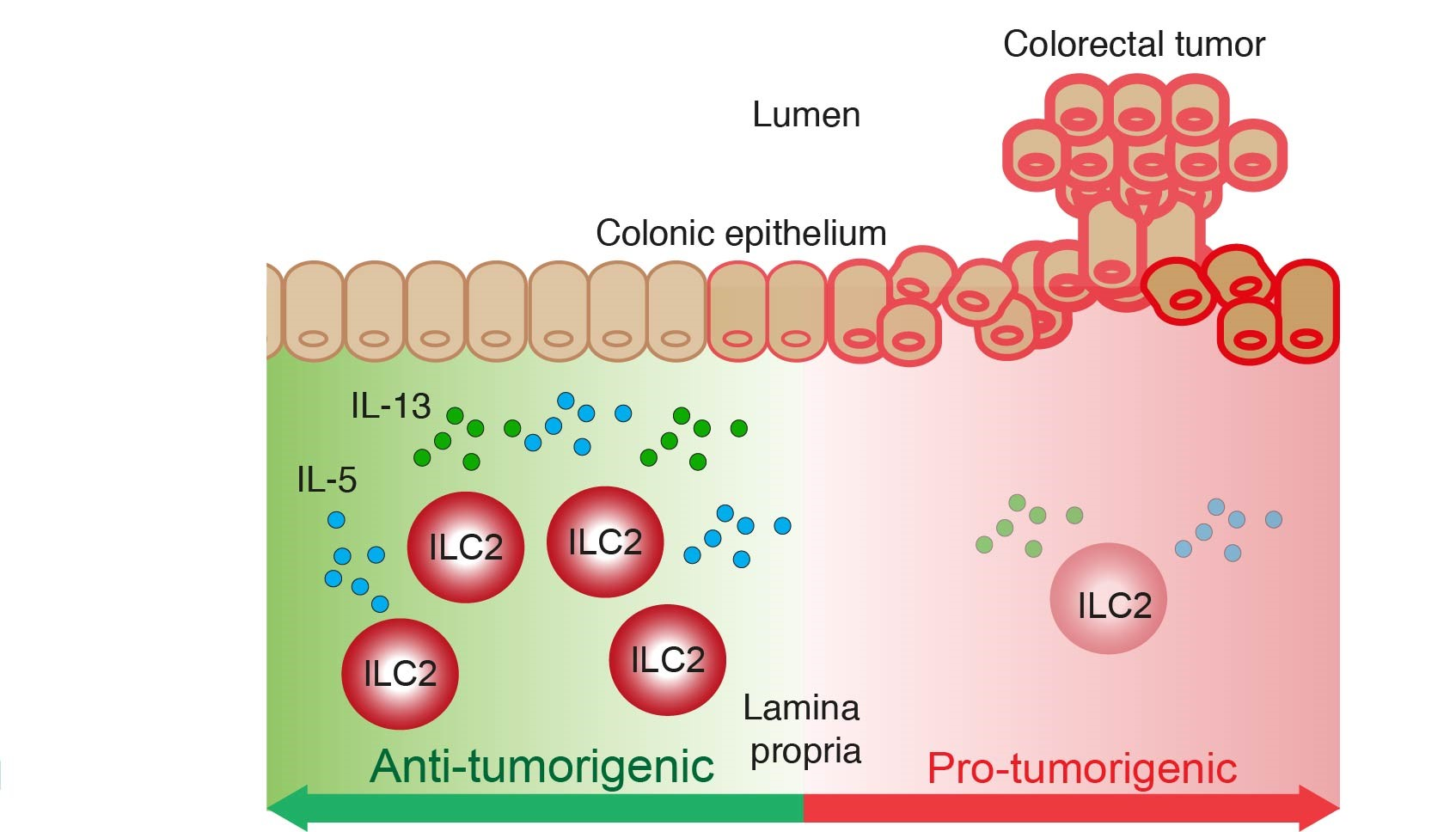
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
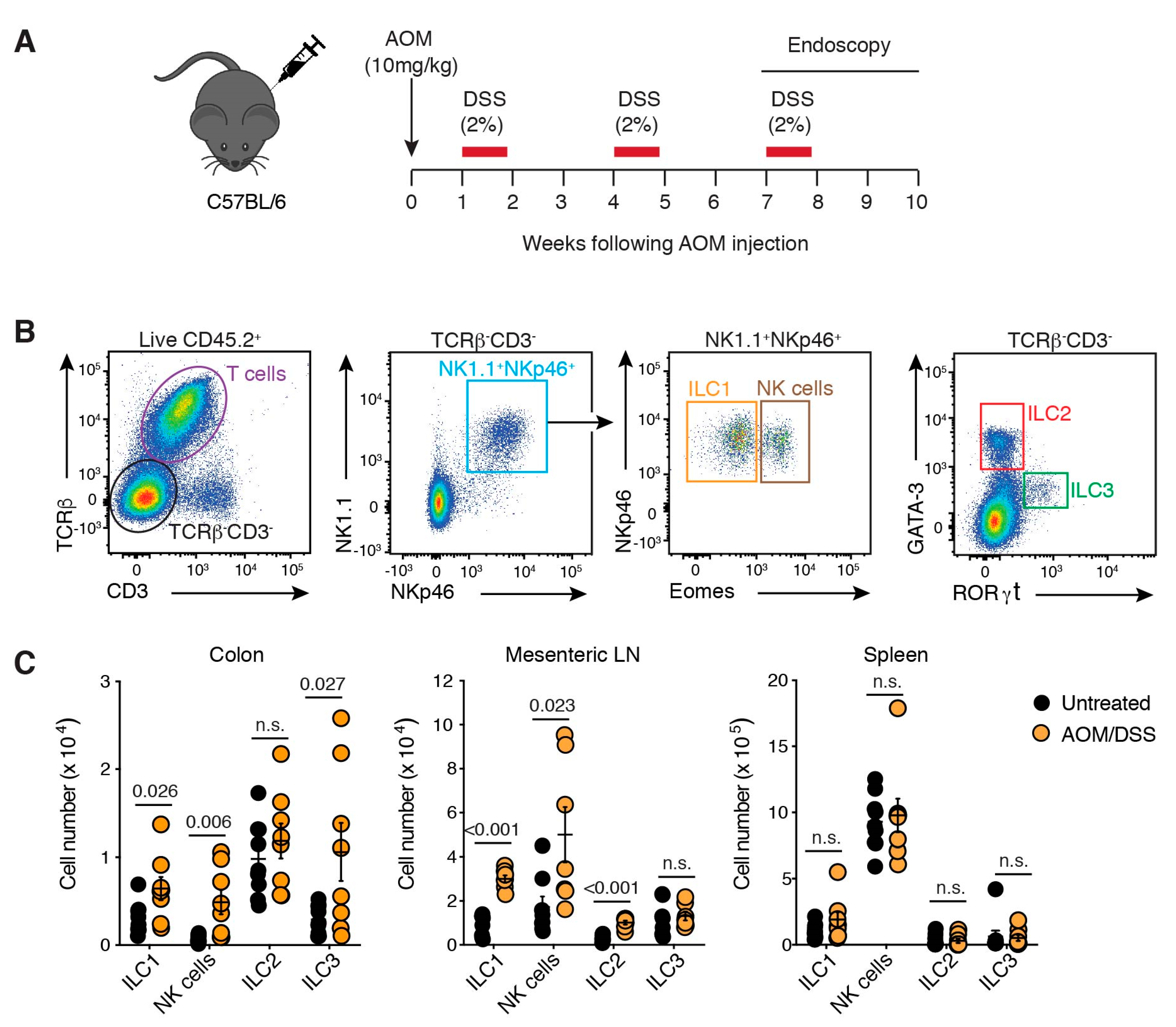
2.1. ILC Are Present in a Murine Model of Colon Cancer and Implicated in Tumour Growth
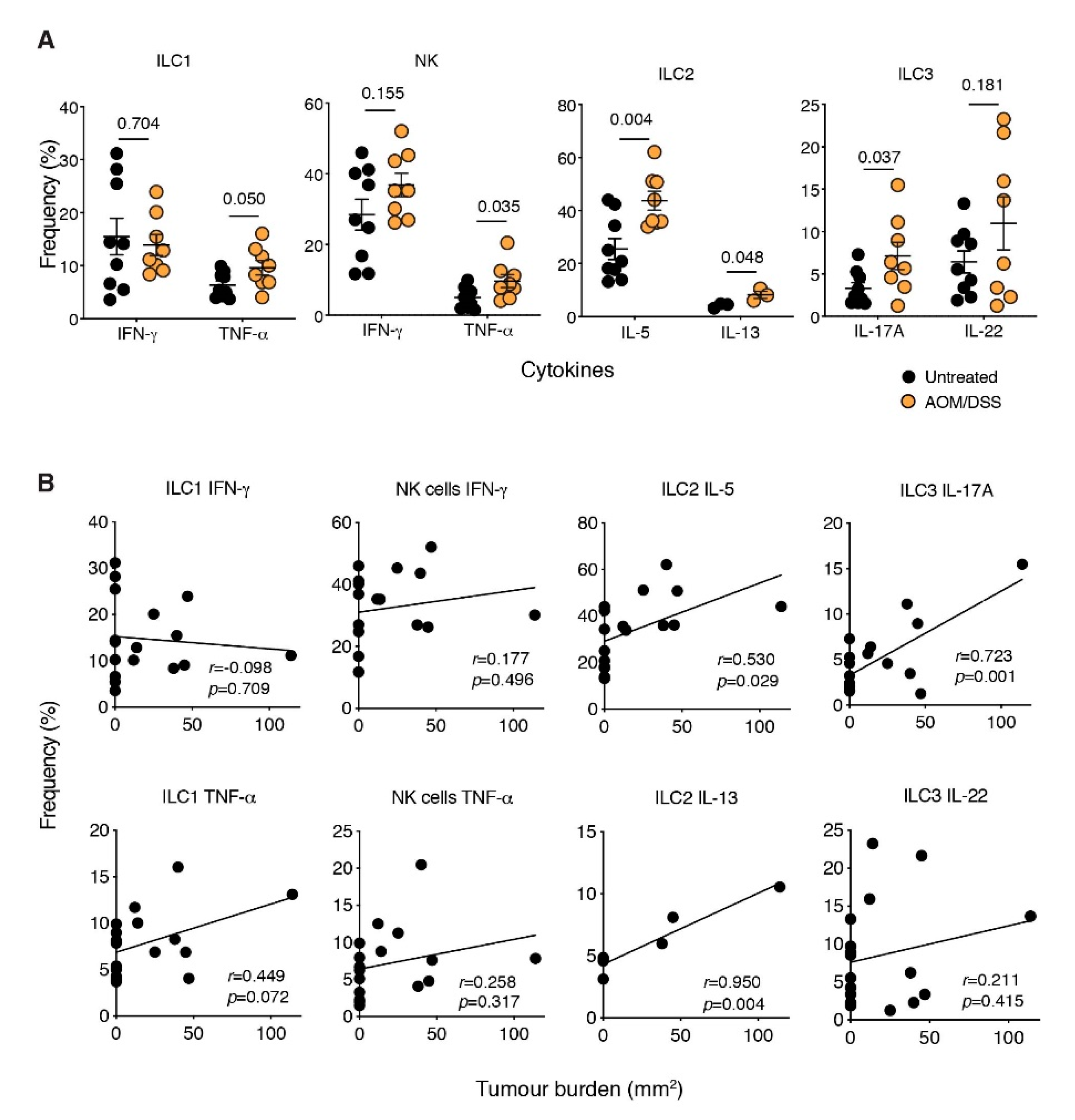
2.2. ILCs Are Increased in Inflammation-Associated Colon Cancer
2.3. The ILC2-Derived Cytokines, IL-5 and IL-13, Are Elevated in CAC
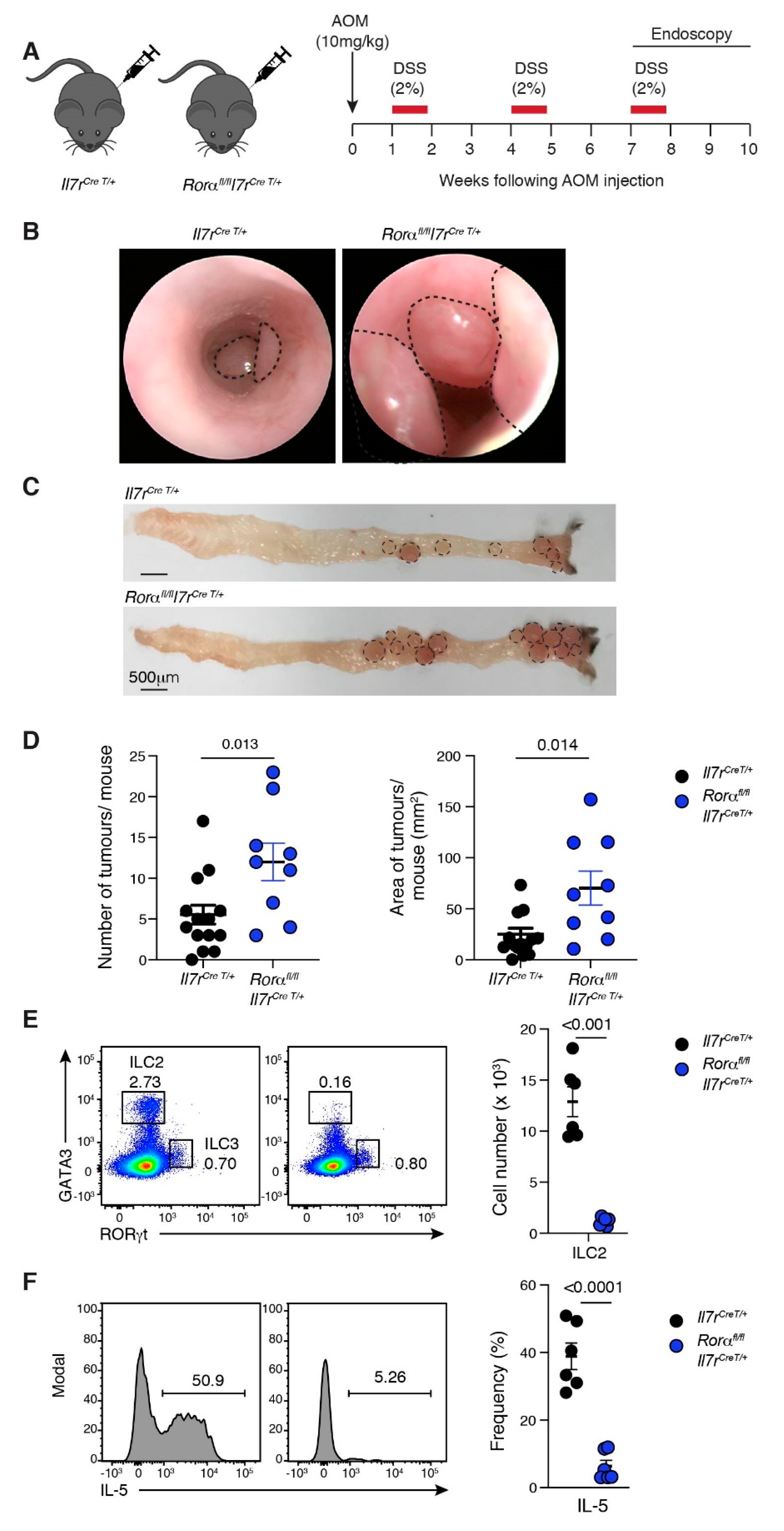
2.4. Loss of ILC2 Results in Increased Tumour Development in CAC
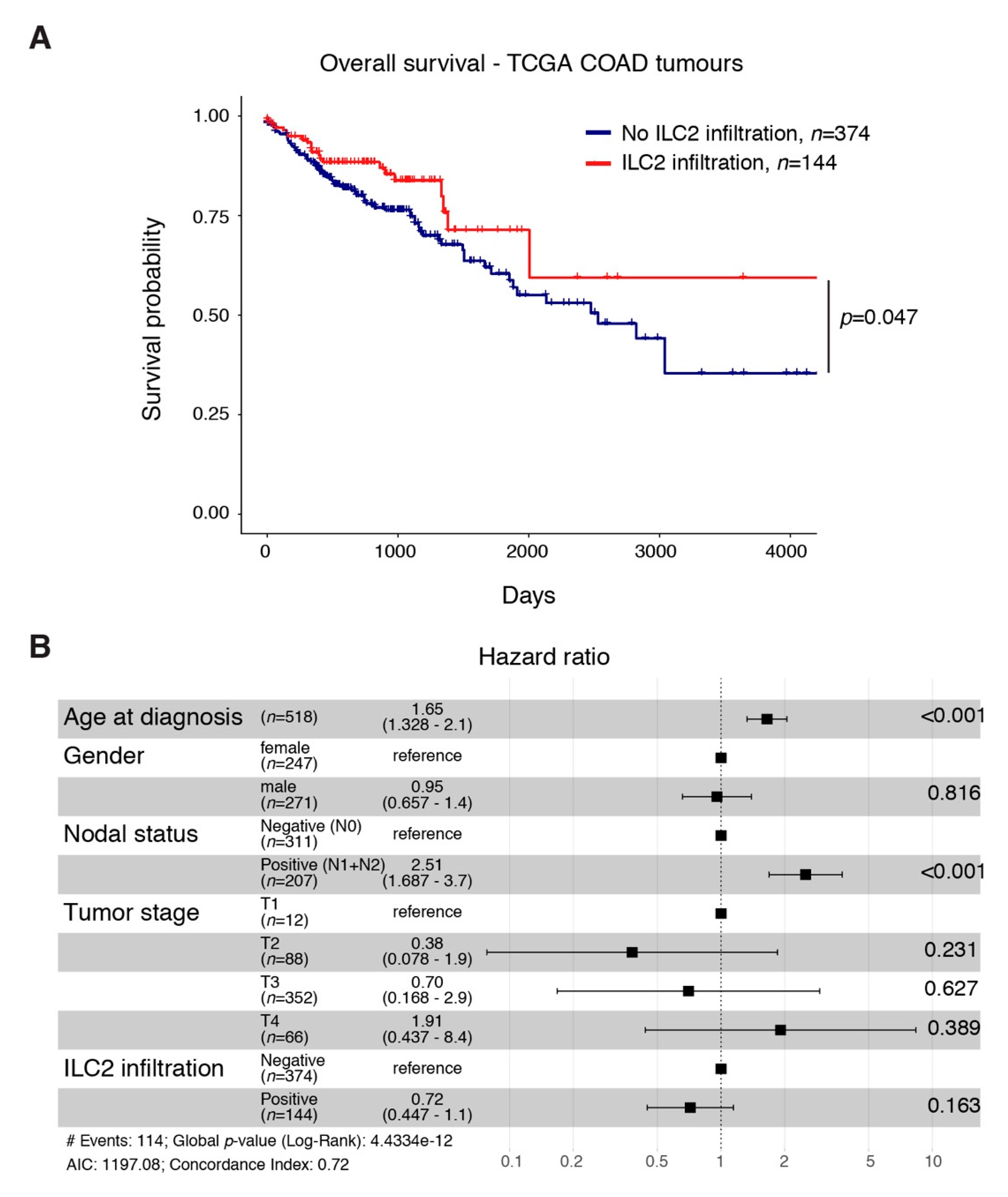
2.5. An ILC2 Gene Signature Is Associated with Increased Overall Survival in CRC
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Cell Culture and Xenograft Establishment
4.3. Isolation of Lymphoid Cells
4.4. Flow Cytometry
4.5. Colitis-Associated Colon Cancer
4.6. Histology
4.7. Training an Intestinal ILC2 Classifier
4.8. Quantification of Infiltrating ILC2 in the TCGA COAD Cohort Using the Intestinal ILC2 Classifier
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mowat, A.M.; Agace, W.W. Regional specialization within the intestinal immune system. Nat. Rev. Immunol. 2014, 14, 667–685. [Google Scholar] [CrossRef] [PubMed]
- Kraus, S.; Arber, N. Inflammation and colorectal cancer. Curr. Opin. Pharm. 2009, 9, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Qu, Y.; Xia, P.; Chen, Y.; Zhu, X.; Zhang, J.; Wang, G.; Tian, Y.; Ying, J.; Fan, Z. Transdifferentiation of tumor infiltrating innate lymphoid cells during progression of colorectal cancer. Cell Res. 2020, 30, 610–622. [Google Scholar] [CrossRef] [PubMed]
- Morson, B.C. Precancer and cancer in inflammatory bowel disease. Pathology 1985, 17, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pages, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [Green Version]
- Pages, F.; Berger, A.; Camus, M.; Sanchez-Cabo, F.; Costes, A.; Molidor, R.; Mlecnik, B.; Kirilovsky, A.; Nilsson, M.; Damotte, D.; et al. Effector memory t cells, early metastasis, and survival in colorectal cancer. N. Engl. J. Med. 2005, 353, 2654–2666. [Google Scholar] [CrossRef]
- Huang, Q.; Cao, W.; Mielke, L.A.; Seillet, C.; Belz, G.T.; Jacquelot, N. Innate lymphoid cells in colorectal cancers: A double-edged sword. Front. Immunol. 2019, 10, 3080. [Google Scholar] [CrossRef] [Green Version]
- Atreya, I.; Kindermann, M.; Wirtz, S. Innate lymphoid cells in intestinal cancer development. Semin. Immunol. 2019, 41, 101267. [Google Scholar] [CrossRef]
- Kirchberger, S.; Royston, D.J.; Boulard, O.; Thornton, E.; Franchini, F.; Szabady, R.L.; Harrison, O.; Powrie, F. Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. J. Exp. Med. 2013, 210, 917–931. [Google Scholar] [CrossRef]
