
Captopril, a Renin-Angiotensin System Inhibitor, Attenuates Features of Tumor Invasion and Down-Regulates C-Myc Expression in a Mouse Model of Colorectal Cancer Liver Metastasis

 , , and
, , and 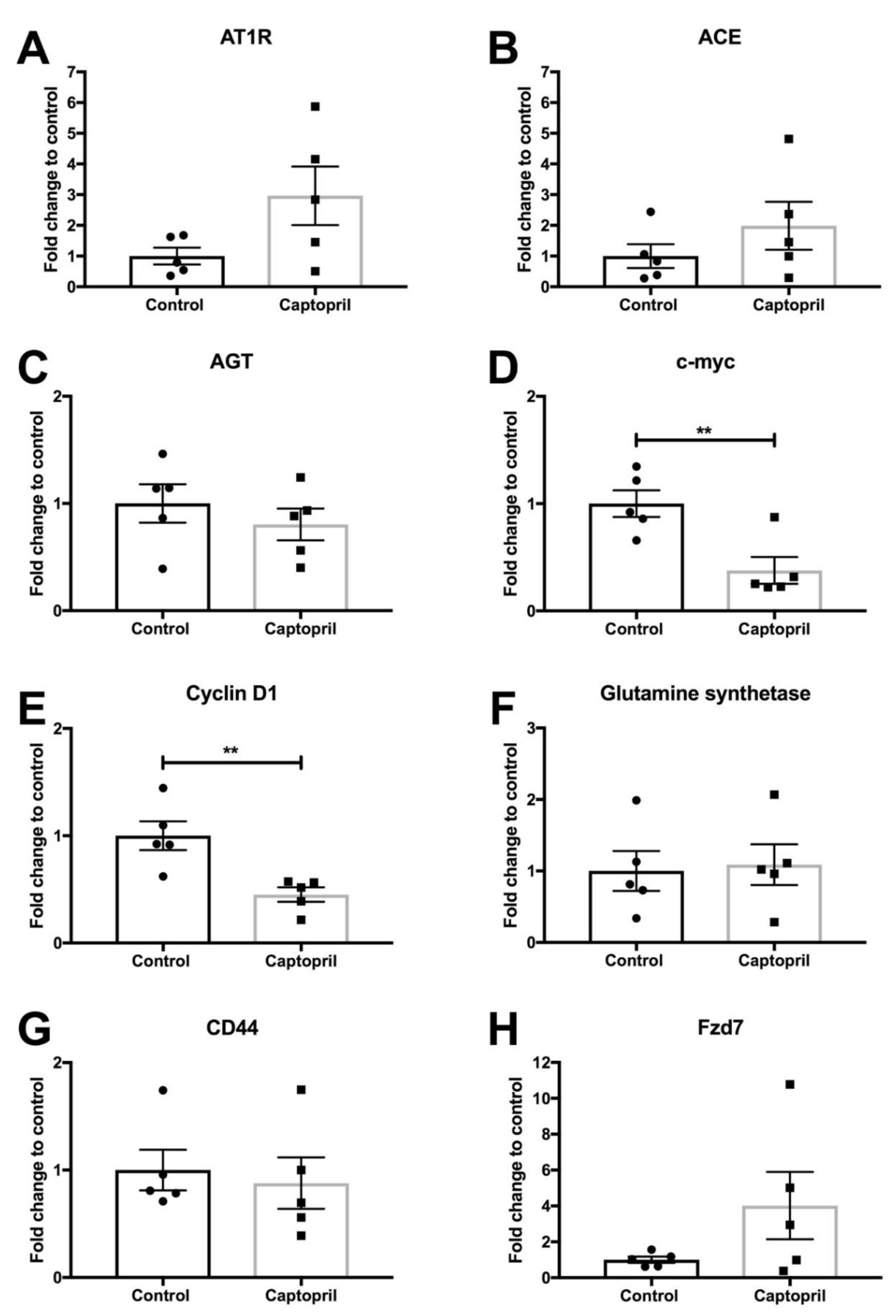{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
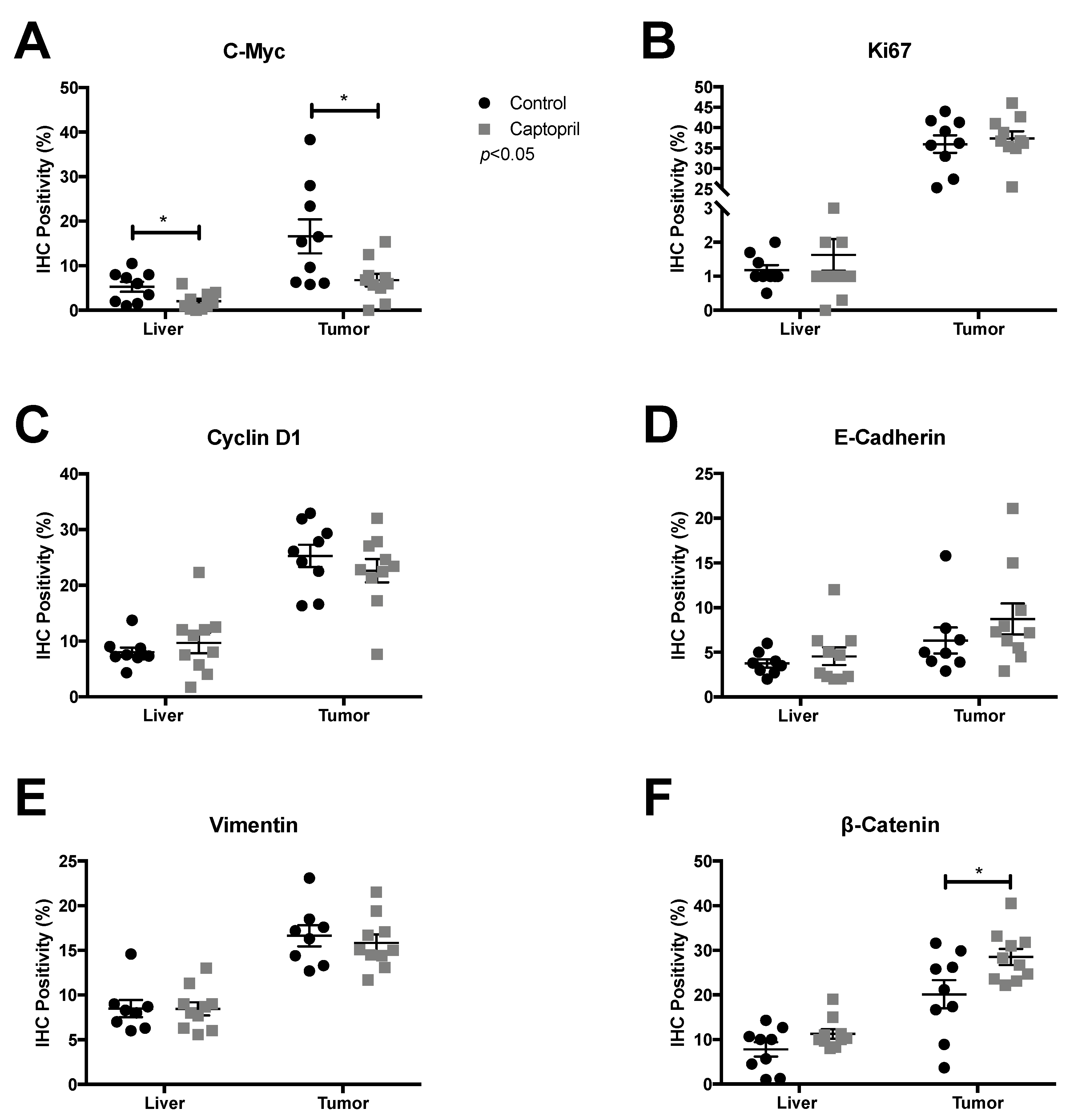
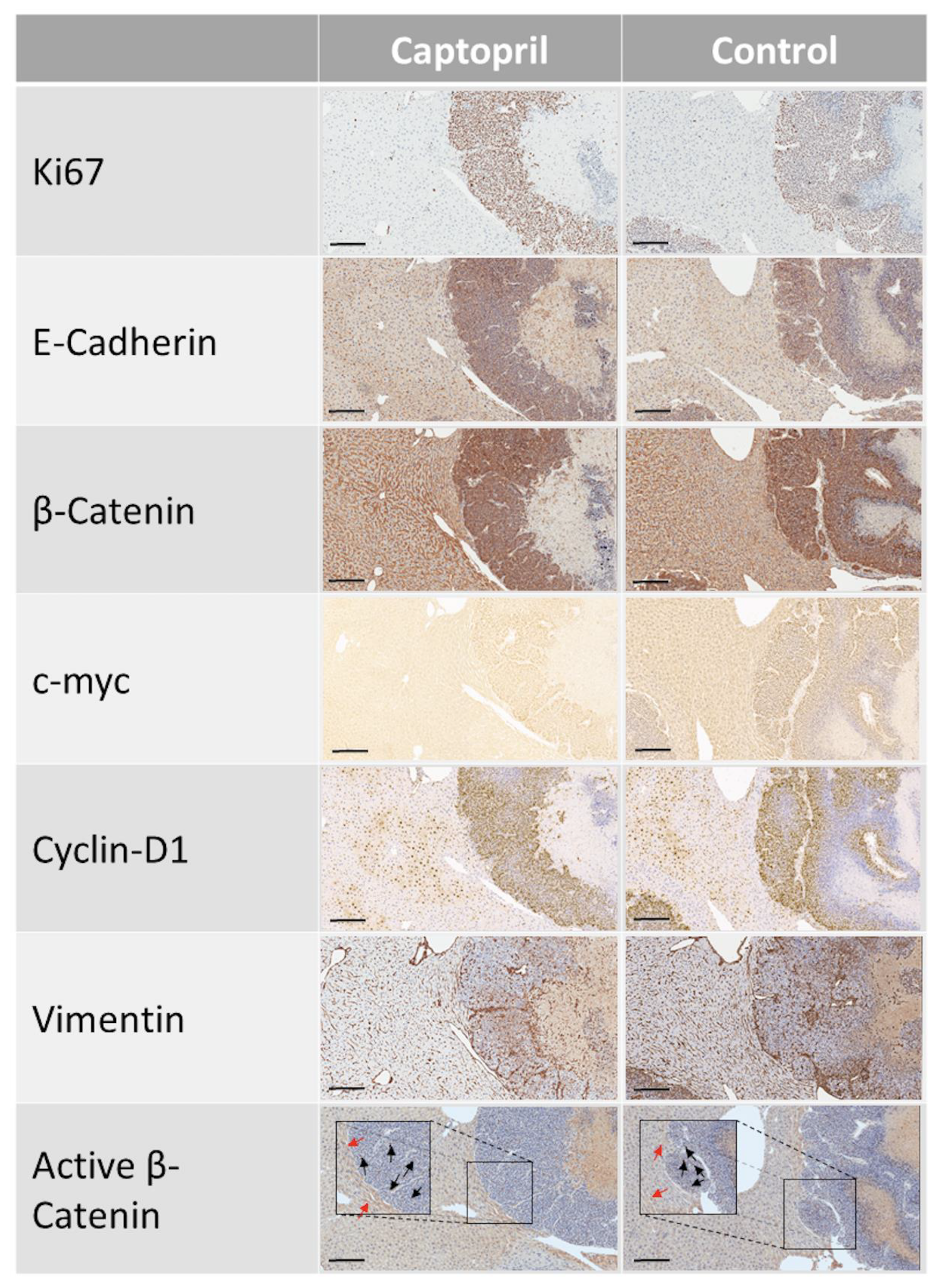
2.1. Captopril Treatment was Associated with a Significant Down-Regulation of c-Myc Expression
2.2. Captopril Treatment was also Associated with a Significant Down-Regulation of Cyclin D1 Expression
2.3. Captopril-Treated Tumors Maintained Ki67 Expression
2.4. Captopril Treatment was Associated with Significantly Greater Cytoplasmic Staining of β-Catenin
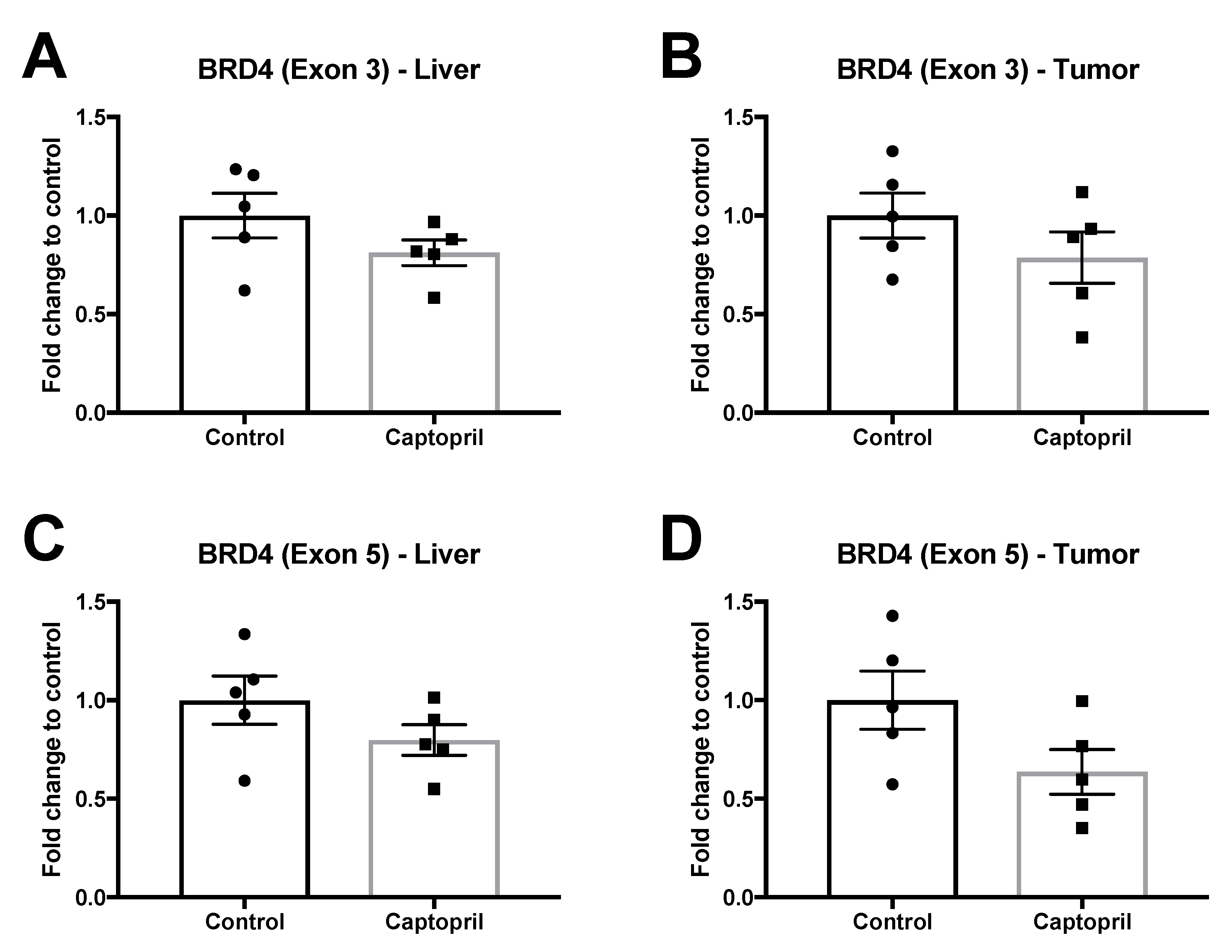
2.5. Captopril Treatment Did Not Significantly Modulate the Expression of Bromodomain and Extra-Terminal Domal Domain (BET) Proteins
3. Discussion
4. Materials and Methods
4.1. Animal Experiments
4.2. qRT-PCR
4.3. Immunohistochemistry
4.4. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Engstrand, J.; Nilsson, H.; Strömberg, C.; Jonas, E.; Freedman, J. Colorectal Cancer Liver Metastases—A Population-Based Study on Incidence, Management and Survival. BMC Cancer 2018, 18, 78. [Google Scholar] [CrossRef]
- Wang, K.; Liu, W.; Yan, X.-L.; Li, J.; Xing, B.-C. Long-Term Postoperative Survival Prediction in Patients with Colorectal Liver Metastasis. Oncotarget 2017, 8, 79927–79934. [Google Scholar] [CrossRef] [Green Version]
- Harun, N.; Nikfarjam, M.; Muralidharan, V.; Christophi, C. Liver Regeneration Stimulates Tumor Metastases. J. Surg. Res. 2007, 138, 284–290. [Google Scholar] [CrossRef]
