
EGFRvIII Promotes Cell Survival during Endoplasmic Reticulum Stress through a Reticulocalbin 1-Dependent Mechanism

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
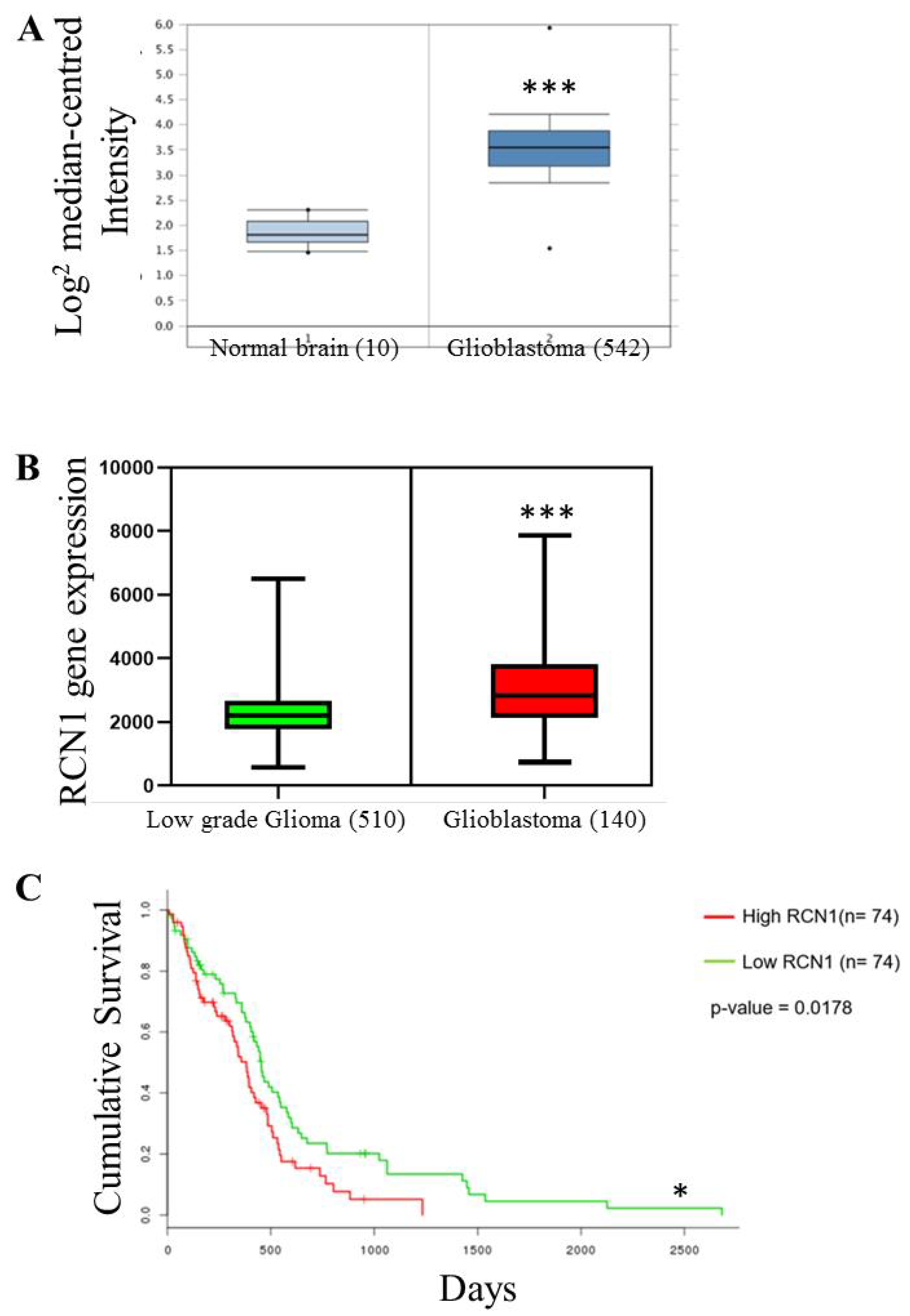
2.1. RCN1 Expression Correlates with Glioma Grade and Glioblastoma Patient Survival
2.2. RCN1 Is Highly Expressed in EGFRvIII Positive Glioblastoma Cells
2.3. RCN1 Expression Correlates with Increased Survival after the Initiation of ER Stress
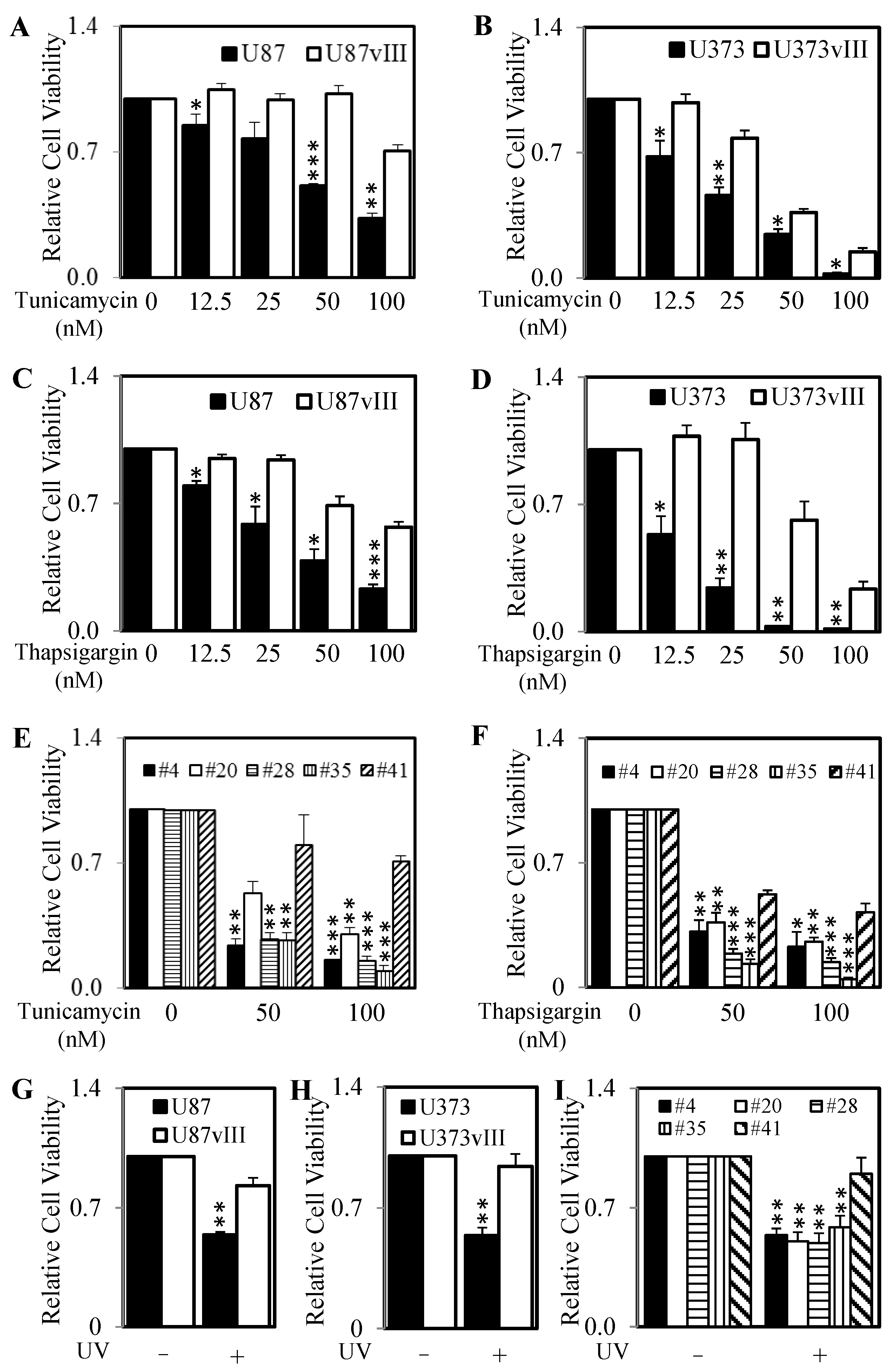
2.4. Knockdown of RCN1 Significantly Decreases Cell Survival and RCN1 Over-Expression Significantly Increases Cell Survival Following the Initiation of ER Stress
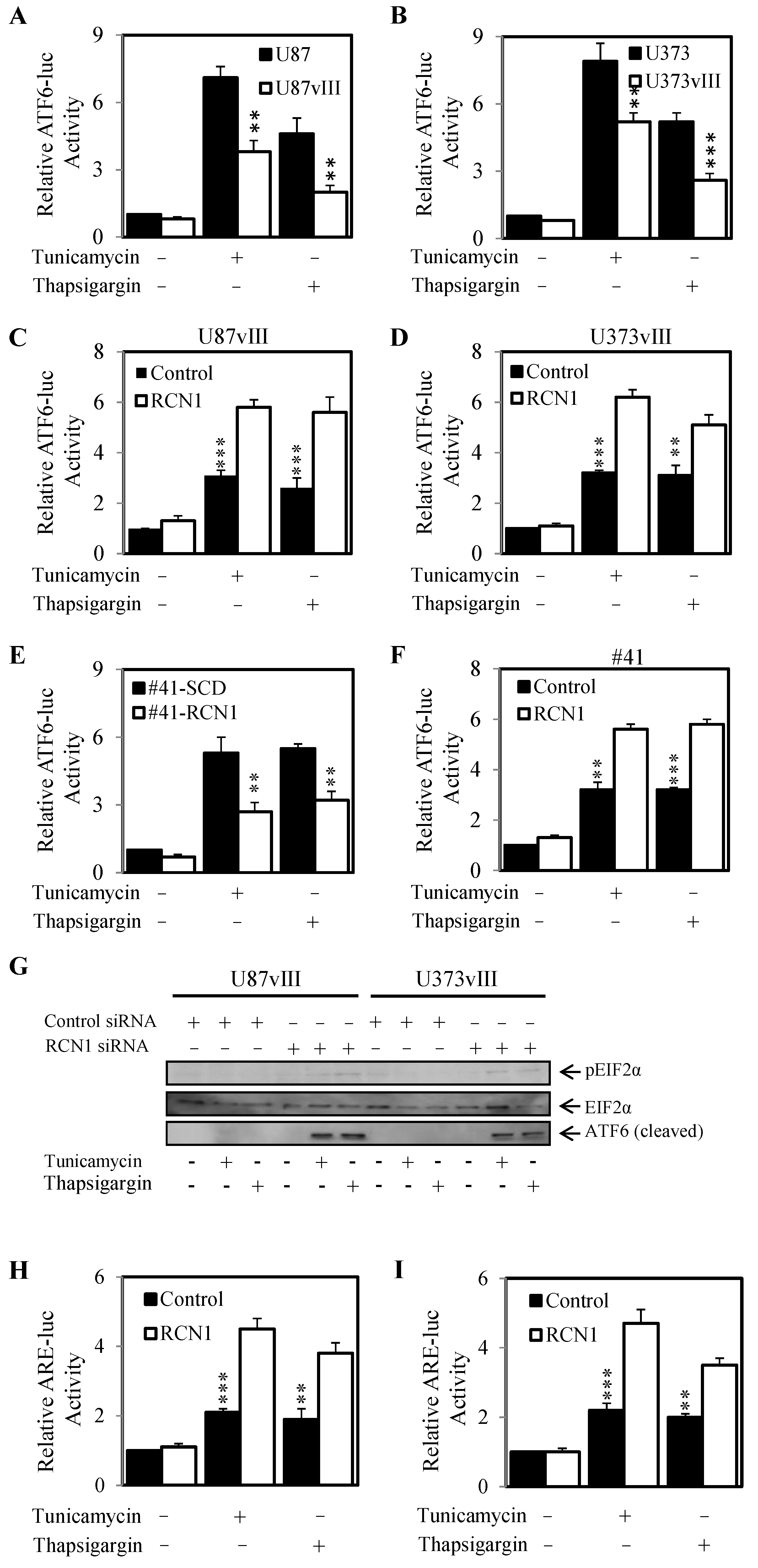
2.5. RCN1 Expression Reduces ATF6 Activity When Challenged with Tunicamycin and Thapsigargin
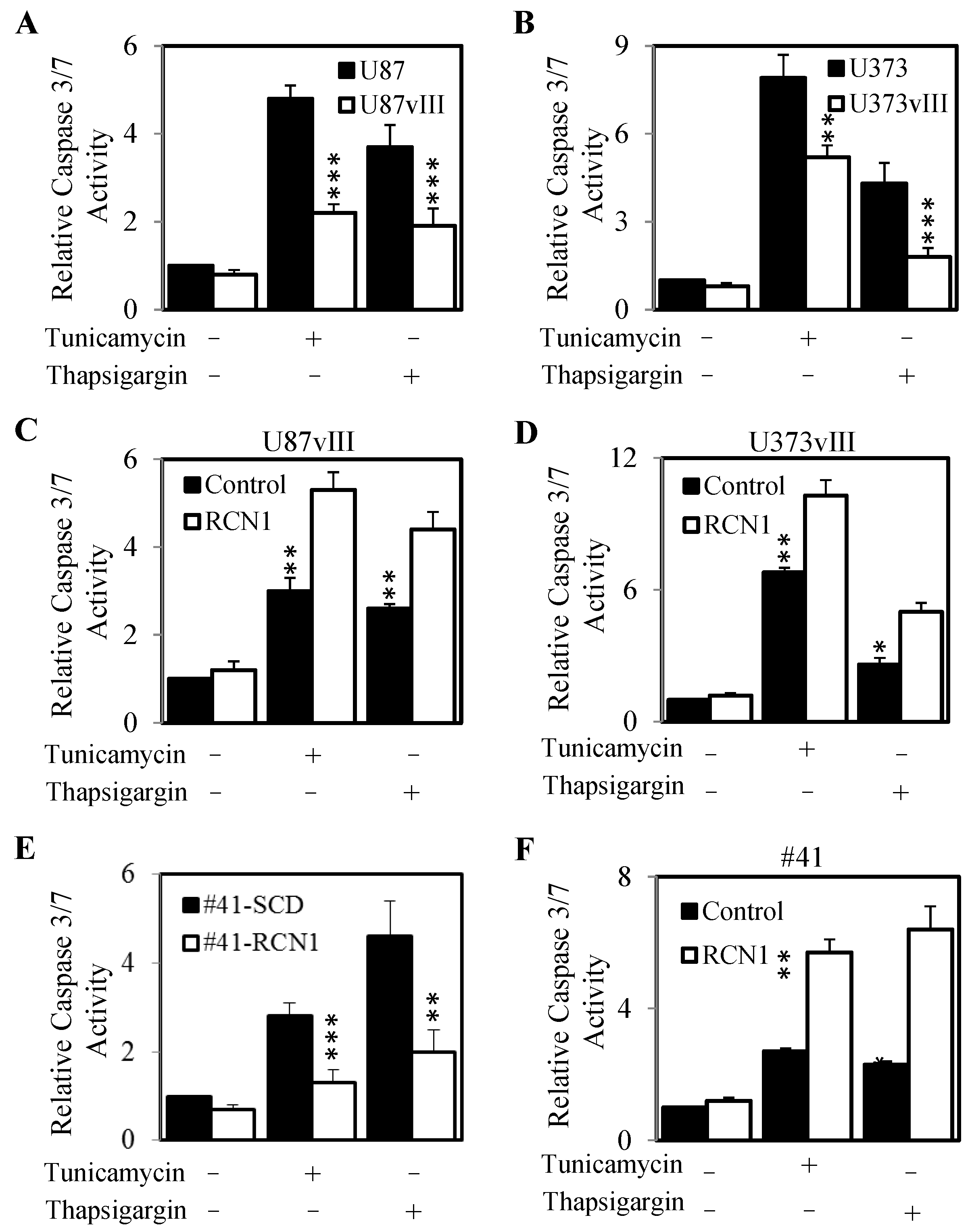
2.6. RCN1 Expression Protects Cells from ER Stress-Mediated Apoptosis
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Cell Culture
4.3. Generation of Cells with Varying Levels of EGFR and EGFRvIII
4.4. Generation of Cells with RCN1 Over-Expression and siRNA Transfection
4.5. Cell Viability Assays
4.6. Western Blotting and Immunoprecipitation
4.7. RNA Extraction and RT-PCR
4.8. ATF6 and ARE-Luc Luciferase Assay
4.9. Caspase3/7 Assay
4.10. OncoLnc (TCGA)
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Davis, F.G.; Freels, S.; Grutsch, J.; Barlas, S.; Brem, S. Survival rates in patients with primary malignant brain tumors stratified by patient age and tumor histological type: An analysis based on Surveillance, Epidemiology, and End Results (SEER) data, 1973–1991. J. Neurosurg. 1998, 88, 1–10. [Google Scholar] [CrossRef]
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, A.J.; Tsui, V.; Stylli, S.S.; Nguyen, H.P.T.; Morokoff, A.P.; Kaye, A.H.; Luwor, R.B. The role of interleukin-6-STAT3 signalling in glioblastoma. Oncol. Lett. 2018, 16, 4095–4104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penaranda Fajardo, N.M.; Meijer, C.; Kruyt, F.A. The endoplasmic reticulum stress/unfolded protein response in gliomagenesis, tumor progression and as a therapeutic target in glioblastoma. Biochem. Pharmacol. 2016, 118, 1–8. [Google Scholar] [CrossRef]
