
DNA Methylation Signatures and the Contribution of Age-Associated Methylomic Drift to Carcinogenesis in Early-Onset Colorectal Cancer

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Participants
2.2. DNA Extraction from FFPE Specimens
2.3. DNA Methylation Array Processing and Bioinformatic Analyses
2.4. Statistical Analysis
2.5. Publicly Available DNA Methylation Dataset of Normal Colonic Mucosa and CRCs
2.6. DNA Methylation-Based Age Estimation
3. Results
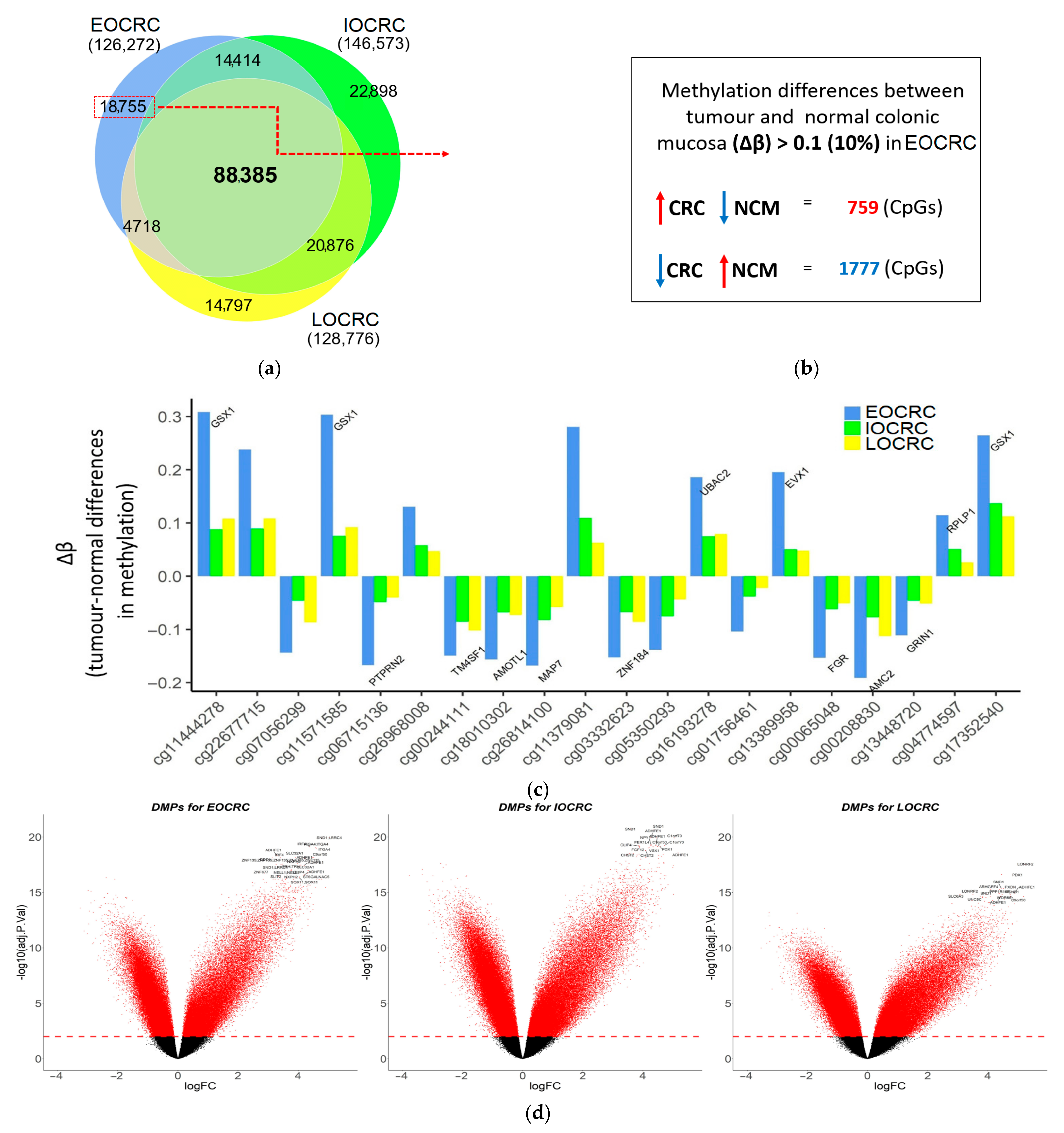
3.1. Differences in Genome-Wide DNA Methylation between Normal Mucosa and CRCs by Age-of-Diagnosis
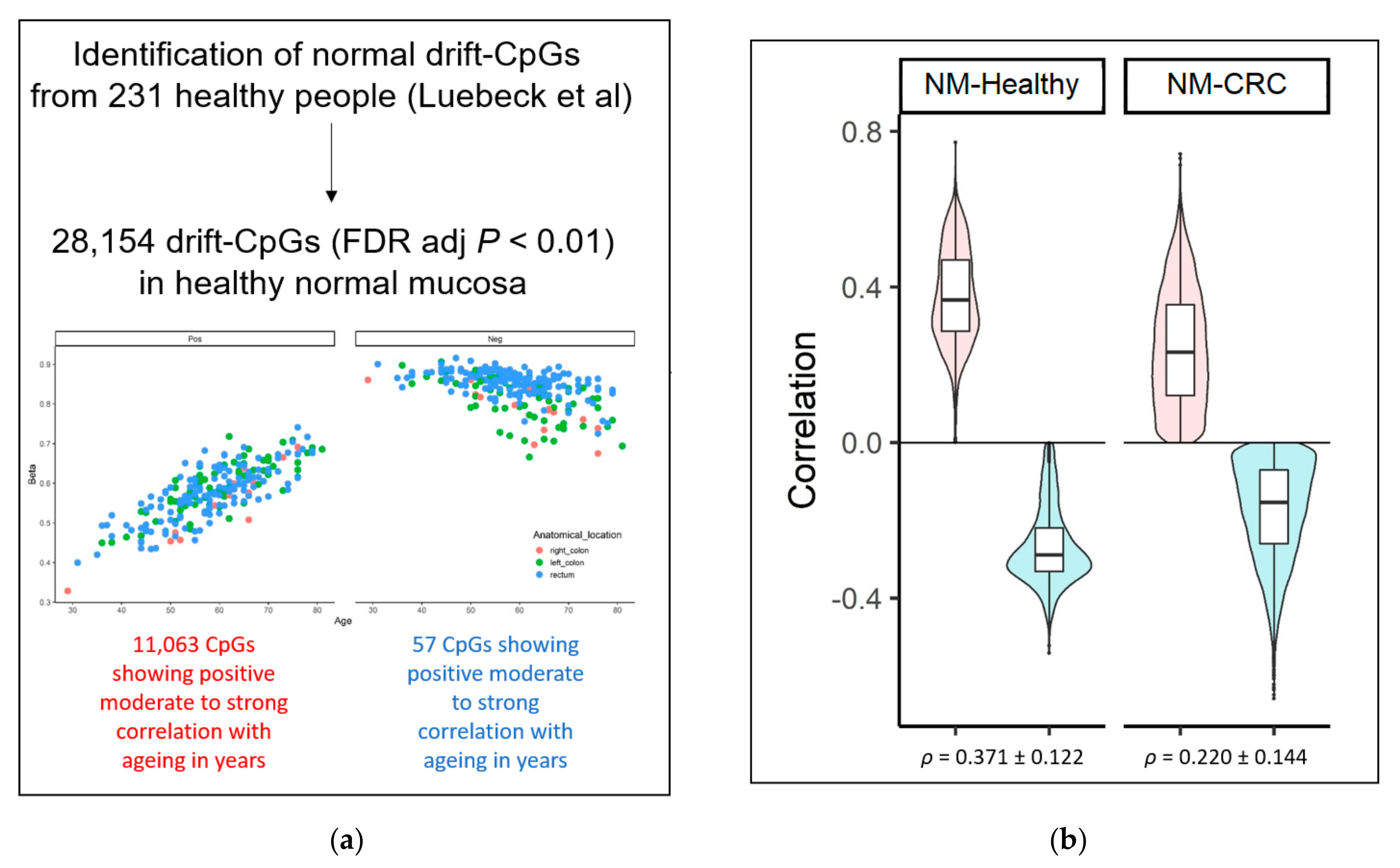
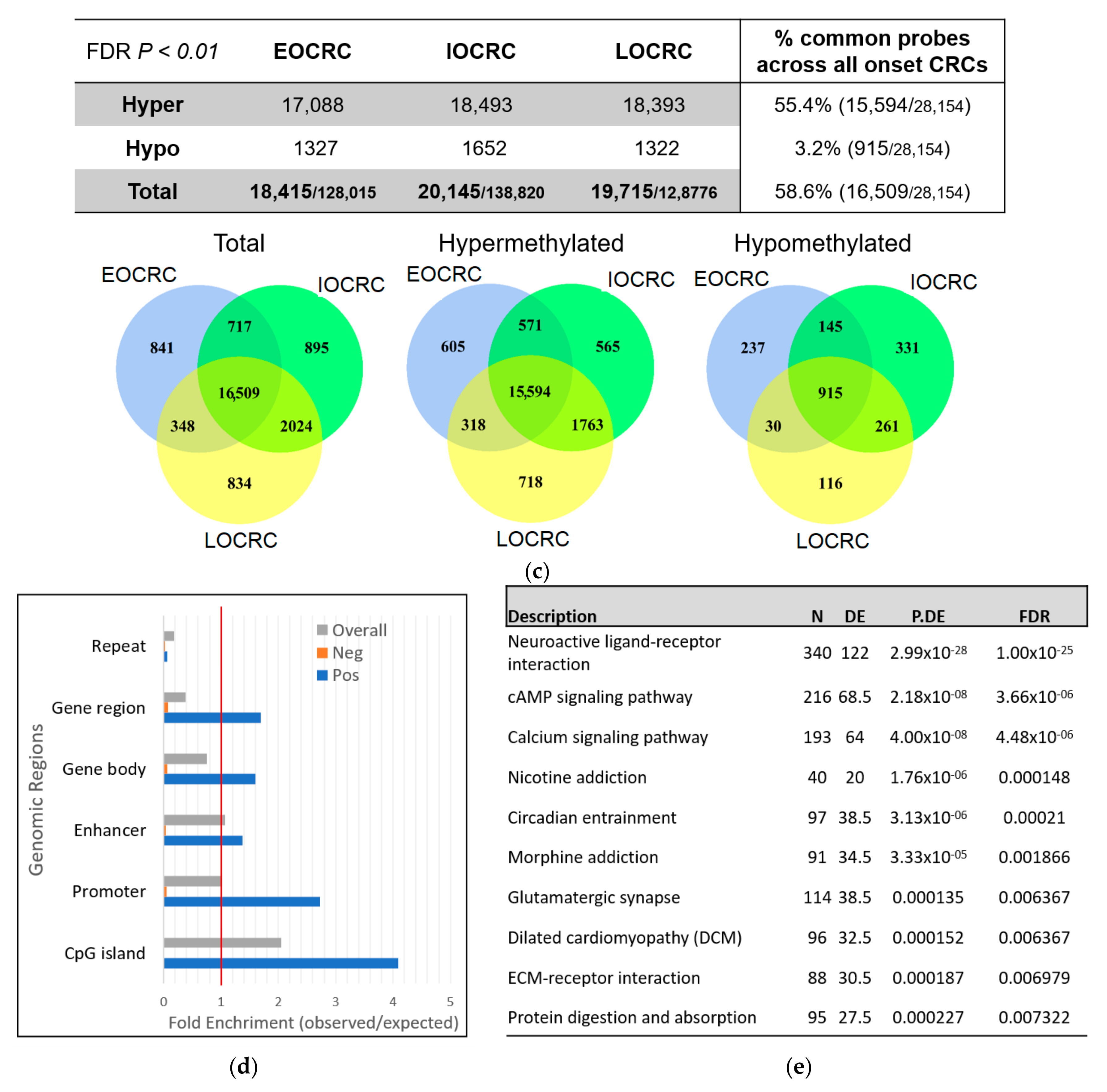
3.2. Methylomic Drift in Normal Colonic Mucosa and Colorectal Carcinogenesis
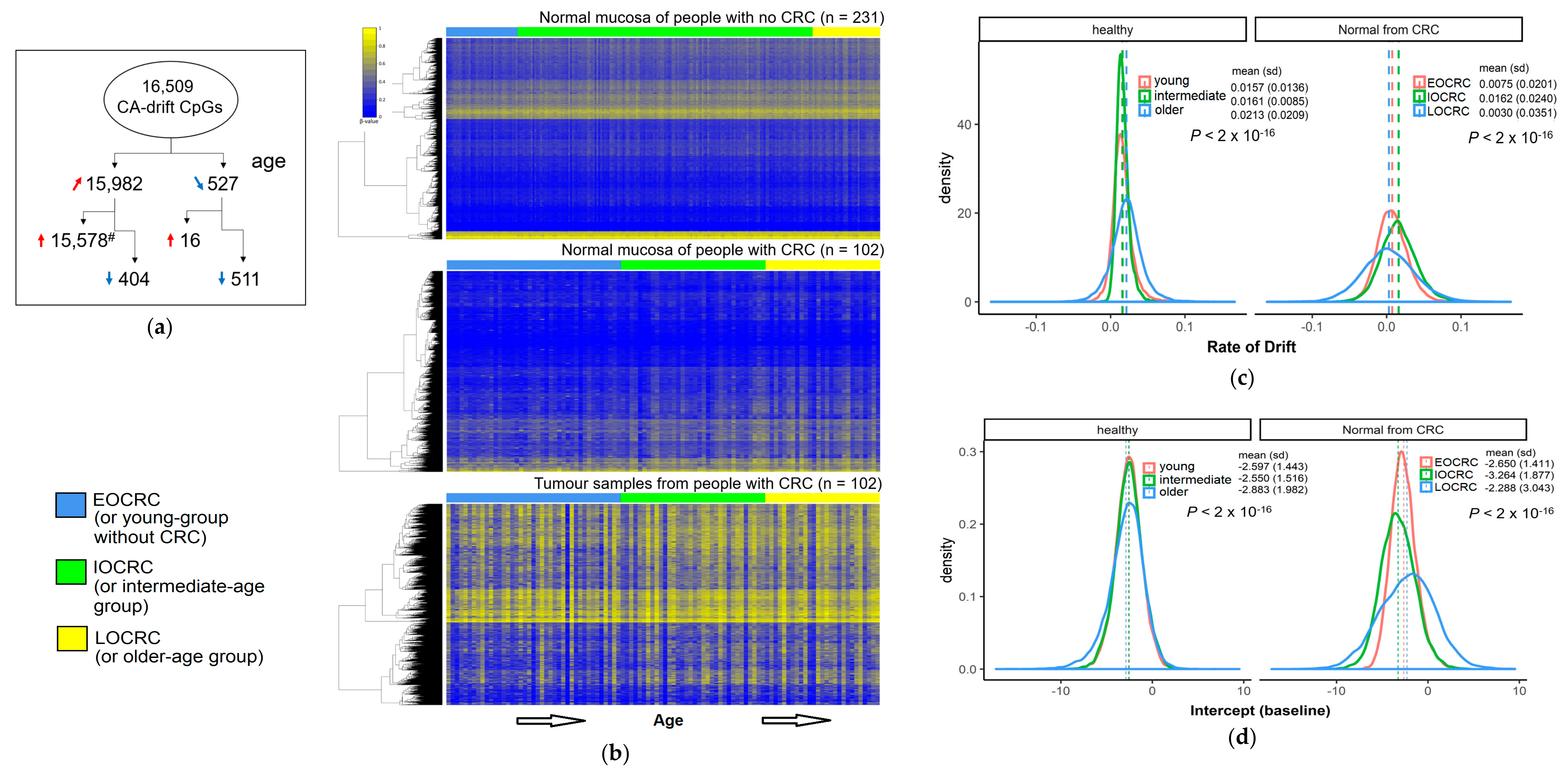
3.3. Rate of Change of DNA Methylation at Cancer-Associated Drift CpGs
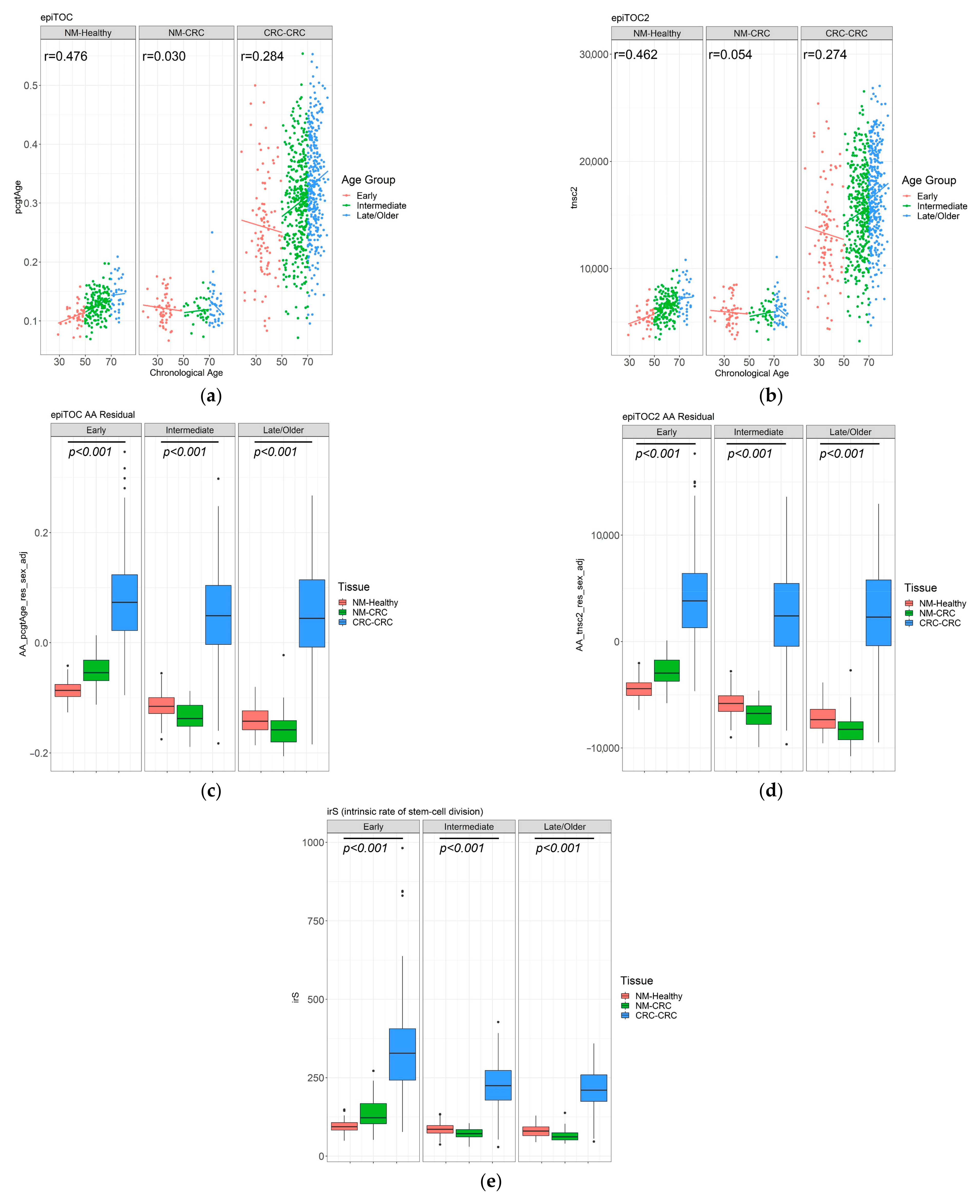
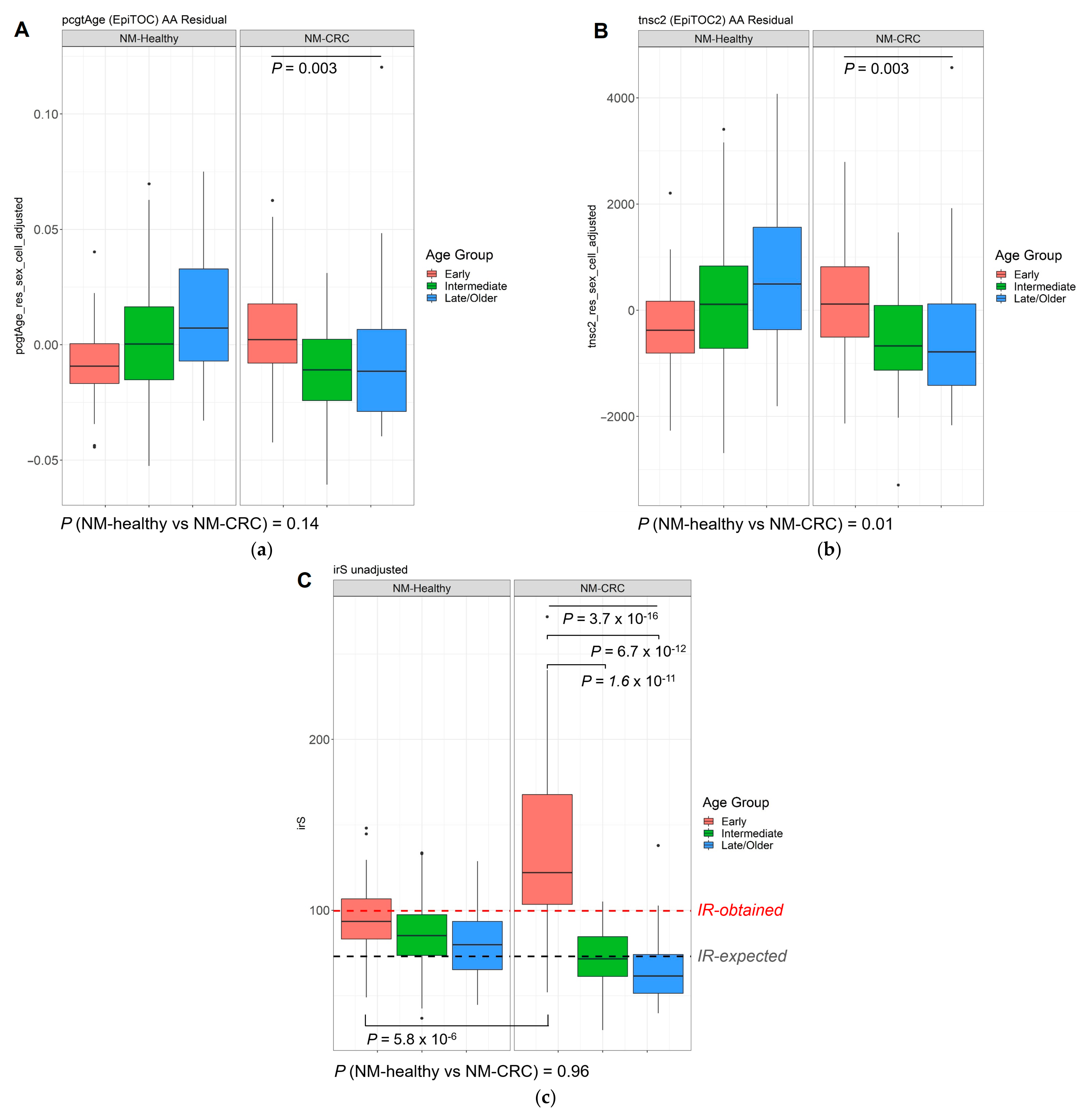
3.4. DNA Methylation-Based Biological Age of the Normal Mucosa and Age Acceleration
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brenner, H.; Chen, C. The colorectal cancer epidemic: Challenges and opportunities for primary, secondary and tertiary prevention. Br. J. Cancer 2018, 119, 785–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenner, H.; Kloor, M.; Pox, C.P. Colorectal cancer. Lancet 2014, 383, 1490–1502. [Google Scholar] [CrossRef]
- Ahnen, D.J.; Wade, S.W.; Jones, W.F.; Sifri, R.; Silveiras, J.M.; Greenamyer, J.; Guiffre, S.; Axilbund, J.; Spiegel, A.; You, Y.N. The Increasing Incidence of Young-Onset Colorectal Cancer: A Call to Action. Mayo Clin. Proc. 2014, 89, 216–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearlman, R.; Frankel, W.L.; Swanson, B.; Zhao, W.; Yilmaz, A.; Miller, K.; Bacher, J.; Bigley, C.; Nelsen, L.; Goodfellow, P.J.; et al. Prevalence and Spectrum of Germline Cancer Susceptibility Gene Mutations Among Patients With Early-Onset Colorectal Cancer. JAMA Oncol. 2017, 3, 464–471. [Google Scholar] [CrossRef]
- Stoffel, E.M.; Koeppe, E.; Everett, J.; Ulintz, P.; Kiel, M.; Osborne, J.; Williams, L.; Hanson, K.; Gruber, S.B.; Rozek, L.S. Germline Genetic Features of Young Individuals With Colorectal Cancer. Gastroenterology 2018, 154, 897–905.e1. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef] [Green Version]