- Mao, R.; Zhao, J.-J.; Bi, X.-Y.; Zhang, Y.-F.; Li, Z.-Y.; Zhou, J.-G.; Wu, X.-L.; Xiao, C.; Zhao, H.; Cai, J.-Q. A Postoperative Scoring System for Post-Hepatectomy Early Recurrence of Colorectal Liver Metastases. Oncotarget 2017, 8, 102531–102539. [Google Scholar] [CrossRef] [Green Version]
- Park, S.-Y.; Nam, J.-S. The Force Awakens: Metastatic Dormant Cancer Cells. Exp. Mol. Med. 2020, 52, 569–581. [Google Scholar] [CrossRef] [Green Version]
- Steinhart, Z.; Angers, S. Wnt Signaling in Development and Tissue Homeostasis. Development 2018, 145, dev146589. [Google Scholar] [CrossRef] [Green Version]
- Wiese, K.E.; Nusse, R.; van Amerongen, R. Wnt Signalling: Conquering Complexity. Development 2018, 145, dev165902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monga, S.P.; Pediaditakis, P.; Mule, K.; Stolz, D.B.; Michalopoulos, G.K. Changes in WNT/Beta-Catenin Pathway During Regulated Growth in Rat Liver Regeneration. Hepatology 2001, 33, 1098–1109. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Mowry, L.E.; Nejak-Bowen, K.N.; Okabe, H.; Diegel, C.R.; Lang, R.A.; Williams, B.O.; Monga, S.P. Β-Catenin Signaling in Murine Liver Zonation and Regeneration: A Wnt-Wnt Situation! Hepatology 2014, 60, 964–976. [Google Scholar] [CrossRef] [Green Version]
- Clevers, H.; Nusse, R. Wnt/Β-Catenin Signaling and Disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef] [Green Version]
- Nusse, R.; Clevers, H. Wnt/Β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef]
- Korinek, V.; Barker, N.; Morin, P.J.; van Wichen, D.; de Weger, R.; Kinzler, K.W.; Vogelstein, B.; Clevers, H. Constitutive Transcriptional Activation by a Beta-Catenin-Tcf Complex in APC-/- Colon Carcinoma. Science 1997, 275, 1784–1787. [Google Scholar] [CrossRef] [Green Version]