- Kim, C.H.; Hashimoto-Hill, S.; Kim, M. Migration and tissue tropism of innate lymphoid cells. Trends Immunol. 2016, 37, 68–79. [Google Scholar] [CrossRef] [Green Version]
- Simoni, Y.; Fehlings, M.; Kloverpris, H.N.; McGovern, N.; Koo, S.L.; Loh, C.Y.; Lim, S.; Kurioka, A.; Fergusson, J.R.; Tang, C.L.; et al. Human innate lymphoid cell subsets possess tissue-type based heterogeneity in phenotype and frequency. Immunity 2017, 46, 148–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vries, N.L.; van Unen, V.; Ijsselsteijn, M.E.; Abdelaal, T.; van der Breggen, R.; Farina Sarasqueta, A.; Mahfouz, A.; Peeters, K.; Hollt, T.; Lelieveldt, B.P.F.; et al. High-dimensional cytometric analysis of colorectal cancer reveals novel mediators of antitumour immunity. Gut 2020, 69, 691–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vivier, E.; van de Pavert, S.A.; Cooper, M.D.; Belz, G.T. The evolution of innate lymphoid cells. Nat. Immunol. 2016, 17, 790–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Q.; Belz, G.T. Parallel worlds of the adaptive and innate immune cell networks. Curr. Opin. Immunol. 2019, 58, 53–59. [Google Scholar] [CrossRef]
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate lymphoid cells: 10 years on. Cell 2018, 174, 1054–1066. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.; Seillet, C.; Belz, G.T. Shaping innate lymphoid cell diversity. Front. Immunol. 2017, 8, 1569. [Google Scholar] [CrossRef] [Green Version]
- Cuturi, M.C.; Anegon, I.; Sherman, F.; Loudon, R.; Clark, S.C.; Perussia, B.; Trinchieri, G. Production of hematopoietic colony-stimulating factors by human natural killer cells. J. Exp. Med. 1989, 169, 569–583. [Google Scholar] [CrossRef]
- Weizman, O.E.; Adams, N.M.; Schuster, I.S.; Krishna, C.; Pritykin, Y.; Lau, C.; Degli-Esposti, M.A.; Leslie, C.S.; Sun, J.C.; O’Sullivan, T.E. Ilc1 confer early host protection at initial sites of viral infection. Cell 2017, 171, 795–808.e12. [Google Scholar] [CrossRef]
- Klose, C.S.N.; Flach, M.; Mohle, L.; Rogell, L.; Hoyler, T.; Ebert, K.; Fabiunke, C.; Pfeifer, D.; Sexl, V.; Fonseca-Pereira, D.; et al. Differentiation of type 1 ilcs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell 2014, 157, 340–356. [Google Scholar] [CrossRef] [Green Version]