- Walters, R.J.; Williamson, E.J.; English, D.R.; Young, J.P.; Rosty, C.; Clendenning, M.; Walsh, M.D.; Parry, S.; Ahnen, D.J.; Baron, J.A.; et al. Association between hypermethylation of DNA repetitive elements in white blood cell DNA and early-onset colorectal cancer. Epigenetics 2013, 8, 748–755. [Google Scholar] [CrossRef] [Green Version]
- Antelo, M.; Balaguer, F.; Shia, J.; Shen, Y.; Hur, K.; Moreira, L.; Cuatrecasas, M.; Bujanda, L.; Giraldez, M.D.; Takahashi, M.; et al. A High Degree of LINE-1 Hypomethylation Is a Unique Feature of Early-Onset Colorectal Cancer. PLoS ONE 2012, 7, e45357. [Google Scholar] [CrossRef]
- Rosato, V.; Bosetti, C.; Levi, F.; Polesel, J.; Zucchetto, A.; Negri, E.; La Vecchia, C. Risk factors for young-onset colorectal cancer. Cancer Causes Control. 2012, 24, 335–341. [Google Scholar] [CrossRef]
- Belshaw, N.J.; Pal, N.; Tapp, H.S.; Dainty, J.R.; Lewis, M.P.; Williams, M.R.; Lund, E.K.; Johnson, I.T. Patterns of DNA methylation in individual colonic crypts reveal aging and cancer-related field defects in the morphologically normal mucosa. Carcinogenesis 2010, 31, 1158–1163. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Yu, M.; Grady, W.M. Field cancerization in the colon: A role for aberrant DNA methylation? Gastroenterol. Rep. 2014, 2, 16–20. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Kondo, Y.; Rosner, G.L.; Xiao, L.; Hernandez, N.S.; Vilaythong, J.; Houlihan, P.S.; Krouse, R.S.; Prasad, A.R.; Einspahr, J.G.; et al. MGMT Promoter Methylation and Field Defect in Sporadic Colorectal Cancer. J. Natl. Cancer Inst. 2005, 97, 1330–1338. [Google Scholar] [CrossRef]
- Issa, J.-P. Aging and epigenetic drift: A vicious cycle. J. Clin. Investig. 2014, 124, 24–29. [Google Scholar] [CrossRef] [Green Version]
- Luebeck, G.E.; Hazelton, W.D.; Curtius, K.; Maden, S.K.; Yu, M.; Carter, K.T.; Burke, W.; Lampe, P.D.; Li, C.I.; Ulrich, C.M.; et al. Implications of Epigenetic Drift in Colorectal Neoplasia. Cancer Res. 2019, 79, 495–504. [Google Scholar] [CrossRef] [Green Version]