- Morin, P.J.; Sparks, A.B.; Korinek, V.; Barker, N.; Clevers, H.; Vogelstein, B.; Kinzler, K.W. Activation of Beta-Catenin-Tcf Signaling in Colon Cancer by Mutations in Beta-Catenin or APC. Science 1997, 275, 1787–1790. [Google Scholar] [CrossRef] [Green Version]
- Brabletz, T.; Jung, A.; Reu, S.; Porzner, M.; Hlubek, F.; Kunz-Schughart, L.A.; Knuechel, R.; Kirchner, T. Variable Beta-Catenin Expression in Colorectal Cancers Indicates Tumor Progression Driven by the Tumor Environment. PNAS 2001, 98, 10356–10361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincan, E.; Darcy, P.K.; Farrelly, C.A.; Faux, M.C.; Brabletz, T.; Ramsay, R.G. Frizzled-7 Dictates Three-Dimensional Organization of Colorectal Cancer Cell Carcinoids. Oncogene 2007, 26, 2340–2352. [Google Scholar] [CrossRef] [Green Version]
- Brabletz, T.; Jung, A.; Hermann, K.; Günther, K.; Hohenberger, W.; Kirchner, T. Nuclear Overexpression of the Oncoprotein Beta-Catenin in Colorectal Cancer Is Localized Predominantly at the Invasion Front. Pathol. Res. Pract. 1998, 194, 701–704. [Google Scholar] [CrossRef]
- Vincan, E.; Darcy, P.K.; Smyth, M.J.; Thompson, E.W.; Thomas, R.J.S.; Phillips, W.A.; Ramsay, R.G. Frizzled-7 Receptor Ectodomain Expression in a Colon Cancer Cell Line Induces Morphological Change and Attenuates Tumor Growth. Differentiation 2005, 73, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P. Epithelial-Mesenchymal Transitions in Tumor Progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Vincan, E.; Flanagan, D.J.; Pouliot, N.; Brabletz, T.; Spaderna, S. Variable FZD7 Expression in Colorectal Cancers Indicates Regulation by the Tumor Microenvironment. Dev. Dyn. 2010, 239, 311–317. [Google Scholar] [PubMed]