- Xu, S.; Xu, Y.; Chen, L.; Fang, Q.; Song, S.; Chen, J.; Teng, J. RCN1 suppresses ER stress-induced apoptosis via calcium homeostasis and PERK-CHOP signaling. Oncogenesis 2017, 6, e304. [Google Scholar] [CrossRef] [Green Version]
- Sansalone, L.; Veliz, E.A.; Myrthil, N.G.; Stathias, V.; Walters, W.; Torrens, I.I.; Schurer, S.C.; Vanni, S.; Leblanc, R.M.; Graham, R.M. Novel Curcumin Inspired Bis-Chalcone Promotes Endoplasmic Reticulum Stress and Glioblastoma Neurosphere Cell Death. Cancers 2019, 11, 357. [Google Scholar] [CrossRef] [Green Version]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef]
- Truscott, K.N.; Bezawork-Geleta, A.; Dougan, D.A. Unfolded protein responses in bacteria and mitochondria: A central role for the ClpXP machine. IUBMB Life 2011, 63, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef]
- Wang, M.; Kaufman, R.J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 2014, 14, 581–597. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Dave, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Madden, E.; Logue, S.E.; Healy, S.J.; Manie, S.; Samali, A. The role of the unfolded protein response in cancer progression: From oncogenesis to chemoresistance. Biol. Cell 2019, 111, 1–17. [Google Scholar] [CrossRef]
- Iurlaro, R.; Munoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef] [Green Version]
- Masud, A.; Mohapatra, A.; Lakhani, S.A.; Ferrandino, A.; Hakem, R.; Flavell, R.A. Endoplasmic reticulum stress-induced death of mouse embryonic fibroblasts requires the intrinsic pathway of apoptosis. J. Biol. Chem. 2007, 282, 14132–14139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCullough, K.D.; Martindale, J.L.; Klotz, L.O.; Aw, T.Y.; Holbrook, N.J. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell Biol. 2001, 21, 1249–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meares, G.P.; Zmijewska, A.A.; Jope, R.S. HSP105 interacts with GRP78 and GSK3 and promotes ER stress-induced caspase-3 activation. Cell Signal. 2008, 20, 347–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozawa, M.; Muramatsu, T. Reticulocalbin, a novel endoplasmic reticulum resident Ca(2+)-binding protein with multiple EF-hand motifs and a carboxyl-terminal HDEL sequence. J. Biol. Chem. 1993, 268, 699–705. [Google Scholar] [CrossRef]
- Giribaldi, G.; Barbero, G.; Mandili, G.; Daniele, L.; Khadjavi, A.; Notarpietro, A.; Ulliers, D.; Prato, M.; Minero, V.G.; Battaglia, A.; et al. Proteomic identification of Reticulocalbin 1 as potential tumor marker in renal cell carcinoma. J. Proteom. 2013, 91, 385–392. [Google Scholar] [CrossRef]
- Liu, Z.; Brattain, M.G.; Appert, H. Differential display of reticulocalbin in the highly invasive cell line, MDA-MB-435, versus the poorly invasive cell line, MCF-7. Biochem. Biophys. Res. Commun. 1997, 231, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Nimmrich, I.; Erdmann, S.; Melchers, U.; Finke, U.; Hentsch, S.; Moyer, M.P.; Hoffmann, I.; Muller, O. Seven genes that are differentially transcribed in colorectal tumor cell lines. Cancer Lett. 2000, 160, 37–43. [Google Scholar] [CrossRef]
- Ekstrand, A.J.; James, C.D.; Cavenee, W.K.; Seliger, B.; Pettersson, R.F.; Collins, V.P. Genes for epidermal growth factor receptor, transforming growth factor alpha, and epidermal growth factor and their expression in human gliomas in vivo. Cancer Res. 1991, 51, 2164–2172. [Google Scholar]
- Libermann, T.A.; Razon, N.; Bartal, A.D.; Yarden, Y.; Schlessinger, J.; Soreq, H. Expression of epidermal growth factor receptors in human brain tumors. Cancer Res. 1984, 44, 753–760. [Google Scholar]