- Noreen, F.; Röösli, M.; Gaj, P.; Pietrzak, J.; Weis, S.; Urfer, P.; Regula, J.; Schär, P.; Truninger, K. Modulation of Age- and Cancer-Associated DNA Methylation Change in the Healthy Colon by Aspirin and Lifestyle. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [Green Version]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.-B.; Gao, Y.; et al. Genome-wide Methylation Profiles Reveal Quantitative Views of Human Aging Rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef] [Green Version]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [Green Version]
- Horvath, S.; Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef]
- Levine, M.E.; Lu, A.T.; Quach, A.; Chen, B.H.; Assimes, T.L.; Bandinelli, S.; Hou, L.; Baccarelli, A.A.; Stewart, J.D.; Li, Y.; et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging 2018, 10, 573–591. [Google Scholar] [CrossRef] [Green Version]
- Teschendorff, A.E. A comparison of epigenetic mitotic-like clocks for cancer risk prediction. Genome Med. 2020, 12, 1–17. [Google Scholar] [CrossRef]
- Yang, Z.; Wong, A.; Kuh, D.; Paul, D.; Rakyan, V.K.; Leslie, R.D.; Zheng, S.C.; Widschwendter, M.; Beck, S.; Teschendorff, A.E. Correlation of an epigenetic mitotic clock with cancer risk. Genome Biol. 2016, 17, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Dinh, H.; Ramjan, Z.; Weisenberger, D.J.; Nicolet, C.M.; Shen, H.; Laird, P.W.; Berman, B.P. DNA methylation loss in late-replicating domains is linked to mitotic cell division. Nat. Genet. 2018, 50, 591–602. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, M.A.; Win, A.K.; Templeton, A.S.; Angelakos, M.S.; Buchanan, D.D.; Cotterchio, M.; Figueiredo, J.C.; Thibodeau, S.N.; Baron, J.A.; Potter, J.D.; et al. Cohort Profile: The Colon Cancer Family Registry Cohort (CCFRC). Int. J. Epidemiol. 2018, 47, 387–388i. [Google Scholar] [CrossRef]
- Newcomb, P.A.; Baron, J.; Cotterchio, M.; Gallinger, S.; Grove, J.; Haile, R.; Hall, D.; Hopper, J.L.; Jass, J.; Le Marchand, L.; et al. Colon Cancer Family Registry: An International Resource for Studies of the Genetic Epidemiology of Colon Cancer. Cancer Epidemiol. Biomark. Prev. 2007, 16, 2331–2343. [Google Scholar] [CrossRef] [Green Version]
- Milne, R.L.; Fletcher, A.S.; MacInnis, R.; Hodge, A.M.; Hopkins, A.H.; Bassett, J.K.; Bruinsma, F.J.; Lynch, B.M.; Dugue, P.-A.; Jayasekara, H.; et al. Cohort Profile: The Melbourne Collaborative Cohort Study (Health 2020). Int. J. Epidemiol. 2017, 46, 1757–1757i. [Google Scholar] [CrossRef]