- Ueno, K.; Hiura, M.; Suehiro, Y.; Hazama, S.; Hirata, H.; Oka, M.; Imai, K.; Dahiya, R.; Hinoda, Y. Frizzled-7 as a Potential Therapeutic Target in Colorectal Cancer. Neoplasia 2008, 10, 697–705. [Google Scholar] [CrossRef] [Green Version]
- Flanagan, D.; Barker, N.; Di Costanzo, N.S.; Mason, E.A.; Gurney, A.; Meniel, V.S.; Koushyar, S.; Austin, C.R.; Pearson, H.B.; Boussioutas, A.; et al. Frizzled-7 Is Required for Wnt Signaling in Gastric Tumors with and Without Apc Mutations. Cancer Res. 2019, 79, 970–981. [Google Scholar] [CrossRef] [Green Version]
- Flanagan, D.J.; Vincan, E.; Phesse, T.J. Wnt Signaling in Cancer: Not a Binary on: OFF Switch. Cancer Res. 2019, 79, 5901–5906. [Google Scholar] [CrossRef]
- Ranjbar, R.; Shafiee, M.; Hesari, A.; Ferns, G.A.; Ghasemi, F.; Avan, A. The Potential Therapeutic Use of Renin-Angiotensin System Inhibitors in the Treatment of Inflammatory Diseases. J. Cell. Physiol. 2018, 168, 123. [Google Scholar] [CrossRef]
- Zhou, L.; Li, Y.; Hao, S.; Zhou, D.; Tan, R.J.; Nie, J.; Hou, F.F.; Kahn, M.; Liu, Y. Multiple Genes of the Renin-Angiotensin System Are Novel Targets of Wnt/Β-Catenin Signaling. J. Am. Soc. Nephrol. 2015, 26, 107–120. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Liu, Y. Wnt/Β-Catenin Signaling and Renin-Angiotensin System in Chronic Kidney Disease. Curr. Opin. Nephrol. Hypertens. 2016, 25, 100–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.; Ager, E.I.; Neo, J.; Christophi, C. Regulation of Colorectal Cancer Cell Epithelial to Mesenchymal Transition by the Renin Angiotensin System. J. Gastroenterol. Hepatol. 2016, 31, 1773–1782. [Google Scholar] [CrossRef] [PubMed]
- George, A.J.; Thomas, W.G.; Hannan, R.D. The Renin-Angiotensin System and Cancer: Old Dog, New Tricks. Nat. Rev. Cancer 2010, 10, 745–759. [Google Scholar] [CrossRef]