- Abt, M.C.; Lewis, B.B.; Caballero, S.; Xiong, H.; Carter, R.A.; Susac, B.; Ling, L.; Leiner, I.; Pamer, E.G. Innate immune defenses mediated by two ilc subsets are critical for protection against acute clostridium difficile infection. Cell Host Microb. 2015, 18, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Chiossone, L.; Dumas, P.Y.; Vienne, M.; Vivier, E. Natural killer cells and other innate lymphoid cells in cancer. Nat. Rev. Immunol. 2018, 18, 671–688. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Soto, A.; Gonzalez, S.; Smyth, M.J.; Galluzzi, L. Control of metastasis by nk cells. Cancer Cell 2017, 32, 135–154. [Google Scholar] [CrossRef] [PubMed]
- Cozar, B.; Greppi, M.; Carpentier, S.; Narni-Mancinelli, E.; Chiossone, L.; Vivier, E. Tumor-infiltrating natural killer cells. Cancer Discov. 2020. [Google Scholar]
- Tallerico, R.; Todaro, M.; Di Franco, S.; Maccalli, C.; Garofalo, C.; Sottile, R.; Palmieri, C.; Tirinato, L.; Pangigadde, P.N.; La Rocca, R.; et al. Human nk cells selective targeting of colon cancer-initiating cells: A role for natural cytotoxicity receptors and mhc class i molecules. J. Immunol. 2013, 190, 2381–2390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seillet, C.; Luong, K.; Tellier, J.; Jacquelot, N.; Shen, R.D.; Hickey, P.; Wimmer, V.C.; Whitehead, L.; Rogers, K.; Smyth, G.K.; et al. The neuropeptide vip confers anticipatory mucosal immunity by regulating ilc3 activity. Nat. Immunol. 2020, 21, 168–177. [Google Scholar] [CrossRef]
- Mielke, L.A.; Groom, J.R.; Rankin, L.C.; Seillet, C.; Masson, F.; Putoczki, T.; Belz, G.T. Tcf-1 controls ilc2 and nkp46+rorgammat+ innate lymphocyte differentiation and protection in intestinal inflammation. J. Immunol. 2013, 191, 4383–4391. [Google Scholar] [CrossRef] [Green Version]
- Rankin, L.C.; Girard-Madoux, M.J.; Seillet, C.; Mielke, L.A.; Kerdiles, Y.; Fenis, A.; Wieduwild, E.; Putoczki, T.; Mondot, S.; Lantz, O.; et al. Complementarity and redundancy of il-22-producing innate lymphoid cells. Nat. Immunol. 2016, 17, 179–186. [Google Scholar] [CrossRef]
- Takatori, H.; Kanno, Y.; Watford, W.T.; Tato, C.M.; Weiss, G.; Ivanov, I.; Littman, D.R.; O’Shea, J.J. Lymphoid tissue inducer-like cells are an innate source of il-17 and il-22. J. Exp. Med. 2009, 206, 35–41. [Google Scholar] [CrossRef] [Green Version]
- Buonocore, S.; Ahern, P.P.; Uhlig, H.H.; Ivanov, I.; Littman, D.R.; Maloy, K.J.; Powrie, F. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature 2010, 464, 1371–1375. [Google Scholar] [CrossRef] [Green Version]
- Oppmann, B.; Lesley, R.; Blom, B.; Timans, J.C.; Xu, Y.; Hunte, B.; Vega, F.; Yu, N.; Wang, J.; Singh, K.; et al. Novel p19 protein engages il-12p40 to form a cytokine, il-23, with biological activities similar as well as distinct from il-12. Immunity 2000, 13, 715–725. [Google Scholar] [CrossRef] [Green Version]