- Buchanan, D.D.; Clendenning, M.; Rosty, C.; Eriksen, S.V.; Walsh, M.D.; Walters, R.J.; Thibodeau, S.N.; Stewart, J.; Preston, S.; Win, A.K.; et al. Tumor testing to identify lynch syndrome in two Australian colorectal cancer cohorts. J. Gastroenterol. Hepatol. 2017, 32, 427–438. [Google Scholar] [CrossRef] [Green Version]
- Wong, E.M.; Joo, J.E.; McLean, C.A.; Baglietto, L.; English, D.R.; Severi, G.; Hopper, J.L.; Milne, R.L.; FitzGerald, L.M.; Giles, G.G.; et al. Tools for translational epigenetic studies involving formalin-fixed paraffin-embedded human tissue: Applying the Infinium HumanMethyation450 Beadchip assay to large population-based studies. BMC Res. Notes 2015, 8, 543. [Google Scholar] [CrossRef] [Green Version]
- Wong, E.M.; Joo, J.H.E.; McLean, C.A.; Baglietto, L.; English, D.R.; Severi, G.; Wu, H.C.; Terry, M.B.; Hopper, J.L.; Milne, R.L.; et al. Analysis of the breast cancer methylome using formalin-fixed paraffin-embedded tumour. Breast Cancer Res. Treat. 2016, 160, 173–180. [Google Scholar] [CrossRef]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [Green Version]
- Fortin, J.-P.; Labbe, A.; Lemire, M.; Zanke, B.W.; Hudson, T.J.; Fertig, E.J.; Greenwood, C.M.; Hansen, K.D. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 2014, 15, 503. [Google Scholar] [CrossRef] [Green Version]
- Triche, T.J., Jr.; Weisenberger, D.J.; Van Den Berg, D.; Laird, P.W.; Siegmund, K.D. Low-level processing of Illumina Infinium DNA Methylation BeadArrays. Nucleic Acids Res. 2013, 41, e90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, P.; Zhang, X.; Huang, C.-C.; Jafari, N.; Kibbe, W.A.; Hou, L.; Lin, S.M. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinform. 2010, 11, 587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Peters, T.J.; Buckley, M.J.; Statham, A.L.; Pidsley, R.; Samaras, K.; Lord, R.V.; Clark, S.J.; Molloy, P.L. De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin 2015, 8, 6. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Phipson, B.; Maksimovic, J.; Oshlack, A. missMethyl: An R package for analyzing data from Illumina’s HumanMethylation450 platform. Bioinformatics 2016, 32, 286–288. [Google Scholar] [CrossRef]