- Vallejo Ardila, D.L.; Walsh, K.A.; Fifis, T.; Paolini, R.; Kastrappis, G.; Christophi, C.; Perini, M.V. Immunomodulatory Effects of Renin-Angiotensin System Inhibitors on T Lymphocytes in Mice with Colorectal Liver Metastases. J. Immunother. Cancer 2020, 8, e000487. [Google Scholar] [CrossRef]
- Perini, M.V.; Dmello, R.S.; Nero, T.L.; Chand, A.L. Evaluating the Benefits of Renin-Angiotensin System Inhibitors as Cancer Treatments. Pharmacol. Ther. 2020, 211, 107527. [Google Scholar] [CrossRef]
- Pinter, M.; Jain, R.K. Targeting the Renin-Angiotensin System to Improve Cancer Treatment: Implications for Immunotherapy. Sci. Transl. Med. 2017, 9, eaan5616. [Google Scholar] [CrossRef] [Green Version]
- Neo, J.H.; Malcontenti-Wilson, C.; Muralidharan, V.; Christophi, C. Effect of ACE Inhibitors and Angiotensin II Receptor Antagonists in a Mouse Model of Colorectal Cancer Liver Metastases. J. Gastroenterol. Hepatol. 2007, 22, 577–584. [Google Scholar] [CrossRef]
- Gerdes, J.; Lemke, H.; Baisch, H.; Wacker, H.H.; Schwab, U.; Stein, H. Cell Cycle Analysis of a Cell Proliferation-Associated Human Nuclear Antigen Defined by the Monoclonal Antibody Ki-67. J. Immunol. 1984, 133, 1710–1715. [Google Scholar]
- van Noort, M.; Meeldijk, J.; van der Zee, R.; Destree, O.; Clevers, H. Wnt Signaling Controls the Phosphorylation Status of Beta-Catenin. J. Biol. Chem. 2002, 277, 17901–17905. [Google Scholar] [CrossRef] [Green Version]
- Lovén, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective Inhibition of Tumor Oncogenes by Disruption of Super-Enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [Green Version]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET Bromodomain Inhibition as a Therapeutic Strategy to Target C-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [Green Version]