- Chan, I.H.; Jain, R.; Tessmer, M.S.; Gorman, D.; Mangadu, R.; Sathe, M.; Vives, F.; Moon, C.; Penaflor, E.; Turner, S.; et al. Interleukin-23 is sufficient to induce rapid de novo gut tumorigenesis, independent of carcinogens, through activation of innate lymphoid cells. Mucosal. Immunol. 2014, 7, 842–856. [Google Scholar] [CrossRef] [PubMed]
- Maoz, A.; Dennis, M.; Greenson, J.K. The crohn’s-like lymphoid reaction to colorectal cancer-tertiary lymphoid structures with immunologic and potentially therapeutic relevance in colorectal cancer. Front. Immunol. 2019, 10, 1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neill, D.R.; Wong, S.H.; Bellosi, A.; Flynn, R.J.; Daly, M.; Langford, T.K.; Bucks, C.; Kane, C.M.; Fallon, P.G.; Pannell, R.; et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 2010, 464, 1367–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mjosberg, J.; Bernink, J.; Golebski, K.; Karrich, J.J.; Peters, C.P.; Blom, B.; te Velde, A.A.; Fokkens, W.J.; van Drunen, C.M.; Spits, H. The transcription factor gata3 is essential for the function of human type 2 innate lymphoid cells. Immunity 2012, 37, 649–659. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.H.; Walker, J.A.; Jolin, H.E.; Drynan, L.F.; Hams, E.; Camelo, A.; Barlow, J.L.; Neill, D.R.; Panova, V.; Koch, U.; et al. Transcription factor roralpha is critical for nuocyte development. Nat. Immunol. 2012, 13, 229–236. [Google Scholar] [CrossRef] [Green Version]
- Oliphant, C.J.; Hwang, Y.Y.; Walker, J.A.; Salimi, M.; Wong, S.H.; Brewer, J.M.; Englezakis, A.; Barlow, J.L.; Hams, E.; Scanlon, S.T.; et al. Mhcii-mediated dialog between group 2 innate lymphoid cells and cd4(+) t cells potentiates type 2 immunity and promotes parasitic helminth expulsion. Immunity 2014, 41, 283–295. [Google Scholar] [CrossRef] [Green Version]
- Smith, K.A.; Loser, S.; Varyani, F.; Harcus, Y.; McSorley, H.J.; McKenzie, A.N.; Maizels, R.M. Concerted il-25r and il-4ralpha signaling drive innate type 2 effector immunity for optimal helminth expulsion. Elife 2018, 7, e38269. [Google Scholar] [CrossRef]
- Monticelli, L.A.; Sonnenberg, G.F.; Abt, M.C.; Alenghat, T.; Ziegler, C.G.; Doering, T.A.; Angelosanto, J.M.; Laidlaw, B.J.; Yang, C.Y.; Sathaliyawala, T.; et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat. Immunol. 2011, 12, 1045–1054. [Google Scholar] [CrossRef]