- Adams, S.V.; Newcomb, P.A.; Burnett-Hartman, A.N.; Wurscher, M.A.; Mandelson, M.; Upton, M.P.; Zhu, L.-C.; Potter, J.D.; Makar, K.W. Rare Circulating MicroRNAs as Biomarkers of Colorectal Neoplasia. PLoS ONE 2014, 9, e108668. [Google Scholar] [CrossRef]
- Barault, L.; Amatu, A.; Siravegna, G.; Ponzetti, A.; Moran, S.; Cassingena, A.; Mussolin, B.; Falcomatà, C.; Binder, A.M.; Cristiano, C.; et al. Discovery of methylated circulating DNA biomarkers for comprehensive non-invasive monitoring of treatment response in metastatic colorectal cancer. Gut 2017, 67, 1995–2005. [Google Scholar] [CrossRef]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef]
- Xu, Z.; Niu, L.; Li, L.; Taylor, J.A. ENmix: A novel background correction method for Illumina HumanMethylation450 BeadChip. Nucleic Acids Res. 2016, 44, e20. [Google Scholar] [CrossRef]
- McEwen, L.M.; Jones, M.J.; Lin, D.T.S.; Edgar, R.D.; Husquin, L.T.; MacIsaac, J.L.; Ramadori, K.E.; Morin, A.M.; Rider, C.F.; Carlsten, C.; et al. Systematic evaluation of DNA methylation age estimation with common preprocessing methods and the Infinium MethylationEPIC BeadChip array. Clin. Epigenetics 2018, 10, 123. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Maden, S.K.; Luebeck, G.E.; Li, C.I.; Newcomb, P.A.; Ulrich, C.M.; Joo, J.-H.E.; Buchanan, D.D.; Milne, R.L.; Southey, M.C.; et al. Dysfunctional epigenetic aging of the normal colon and colorectal cancer risk. Clin. Epigenetics 2020, 12, 1–9. [Google Scholar] [CrossRef]
- Zheng, S.C.; Breeze, C.E.; Beck, S.; Teschendorff, A.E. Identification of differentially methylated cell types in epigenome-wide association studies. Nat. Methods 2018, 15, 1059–1066. [Google Scholar] [CrossRef]
- Tomasetti, C.; Vogelstein, B. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 2015, 347, 78–81. [Google Scholar] [CrossRef] [Green Version]
- Crujeiras, A.B.; Morcillo, S.; Diaz-Lagares, A.; Sandoval, J.; Castellano-Castillo, D.; Torres, E.; Hervas, D.; Moran, S.; Esteller, M.; Macias-Gonzalez, M.; et al. Identification of an episignature of human colorectal cancer associated with obesity by genome-wide DNA methylation analysis. Int. J. Obes. 2018, 43, 176–188. [Google Scholar] [CrossRef]
- Graham, R.P.; Shrestha, B.; Caron, B.L.; Smyrk, T.C.; Grogg, K.L.; Lloyd, R.V.; Zhang, L. Islet-1 Is a Sensitive But Not Entirely Specific Marker for Pancreatic Neuroendocrine Neoplasms and Their Metastases. Am. J. Surg. Pathol. 2013, 37, 399–405. [Google Scholar] [CrossRef]