- Mertz, J.A.; Conery, A.R.; Bryant, B.M.; Sandy, P.; Balasubramanian, S.; Mele, D.A.; Bergeron, L.; Sims, R.J. Targeting MYC Dependence in Cancer by Inhibiting BET Bromodomains. Proc. Natl. Acad. Sci. USA 2011, 108, 16669–16674. [Google Scholar] [CrossRef] [Green Version]
- Zuber, J.; Shi, J.; Wang, E.; Rappaport, A.R.; Herrmann, H.; Sison, E.A.; Magoon, D.; Qi, J.; Blatt, K.; Wunderlich, M.; et al. RNAi Screen Identifies Brd4 as a Therapeutic Target in Acute Myeloid Leukaemia. Nature 2011, 478, 524–528. [Google Scholar] [CrossRef] [Green Version]
- Shao, Q.; Kannan, A.; Lin, Z.; Stack, B.C.; Suen, J.Y.; Gao, L. BET Protein Inhibitor JQ1 Attenuates Myc-Amplified MCC Tumor Growth in Vivo. Cancer Res. 2014, 74, 7090–7102. [Google Scholar] [CrossRef] [Green Version]
- Ding, N.; Hah, N.; Yu, R.T.; Sherman, M.H.; Benner, C.; Leblanc, M.; He, M.; Liddle, C.; Downes, M.; Evans, R.M. BRD4 Is a Novel Therapeutic Target for Liver Fibrosis. Proc. Natl. Acad. Sci. USA 2015, 112, 15713–15718. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Senapati, P.; Chen, Z.; Reddy, M.A.; Ganguly, R.; Lanting, L.; Mandi, V.; Bansal, A.; Leung, A.; Zhang, S.; et al. Regulation of Angiotensin II Actions by Enhancers and Super-Enhancers in Vascular Smooth Muscle Cells. Nat. Commun. 2017, 8, 1467. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Network. Comprehensive Molecular Characterization of Human Colon and Rectal Cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Fearon, E.R. Molecular Genetics of Colorectal Cancer. Annu. Rev. Pathol. 2011, 6, 479–507. [Google Scholar] [CrossRef]
- Wilkins, J.A.; Sansom, O.J. C-Myc Is a Critical Mediator of the Phenotypes of Apc Loss in the Intestine. Cancer Res. 2008, 68, 4963–4966. [Google Scholar] [CrossRef] [Green Version]