- Barderas, R.; Bartolome, R.A.; Fernandez-Acenero, M.J.; Torres, S.; Casal, J.I. High expression of il-13 receptor alpha2 in colorectal cancer is associated with invasion, liver metastasis, and poor prognosis. Cancer Res. 2012, 72, 2780–2790. [Google Scholar] [CrossRef] [Green Version]
- Trabanelli, S.; Chevalier, M.F.; Martinez-Usatorre, A.; Gomez-Cadena, A.; Salome, B.; Lecciso, M.; Salvestrini, V.; Verdeil, G.; Racle, J.; Papayannidis, C.; et al. Tumour-derived pgd2 and nkp30-b7h6 engagement drives an immunosuppressive ilc2-mdsc axis. Nat. Commun. 2017, 8, 593. [Google Scholar] [CrossRef] [Green Version]
- Chevalier, M.F.; Trabanelli, S.; Racle, J.; Salome, B.; Cesson, V.; Gharbi, D.; Bohner, P.; Domingos-Pereira, S.; Dartiguenave, F.; Fritschi, A.S.; et al. Ilc2-modulated t cell-to-mdsc balance is associated with bladder cancer recurrence. J. Clin. Invest. 2017, 127, 2916–2929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nussbaum, J.C.; Van Dyken, S.J.; von Moltke, J.; Cheng, L.E.; Mohapatra, A.; Molofsky, A.B.; Thornton, E.E.; Krummel, M.F.; Chawla, A.; Liang, H.E.; et al. Type 2 innate lymphoid cells control eosinophil homeostasis. Nature 2013, 502, 245–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, Y.; Hayashi, Y.; Sugama, Y.; Miura, Y.; Kasahara, T.; Kitamura, S.; Torisu, M.; Mita, S.; Tominaga, A.; Takatsu, K. Highly purified murine interleukin 5 (il-5) stimulates eosinophil function and prolongs in vitro survival. Il-5 as an eosinophil chemotactic factor. J. Exp. Med. 1988, 167, 1737–1742. [Google Scholar] [CrossRef] [PubMed]
- Reichman, H.; Itan, M.; Rozenberg, P.; Yarmolovski, T.; Brazowski, E.; Varol, C.; Gluck, N.; Shapira, S.; Arber, N.; Qimron, U.; et al. Activated eosinophils exert antitumorigenic activities in colorectal cancer. Cancer Immunol. Res. 2019, 7, 388–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, I.C.; Artola-Boran, M.; Gurtner, A.; Bertram, K.; Bauer, M.; Frangez, Z.; Becher, B.; Kopf, M.; Yousefi, S.; Simon, H.U.; et al. The gm-csf-irf5 signaling axis in eosinophils promotes antitumor immunity through activation of type 1 t cell responses. J. Exp. Med. 2020, 217, e20190706. [Google Scholar] [CrossRef]
- Grisaru-Tal, S.; Itan, M.; Klion, A.D.; Munitz, A. A new dawn for eosinophils in the tumour microenvironment. Nat. Rev. Cancer 2020, 20, 594–6007. [Google Scholar] [CrossRef]
- Harbaum, L.; Pollheimer, M.J.; Kornprat, P.; Lindtner, R.A.; Bokemeyer, C.; Langner, C. Peritumoral eosinophils predict recurrence in colorectal cancer. Mod. Pathol. 2015, 28, 403–413. [Google Scholar] [CrossRef] [Green Version]