- Ropponen, K.M.; Kellokoski, J.K.; Pirinen, R.T.; Moisio, K.I.; Eskelinen, M.J.; Alhava, E.M.; Kosma, V.-M. Expression of transcription factor AP-2 in colorectal adenomas and adenocarcinomas; comparison of immunohistochemistry and in situ hybridisation. J. Clin. Pathol. 2001, 54, 533–538. [Google Scholar] [CrossRef] [Green Version]
- Araghi, M.; Fidler, M.M.; Arnold, M.; Jemal, A.; Bray, F.; Soerjomataram, I. The Future Burden of Colorectal Cancer Among US Blacks and Whites. J. Natl. Cancer Inst. 2018, 110, 791–793. [Google Scholar] [CrossRef]
- Yuhara, H.; Steinmaus, C.; Cohen, S.E.; Corley, D.A.; Tei, Y.; Buffler, P.A. Is Diabetes Mellitus an Independent Risk Factor for Colon Cancer and Rectal Cancer? Am. J. Gastroenterol. 2011, 106, 1911–1921. [Google Scholar] [CrossRef] [Green Version]
- Young, J.P.; Win, A.K.; Rosty, C.; Flight, I.; Roder, D.; Young, G.P.; Frank, O.; Suthers, G.K.; Hewett, P.J.; Ruszkiewicz, A.; et al. Rising incidence of early-onset colorectal cancer in Australia over two decades: Report and review. J. Gastroenterol. Hepatol. 2015, 30, 6–13. [Google Scholar] [CrossRef]
- Gausman, V.; Dornblaser, D.; Anand, S.; Hayes, R.B.; O’Connell, K.; Du, M.; Liang, P.S. Risk Factors Associated With Ear-ly-Onset Colorectal Cancer. Clin. Gastroenterol. Hepatol. 2019, 18, 2752–2759. [Google Scholar] [CrossRef] [PubMed]
- Anık, A.; Çatlı, G.; Abacı, A.; Böber, E. Maturity-onset diabetes of the young (MODY): An update. J. Pediatr. Endocrinol. Metab. 2015, 28, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Thaiss, C.A.; Levy, M.; Grosheva, I.; Zheng, D.; Soffer, E.; Blacher, E.; Braverman, S.; Tengeler, A.C.; Barak, O.; Elazar, M.; et al. Hyperglycemia drives intestinal barrier dysfunction and risk for enteric infection. Science 2018, 359, 1376–1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Liping, S.; Sun, L.; Wang, Z.; Xing, C.; Yuan, Y. Aberrantly methylated-differentially expressed genes and pathways in colorectal cancer. Cancer Cell Int. 2017, 17, 75. [Google Scholar] [CrossRef] [Green Version]
- Ali, O.; Cerjak, D.; Kent, J.W.; James, R.; Blangero, J.; Carless, M.A.; Zhang, Y. An epigenetic map of age-associated autosomal loci in northern European families at high risk for the metabolic syndrome. Clin. Epigenetics 2015, 7, 12. [Google Scholar] [CrossRef] [Green Version]