- Wong, A.J.; Ruppert, J.M.; Bigner, S.H.; Grzeschik, C.H.; Humphrey, P.A.; Bigner, D.S.; Vogelstein, B. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc. Natl. Acad. Sci. USA 1992, 89, 2965–2969. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, R.A.; Luwor, R.B.; Burgess, A.W. Epidermal growth factor receptor: Structure-function informing the design of anticancer therapeutics. Exp. Cell Res. 2018, 371, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.K.; Kaye, A.H.; Luwor, R.B. The EGFRvIII variant in glioblastoma multiforme. J. Clin. Neurosci. 2009, 16, 748–754. [Google Scholar] [CrossRef]
- Luwor, R.B.; Zhu, H.J.; Walker, F.; Vitali, A.A.; Perera, R.M.; Burgess, A.W.; Scott, A.M.; Johns, T.G. The tumor-specific de2-7 epidermal growth factor receptor (EGFR) promotes cells survival and heterodimerizes with the wild-type EGFR. Oncogene 2004, 23, 6095–6104. [Google Scholar] [CrossRef] [Green Version]
- Garcia de Palazzo, I.E.; Adams, G.P.; Sundareshan, P.; Wong, A.J.; Testa, J.R.; Bigner, D.D.; Weiner, L.M. Expression of mutated epidermal growth factor receptor by non-small cell lung carcinomas. Cancer Res. 1993, 53, 3217–3220. [Google Scholar]
- Moscatello, D.K.; Holgado-Madruga, M.; Godwin, A.K.; Ramirez, G.; Gunn, G.; Zoltick, P.W.; Biegel, J.A.; Hayes, R.L.; Wong, A.J. Frequent expression of a mutant epidermal growth factor receptor in multiple human tumors. Cancer Res. 1995, 55, 5536–5539. [Google Scholar] [PubMed]
- Ekstrand, A.J.; Sugawa, N.; James, C.D.; Collins, V.P. Amplified and rearranged epidermal growth factor receptor genes in human glioblastomas reveal deletions of sequences encoding portions of the N- and/or C-terminal tails. Proc. Natl. Acad. Sci. USA 1992, 89, 4309–4313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugawa, N.; Ekstrand, A.J.; James, C.D.; Collins, V.P. Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proc. Natl. Acad. Sci. USA 1990, 87, 8602–8606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Rourke, D.M.; Nute, E.J.; Davis, J.G.; Wu, C.; Lee, A.; Murali, R.; Zhang, H.T.; Qian, X.; Kao, C.C.; Greene, M.I. Inhibition of a naturally occurring EGFR oncoprotein by the p185neu ectodomain: Implications for subdomain contributions to receptor assembly. Oncogene 1998, 16, 1197–1207. [Google Scholar] [CrossRef] [Green Version]
- Voldborg, B.R.; Damstrup, L.; Spang-Thomsen, M.; Poulsen, H.S. Epidermal growth factor receptor (EGFR) and EGFR mutations, function and possible role in clinical trials. Ann. Oncol. 1997, 8, 1197–1206. [Google Scholar] [CrossRef]
- Nagane, M.; Coufal, F.; Lin, H.; Bogler, O.; Cavenee, W.K.; Huang, H.J. A common mutant epidermal growth factor receptor confers enhanced tumorigenicity on human glioblastoma cells by increasing proliferation and reducing apoptosis. Cancer Res. 1996, 56, 5079–5086. [Google Scholar]
- Lammering, G.; Valerie, K.; Lin, P.S.; Hewit, T.H.; Schmidt-Ullrich, R.K. Radiation-induced activation of a common variant of EGFR confers enhanced radioresistance. Radiother. Oncol. 2004, 72, 267–273. [Google Scholar] [CrossRef]
- Nagane, M.; Levitzki, A.; Gazit, A.; Cavenee, W.K.; Huang, H.J. Drug resistance of human glioblastoma cells conferred by a tumor-specific mutant epidermal growth factor receptor through modulation of Bcl-XL and caspase-3-like proteases. Proc. Natl. Acad. Sci. USA 1998, 95, 5724–5729. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Shao, W.; Huang, H.; Feng, X.; Yao, S.; Ke, H. Overexpression of RCN1 correlates with poor prognosis and progression in non-small cell lung cancer. Hum. Pathol. 2019, 83, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, N.; Wang, D.; Zhu, D.; Yuan, Q.; Zhang, X.; Qian, L.; Niu, H.; Lu, Y.; Ren, G.; et al. Downregulation of reticulocalbin-1 differentially facilitates apoptosis and necroptosis in human prostate cancer cells. Cancer Sci. 2018, 109, 