- Efremova, M.; Rieder, D.; Klepsch, V.; Charoentong, P.; Finotello, F.; Hackl, H.; Hermann-Kleiter, N.; Lower, M.; Baier, G.; Krogsdam, A.; et al. Targeting immune checkpoints potentiates immunoediting and changes the dynamics of tumor evolution. Nat. Commun. 2018, 9, 32. [Google Scholar] [CrossRef]
- Christiansen, A.J.; West, A.; Banks, K.M.; Haynes, N.M.; Teng, M.W.; Smyth, M.J.; Johnstone, R.W. Eradication of solid tumors using histone deacetylase inhibitors combined with immune-stimulating antibodies. Proc. Natl. Acad. Sci. USA 2011, 108, 4141–4146. [Google Scholar] [CrossRef] [Green Version]
- Ghaedi, M.; Shen, Z.Y.; Orangi, M.; Martinez-Gonzalez, I.; Wei, L.; Lu, X.; Das, A.; Heravi-Moussavi, A.; Marra, M.A.; Bhandoola, A.; et al. Single-cell analysis of roralpha tracer mouse lung reveals ilc progenitors and effector ilc2 subsets. J. Exp. Med. 2020, 217, e20182293. [Google Scholar] [CrossRef]
- Halim, T.Y.; MacLaren, A.; Romanish, M.T.; Gold, M.J.; McNagny, K.M.; Takei, F. Retinoic-acid-receptor-related orphan nuclear receptor alpha is required for natural helper cell development and allergic inflammation. Immunity 2012, 37, 463–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas, N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Eeghen, E.E.; Bakker, S.D.; van Bochove, A.; Loffeld, R.J. Impact of age and comorbidity on survival in colorectal cancer. J. Gastrointest. Oncol. 2015, 6, 605–612. [Google Scholar]
- Wang, C.B.; Shahjehan, F.; Merchea, A.; Li, Z.; Bekaii-Saab, T.S.; Grothey, A.; Colibaseanu, D.T.; Kasi, P.M. Impact of tumor location and variables associated with overall survival in patients with colorectal cancer: A mayo clinic colon and rectal cancer registry study. Front. Oncol. 2019, 9, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melo-Gonzalez, F.; Kammoun, H.; Evren, E.; Dutton, E.E.; Papadopoulou, M.; Bradford, B.M.; Tanes, C.; Fardus-Reid, F.; Swann, J.R.; Bittinger, K.; et al. Antigen-presenting ilc3 regulate t cell-dependent iga responses to colonic mucosal bacteria. J. Exp. Med. 2019, 216, 728–742. [Google Scholar] [CrossRef]
- Cho, H.S.; Reboldi, A.; Hall, J.A.; Berg, L.J. The tec kinase itk is essential for ilc2 survival and epithelial integrity in the intestine. Nat. Commun. 2019, 10, 784. [Google Scholar] [CrossRef]
- Maywald, R.L.; Doerner, S.K.; Pastorelli, L.; De Salvo, C.; Benton, S.M.; Dawson, E.P.; Lanza, D.G.; Berger, N.A.; Markowitz, S.D.; Lenz, H.J.; et al. Il-33 activates tumor stroma to promote intestinal polyposis. Proc. Natl. Acad. Sci. USA 2015, 112, E2487–E2496. [Google Scholar] [CrossRef] [Green Version]