- Ahuja, N.; Li, Q.; Mohan, A.L.; Baylin, S.B.; Issa, J.-P. Aging and DNA methylation in colorectal mucosa and cancer. Cancer Res. 1998, 58, 5489–5494. [Google Scholar]
- Maegawa, S.; Hinkal, G.; Kim, H.S.; Shen, L.; Zhang, L.; Zhang, J.; Zhang, N.; Liang, S.; Donehower, L.A.; Issa, J.-P. Widespread and tissue specific age-related DNA methylation changes in mice. Genome Res. 2010, 20, 332–340. [Google Scholar] [CrossRef] [Green Version]
- Nejman, D.; Straussman, R.; Steinfeld, I.; Ruvolo, M.; Roberts, D.; Yakhini, Z.; Cedar, H. Molecular Rules Governing De Novo Methylation in Cancer. Cancer Res. 2014, 74, 1475–1483. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Hazelton, W.D.; Luebeck, G.E.; Grady, W.M. Epigenetic Aging: More Than Just a Clock When It Comes to Cancer. Cancer Res. 2020, 80, 367–374. [Google Scholar] [CrossRef] [Green Version]
- Weisenberger, D.J.; Liang, G.; Lenz, H.-J. DNA methylation aberrancies delineate clinically distinct subsets of colorectal cancer and provide novel targets for epigenetic therapies. Oncogene 2018, 37, 566–577. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Onset of Disease | Early (EOCRC) | Intermediate (IOCRC) | Late (LOCRC) | ||||
|---|---|---|---|---|---|---|---|
| Ages of onset | <50 years | ≥50 and <70 years | ≥70 years | ||||
| Mean (sd) | 36.2 (6.2) years | 62.6 (5.2) years | 74.8 (3.5) years | ||||
| Total number of cases (matched tumour/normal pairs) | 110 (41) | 334 (34) | 325 (27) | ||||
| Sex (%) | F | M | F | M | F | M | |
| 64 (58) | 46 (42) | 158 (47) | 176 (53) | 142 (43) | 183 (57) | ||
| Tissue type | Tumour | Normal | Tumour | Normal | Tumour | Normal | |
| 97 | 54 | 343 | 35 | 318 | 40 | ||
| Anatomical site (%) | Right colon | 20 (20.5) | 11 (21) | 86 (25) | 8 (23) | 107 (34) | 14 (35) |
| Left colon | 32 (33.5) | 13 (24) | 100 (29) | 8 (23) | 91 (29) | 9 (22.5) | |
| Rectum | 40 (41) | 19 (35) | 138 (40) | 8 (23) | 114 (36) | 7 (17.5) | |
| unknown | 5 (5) | 11 (20) | 19 (6) | 11 (31) | 6 (2) | 10 (25) | |
| TNM (AJCC) stage (%) | Stage I | 17 (18) | - | 70 (20) | - | 75 (24) | - |
| Stage II | 25 (26) | - | 81 (24) | - | 81 (25) | - | |
| Stage III | 43 (44) | - | 101 (30) | - | 87 (27) | - | |