1147–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.R.; Zeng, R.; Shao, X.X.; Wang, N.; Xu, Y.H.; Xia, Q.C. Identification of differentially expressed proteins between human hepatoma and normal liver cell lines by two-dimensional electrophoresis and liquid chromatography-ion trap mass spectrometry. Electrophoresis 2000, 21, 3058–3068. [Google Scholar] [CrossRef]
- Huang, Z.H.; Qiao, J.; Feng, Y.Y.; Qiu, M.T.; Cheng, T.; Wang, J.; Zheng, C.F.; Lv, Z.Q.; Wang, C.H. Reticulocalbin-1 knockdown increases the sensitivity of cells to Adriamycin in nasopharyngeal carcinoma and promotes endoplasmic reticulum stress-induced cell apoptosis. Cell Cycle 2020, 19, 1576–1589. [Google Scholar] [CrossRef]
- Coker, K.J.; Staros, J.V.; Guyer, C.A. A kinase-negative epidermal growth factor receptor that retains the capacity to stimulate DNA synthesis. Proc. Natl. Acad. Sci. USA 1994, 91, 6967–6971. [Google Scholar] [CrossRef] [Green Version]
- Ewald, J.A.; Wilkinson, J.C.; Guyer, C.A.; Staros, J.V. Ligand- and kinase activity-independent cell survival mediated by the epidermal growth factor receptor expressed in 32D cells. Exp. Cell Res. 2003, 282, 121–131. [Google Scholar] [CrossRef]
- Weihua, Z.; Tsan, R.; Huang, W.C.; Wu, Q.; Chiu, C.H.; Fidler, I.J.; Hung, M.C. Survival of cancer cells is maintained by EGFR independent of its kinase activity. Cancer Cell 2008, 13, 385–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, X.; Zhu, H.; Ali-Osman, F.; Lo, H.W. EGFR and EGFRvIII undergo stress- and EGFR kinase inhibitor-induced mitochondrial translocalization: A potential mechanism of EGFR-driven antagonism of apoptosis. Mol. Cancer 2011, 10, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cvrljevic, A.N.; Akhavan, D.; Wu, M.; Martinello, P.; Furnari, F.B.; Johnston, A.J.; Guo, D.; Pike, L.; Cavenee, W.K.; Scott, A.M.; et al. Activation of Src induces mitochondrial localisation of de2-7EGFR (EGFRvIII) in glioma cells: Implications for glucose metabolism. J. Cell Sci. 2011, 124, 