- Sansom, O.J.; Meniel, V.S.; Muncan, V.; Phesse, T.J.; Wilkins, J.A.; Reed, K.R.; Vass, J.K.; Athineos, D.; Clevers, H.; Clarke, A.R. Myc Deletion Rescues Apc Deficiency in the Small Intestine. Nature 2007, 446, 676–679. [Google Scholar] [CrossRef]
- Arvanitis, C.; Felsher, D.W. Conditional Transgenic Models Define How MYC Initiates and Maintains Tumorigenesis. Semin. Cancer Biol. 2006, 16, 313–317. [Google Scholar] [CrossRef]
- Chen, H.; Liu, H.; Qing, G. Targeting Oncogenic Myc as a Strategy for Cancer Treatment. Signal Transduct. Target. Ther. 2018, 3, 5. [Google Scholar] [CrossRef] [Green Version]
- He, T.C.; Sparks, A.B.; Rago, C.; Hermeking, H.; Zawel, L.; da Costa, L.T.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Identification of C-MYC as a Target of the APC Pathway. Science 1998, 281, 1509–1512. [Google Scholar] [CrossRef]
- Riddiough, G.E.; Fifis, T.; Muralidharan, V.; Perini, M.V.; Christophi, C. Searching for the Link; Mechanisms Underlying Liver Regeneration and Recurrence of Colorectal Liver Metastasis Post Partial Hepatectomy. J. Gastroenterol. Hepatol. 2019, 34, 1276–1286. [Google Scholar] [CrossRef] [Green Version]
- Prochownik, E.V.; Vogt, P.K. Therapeutic Targeting of Myc. Genes Cancer 2010, 1, 650–659. [Google Scholar] [CrossRef] [Green Version]
- Meyer, N.; Penn, L.Z. Reflecting on 25 Years with MYC. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar] [CrossRef] [PubMed]
- Willert, J.; Epping, M.; Pollack, J.R.; Brown, P.O.; Nusse, R. A Transcriptional Response to Wnt Protein in Human Embryonic Carcinoma Cells. BMC Dev. Biol. 2002, 2, 8. [Google Scholar] [CrossRef] [Green Version]