| Stage IV | 9 (9) | - | 54 (16) | - | 55 (17) | - | |
| unknown | 3 | - | 37 | - | 20 | - | |
| CIMP group | CIMP + | 0 | - | 19 | - | 34 | - |
| CIMP - | 93 | - | 320 | - | 280 | - | |
| unknown | 4 | - | 4 | - | 4 | - | |
| Early-Onset (EOCRC) | Intermediate-Onset (IOCRC) | Late-Onset (LOCRC) | p-Value | ||
|---|---|---|---|---|---|
| Age of Dx | <50 years | ≥50 and <70 years | ≥70 years | ||
| N of tumour and matched normal pairs | 41 | 34 | 27 | ||
| Number of DMPs * | Hypermethylated | 49,044 (38.8%) | 59,125 (40.3%) | 57,970 (45.1%) | <0.001 # |
| Hypomethylated | 77,228 (61.2%) | 87,448 (59.7%) | 70,806 (54.9%) | ||
| Total | 126,272 (29.2% of all CpGs included in the analysis) | 146,573 (33.9%) | 128,776 (29.8%) | ||
| Number of DMRs | Hypermethylated | 7102 (40%) | 7985 (39.5%) | 8407 (47.3%) | <0.001 # |
| Hypomethylated | 10,564 (60%) | 12,236 (60.5%) | 9362 (52.7%) | ||
| Total | 17,666 | 20,221 | 17,769 | ||
| Number of DMPs * that are only presented in each group | Hypermethylated | 7369 (39.3%) | 9171 (40%) | 10,216 (69%) | <0.001 # |
| Hypomethylated | 11,386 (60.7%) | 13,727 (60%) | 4581 (31%) | ||
| Total | 18,755 (4.3%) | 22,898 (5.3%) | 14,797 (3.4%) | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joo, J.E.; Clendenning, M.; Wong, E.M.; Rosty, C.; Mahmood, K.; Georgeson, P.; Winship, I.M.; Preston, S.G.; Win, A.K.; Dugué, P.-A.; et al. DNA Methylation Signatures and the Contribution of Age-Associated Methylomic Drift to Carcinogenesis in Early-Onset Colorectal Cancer. Cancers 2021, 13, 2589. https://doi.org/10.3390/cancers13112589
Joo JE, Clendenning M, Wong EM, Rosty C, Mahmood K, Georgeson P, Winship IM, Preston SG, Win AK, Dugué P-A, et al. DNA Methylation Signatures and the Contribution of Age-Associated Methylomic Drift to Carcinogenesis in Early-Onset Colorectal Cancer. Cancers. 2021; 13(11):2589. https://doi.org/10.3390/cancers13112589
Chicago/Turabian StyleJoo, Jihoon E., Mark Clendenning, Ee Ming Wong, Christophe Rosty, Khalid Mahmood, Peter Georgeson, Ingrid M. Winship, Susan G. Preston, Aung Ko Win, Pierre-Antoine Dugué, and et al. 2021. "DNA Methylation Signatures and the Contribution of Age-Associated Methylomic Drift to Carcinogenesis in Early-Onset Colorectal Cancer" Cancers 13, no. 11: 2589. https://doi.org/10.3390/cancers13112589