2938–2950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gururaj, A.E.; Gibson, L.; Panchabhai, S.; Bai, M.; Manyam, G.; Lu, Y.; Latha, K.; Rojas, M.L.; Hwang, Y.; Liang, S.; et al. Access to the nucleus and functional association with c-Myc is required for the full oncogenic potential of DeltaEGFR/EGFRvIII. J. Biol. Chem. 2013, 288, 3428–3438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, H.W.; Cao, X.; Zhu, H.; Ali-Osman, F. Cyclooxygenase-2 is a novel transcriptional target of the nuclear EGFR-STAT3 and EGFRvIII-STAT3 signaling axes. Mol. Cancer Res. 2010, 8, 232–245. [Google Scholar] [CrossRef] [Green Version]
- Oldrini, B.; Hsieh, W.Y.; Erdjument-Bromage, H.; Codega, P.; Carro, M.S.; Curiel-Garcia, A.; Campos, C.; Pourmaleki, M.; Grommes, C.; Vivanco, I.; et al. EGFR feedback-inhibition by Ran-binding protein 6 is disrupted in cancer. Nat. Commun. 2017, 8, 2035. [Google Scholar] [CrossRef] [Green Version]
- Mandic, A.; Hansson, J.; Linder, S.; Shoshan, M.C. Cisplatin induces endoplasmic reticulum stress and nucleus-independent apoptotic signaling. J. Biol. Chem. 2003, 278, 9100–9106. [Google Scholar] [CrossRef] [Green Version]
- Martins, I.; Kepp, O.; Schlemmer, F.; Adjemian, S.; Tailler, M.; Shen, S.; Michaud, M.; Menger, L.; Gdoura, A.; Tajeddine, N.; et al. Restoration of the immunogenicity of cisplatin-induced cancer cell death by endoplasmic reticulum stress. Oncogene 2011, 30, 1147–1158. [Google Scholar] [CrossRef] [Green Version]
- Xie, H.; Yang, J.; Xing, W.; Dong, Y.; Ren, H. Nuclear EGFRvIII resists hypoxic microenvironment induced apoptosis via recruiting ERK1/2 nuclear translocation. Biochem. Biophys. Res. Commun. 2016, 470, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Flavahan, W.A.; Wu, Q.; Hitomi, M.; Rahim, N.; Kim, Y.; Sloan, A.E.; Weil, R.J.; Nakano, I.; Sarkaria, J.N.; Stringer, B.W.; et al. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat. Neurosci. 2013, 16, 1373–1382. [Google Scholar] [CrossRef]