- Pastille, E.; Wasmer, M.H.; Adamczyk, A.; Vu, V.P.; Mager, L.F.; Phuong, N.N.T.; Palmieri, V.; Simillion, C.; Hansen, W.; Kasper, S.; et al. The il-33/st2 pathway shapes the regulatory t cell phenotype to promote intestinal cancer. Mucosal. Immunol. 2019, 12, 990–1003. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, C.; Mahmoud, A.; Keane, J.; Murphy, C.; White, D.; Carey, S.; O’Riordain, M.; Bennett, M.W.; Brint, E.; Houston, A. An antitumorigenic role for the il-33 receptor, st2l, in colon cancer. Br. J. Cancer 2016, 114, 37–43. [Google Scholar] [CrossRef] [Green Version]
- Andre, T.; Shiu, K.K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in microsatellite-instability-high advanced colorectal cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef]
- Mlecnik, B.; Bifulco, C.; Bindea, G.; Marliot, F.; Lugli, A.; Lee, J.J.; Zlobec, I.; Rau, T.T.; Berger, M.D.; Nagtegaal, I.D.; et al. Multicenter international society for immunotherapy of cancer study of the consensus immunoscore for the prediction of survival and response to chemotherapy in stage iii colon cancer. J. Clin. Oncol. 2020, 38, 3638–3651. [Google Scholar] [CrossRef] [PubMed]
- Pages, F.; Andre, T.; Taieb, J.; Vernerey, D.; Henriques, J.; Borg, C.; Marliot, F.; Ben Jannet, R.; Louvet, C.; Mineur, L.; et al. Prognostic and predictive value of the immunoscore in stage iii colon cancer patients treated with oxaliplatin in the prospective idea france prodige-gercor cohort study. Ann. Oncol. 2020, 31, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Garcia, S.; DiSanto, J.; Stockinger, B. Following the development of a cd4 t cell response in vivo: From activation to memory formation. Immunity 1999, 11, 163–171. [Google Scholar] [CrossRef] [Green Version]
- Schlenner, S.M.; Madan, V.; Busch, K.; Tietz, A.; Laufle, C.; Costa, C.; Blum, C.; Fehling, H.J.; Rodewald, H.R. Fate mapping reveals separate origins of t cells and myeloid lineages in the thymus. Immunity 2010, 32, 426–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rankin, L.; Groom, J.; Mielke, L.A.; Seillet, C.; Belz, G.T. Diversity, function, and transcriptional regulation of gut innate lymphocytes. Front. Immunol. 2013, 4, 22. [Google Scholar] [CrossRef] [Green Version]
- Mielke, L.; Preaudet, A.; Belz, G.; Putoczki, T. Confocal laser endomicroscopy to monitor the colonic mucosa of mice. J. Immunol. Methods 2015, 421, 81–88. [Google Scholar] [CrossRef] [Green Version]