- De la Iglesia Inigo, S.; Lopez-Jorge, C.E.; Gomez-Casares, M.T.; Castellano, A.L.; Cabrera, P.M.; Brito, J.L.; Cabrera, A.S.; Labarta, T.M. Induction of apoptosis in leukemic cell lines treated with captopril, trandolapril and losartan: A new role in the treatment of leukaemia for these agents. Leuk. Res. 2009, 33, 810–816. [Google Scholar] [CrossRef]
- Koh, S.L.; Ager, E.I.; Costa, P.L.N.; Malcontenti-Wilson, C.; Muralidharan, V.; Christophi, C. Blockade of the Renin-Angiotensin System Inhibits Growth of Colorectal Cancer Liver Metastases in the Regenerating Liver. Clin. Exp. Metastasis 2014, 31, 395–405. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riddiough, G.E.; Fifis, T.; Walsh, K.A.; Muralidharan, V.; Christophi, C.; Tran, B.M.; Vincan, E.; Perini, M.V. Captopril, a Renin-Angiotensin System Inhibitor, Attenuates Features of Tumor Invasion and Down-Regulates C-Myc Expression in a Mouse Model of Colorectal Cancer Liver Metastasis. Cancers 2021, 13, 2734. https://doi.org/10.3390/cancers13112734
Riddiough GE, Fifis T, Walsh KA, Muralidharan V, Christophi C, Tran BM, Vincan E, Perini MV. Captopril, a Renin-Angiotensin System Inhibitor, Attenuates Features of Tumor Invasion and Down-Regulates C-Myc Expression in a Mouse Model of Colorectal Cancer Liver Metastasis. Cancers. 2021; 13(11):2734. https://doi.org/10.3390/cancers13112734
Chicago/Turabian StyleRiddiough, Georgina E., Theodora Fifis, Katrina A. Walsh, Vijayaragavan Muralidharan, Christopher Christophi, Bang M. Tran, Elizabeth Vincan, and Marcos V. Perini. 2021. "Captopril, a Renin-Angiotensin System Inhibitor, Attenuates Features of Tumor Invasion and Down-Regulates C-Myc Expression in a Mouse Model of Colorectal Cancer Liver Metastasis" Cancers 13, no. 11: 2734. https://doi.org/10.3390/cancers13112734