- Noack, J.; Choi, J.; Richter, K.; Kopp-Schneider, A.; Regnier-Vigouroux, A. A sphingosine kinase inhibitor combined with temozolomide induces glioblastoma cell death through accumulation of dihydrosphingosine and dihydroceramide, endoplasmic reticulum stress and autophagy. Cell Death Dis. 2014, 5, e1425. [Google Scholar] [CrossRef] [Green Version]
- Dadey, D.Y.A.; Kapoor, V.; Khudanyan, A.; Thotala, D.; Hallahan, D.E. PERK Regulates Glioblastoma Sensitivity to ER Stress Although Promoting Radiation Resistance. Mol. Cancer Res. 2018, 16, 1447–1453. [Google Scholar] [CrossRef] [Green Version]
- Yoshimoto, K.; Dang, J.; Zhu, S.; Nathanson, D.; Huang, T.; Dumont, R.; Seligson, D.B.; Yong, W.H.; Xiong, Z.; Rao, N.; et al. Development of a real-time RT-PCR assay for detecting EGFRvIII in glioblastoma samples. Clin. Cancer Res. 2008, 14, 488–493. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomez, J.; Areeb, Z.; Stuart, S.F.; Nguyen, H.P.T.; Paradiso, L.; Zulkifli, A.; Madan, S.; Rajagopal, V.; Montgomery, M.K.; Gan, H.K.; et al. EGFRvIII Promotes Cell Survival during Endoplasmic Reticulum Stress through a Reticulocalbin 1-Dependent Mechanism. Cancers 2021, 13, 1198. https://doi.org/10.3390/cancers13061198
Gomez J, Areeb Z, Stuart SF, Nguyen HPT, Paradiso L, Zulkifli A, Madan S, Rajagopal V, Montgomery MK, Gan HK, et al. EGFRvIII Promotes Cell Survival during Endoplasmic Reticulum Stress through a Reticulocalbin 1-Dependent Mechanism. Cancers. 2021; 13(6):1198. https://doi.org/10.3390/cancers13061198
Chicago/Turabian StyleGomez, Juliana, Zammam Areeb, Sarah F. Stuart, Hong P. T. Nguyen, Lucia Paradiso, Ahmad Zulkifli, Sonakshi Madan, Vijay Rajagopal, Magdalene K. Montgomery, Hui K. Gan, and et al. 2021. "EGFRvIII Promotes Cell Survival during Endoplasmic Reticulum Stress through a Reticulocalbin 1-Dependent Mechanism" Cancers 13, no. 6: 1198. https://doi.org/10.3390/cancers13061198