- Gury-BenAri, M.; Thaiss, C.A.; Serafini, N.; Winter, D.R.; Giladi, A.; Lara-Astiaso, D.; Levy, M.; Salame, T.M.; Weiner, A.; David, E.; et al. The spectrum and regulatory landscape of intestinal innate lymphoid cells are shaped by the microbiome. Cell 2016, 166, 1231–1246.e13. [Google Scholar] [CrossRef]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of rna-seq data. Genome Biol 2010, 11, R25. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. Edger: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Bjorklund, A.K.; Forkel, M.; Picelli, S.; Konya, V.; Theorell, J.; Friberg, D.; Sandberg, R.; Mjosberg, J. The heterogeneity of human cd127(+) innate lymphoid cells revealed by single-cell rna sequencing. Nat. Immunol. 2016, 17, 451–460. [Google Scholar] [CrossRef]
- Chen, T.; Guestrin, C. Xgboost: A scalable tree boosting system. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; pp. 785–794. [Google Scholar]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. Tcgabiolinks: An r/bioconductor package for integrative analysis of tcga data. Nucleic. Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef] [PubMed]
- Terry, M.; Therneau, P.M.G. Modeling Survival Data: Extending the Cox Model; Springer: New York, NY, USA, 2000. [Google Scholar]
- Kassambara, A.; Kosinski, M.; Biecek, P.; Fabian, S. Survminer: Drawing Survival Curves Using ‘ggplot2’; R Package Version 0.31; R foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- Enot, D.P.; Vacchelli, E.; Jacquelot, N.; Zitvogel, L.; Kroemer, G. Tumgrowth: An open-access web tool for the statistical analysis of tumor growth curves. Oncoimmunology 2018, 7, e1462431. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Q.; Jacquelot, N.; Preaudet, A.; Hediyeh-zadeh, S.; Souza-Fonseca-Guimaraes, F.; McKenzie, A.N.J.; Hansbro, P.M.; Davis, M.J.; Mielke, L.A.; Putoczki, T.L.; et al. Type 2 Innate Lymphoid Cells Protect against Colorectal Cancer Progression and Predict Improved Patient Survival. Cancers 2021, 13, 559. https://doi.org/10.3390/cancers13030559
Huang Q, Jacquelot N, Preaudet A, Hediyeh-zadeh S, Souza-Fonseca-Guimaraes F, McKenzie ANJ, Hansbro PM, Davis MJ, Mielke LA, Putoczki TL, et al. Type 2 Innate Lymphoid Cells Protect against Colorectal Cancer Progression and Predict Improved Patient Survival. Cancers. 2021; 13(3):559. https://doi.org/10.3390/cancers13030559
Chicago/Turabian StyleHuang, Qiutong, Nicolas Jacquelot, Adele Preaudet, Soroor Hediyeh-zadeh, Fernando Souza-Fonseca-Guimaraes, Andrew N. J. McKenzie, Philip M. Hansbro, Melissa J. Davis, Lisa A. Mielke, Tracy L. Putoczki, and et al. 2021. "Type 2 Innate Lymphoid Cells Protect against Colorectal Cancer Progression and Predict Improved Patient Survival" Cancers 13, no. 3: 559. https://doi.org/10.3390/cancers13030559