
Regulation of mRNA Translation by Hormone Receptors in Breast and Prostate Cancer


1
Flinders Health and Medical Research Institute, Flinders University, Bedford Park, SA 5042, Australia
2
Translational Prostate Cancer Research, Peter MacCallum Cancer Centre, Melbourne, VIC 3000, Australia
3
Cancer Program, Biomedicine Discovery Institute and Department of Anatomy and Developmental Biology, Monash University, Clayton, VIC 3800, Australia
4
Sir Peter MacCallum Department of Oncology, University of Melbourne, Parkville, VIC 3010, Australia
5
Freemasons Centre for Male Health and Wellbeing, Flinders University, Bedford Park, SA 5042, Australia
6
Adelaide Medical School, University of Adelaide, Adelaide, SA 5005, Australia
*
Authors to whom correspondence should be addressed.
†
Authors contributed equally to this work.
Cancers 2021, 13(13), 3254; https://doi.org/10.3390/cancers13133254
Submission received: 12 May 2021
/
Revised: 23 June 2021
/
Accepted: 24 June 2021
/
Published: 29 June 2021
(This article belongs to the Special Issue Nuclear Receptors and Cancer)
Abstract
:Simple Summary
The estrogen and androgen receptors (ER, AR) are key oncogenic drivers and therapeutic targets in breast and prostate cancer, respectively. These receptors bind to DNA and regulate gene expression but emerging evidence indicates that they also play important roles in controlling the process of mRNA translation, which dictates cellular protein production. Here, we review the mechanisms by which abnormal activities of ER and AR can dysregulate mRNA translation in breast and prostate cancer cells. Specifically, we explore how the intricate cellular signalling pathways that keep mRNA translation in check are perturbed by aberrant ER and AR signalling, which can lead to enhanced cancer cell growth. We also discuss the potential of targeting mRNA translation as a strategy to treat patients with breast and prostate cancer.
Abstract
Breast and prostate cancer are the second and third leading causes of death amongst all cancer types, respectively. Pathogenesis of these malignancies is characterised by dysregulation of sex hormone signalling pathways, mediated by the estrogen receptor-α (ER) in breast cancer and androgen receptor (AR) in prostate cancer. ER and AR are transcription factors whose aberrant function drives oncogenic transcriptional programs to promote cancer growth and progression. While ER/AR are known to stimulate cell growth and survival by modulating gene transcription, emerging findings indicate that their effects in neoplasia are also mediated by dysregulation of protein synthesis (i.e., mRNA translation). This suggests that ER/AR can coordinately perturb both transcriptional and translational programs, resulting in the establishment of proteomes that promote malignancy. In this review, we will discuss relatively understudied aspects of ER and AR activity in regulating protein synthesis as well as the potential of targeting mRNA translation in breast and prostate cancer.
1. Parallels between Aberrant Hormone Receptor Signalling in Breast and Prostate Cancer
Breasts and prostate are accessory sex organs that are exquisitely sensitive to sex hormones (i.e., estrogens and androgens). Malignancies arising from these organs are frequently driven by abnormal activity of the receptors of these sex hormones: indeed, >90% of prostate cancers (PC) are driven by the androgen receptor (AR) and ~70% of breast cancers (BC) are driven by the estrogen receptor-α (ER) [1]. As such, hormone deprivation therapies are the mainstay of treatment for locally progressive and advanced BC and PC. Although initially effective in most patients, these therapies are never curative and the resultant treatment-resistant tumours are aggressive and often fatal. Gaining a more complete understanding of the oncogenic activities of AR and ER is imperative for the development of more effective targeting strategies.
AR and ER are transcription factors that regulate the expression of genes involved in cancer development and progression, but also genes that are essential for the normal functioning of the prostate and breast glands. Both are members of the nuclear receptor superfamily and possess modular structures composed of N-terminal transactivation domains (NTD), DNA-binding domains (DBD), hinge regions and ligand binding domains (LBD) (Figure 1). Upon binding to their hormone ligands, AR/ER bind to chromatin at androgen/estrogen response elements and mediate coordinated recruitment of transcriptional co-regulators, resulting in activation or, more rarely, repression of gene expression [2,3]. In normal prostate and breast tissues, the transcriptional programs regulated by AR/ER in epithelial cells are responsible for promoting differentiation and regulating metabolism and the production of secreted proteins, whereas the AR/ER-regulated transcriptional programs in tumour cells promote cell growth and survival [4]. This switch in activities is mediated at least in part by altered expression of AR/ER co-regulator proteins and changes to the epigenome, resulting in altered AR/ER DNA binding profiles [5,6].
The similarities between AR/ER signalling in prostate and breast cancer extend to mechanisms of therapy resistance; indeed, most therapy-resistant cases of BC and PC exhibit alterations to the AR/ER signalling axes that enhance oncogenic signalling [1,7,8]. One key mechanism is mutation of the AR/ER LBD, which enhances the promiscuity of the receptor such that it can be activated by additional non-canonical ligands or even by antagonist drugs [7,8]. Other resistance mechanisms shared by AR/ER include the generation of constitutively active splice variants of the receptors, increased expression and/or activity of a plethora of transcriptional coactivators (e.g., steroid receptor coactivators, forkhead box protein A, GATA binding proteins, cAMP responsive element binding protein-binding protein/p300, switch/sucrose non-fermentable complex, E3 ubiquitin-protein ligases and steroid RNA activator, among others), and dampening of the activities of corepressors (e.g., nuclear receptor co-repressors, speckle type BTB/POZ protein and many others [9,10]. Collectively, these alterations to AR/ER signalling confer insensitivity to conventional hormone deprivation therapies (reviewed in [7,11]) and highlight the dependency of tumours on these pathways.
2. Dysregulation of Translation Is a Common Feature of Cancer
2.1. Translation Initiation
Translation of mRNAs can be broadly divided into four steps: initiation, elongation, termination and ribosome recycling [12]. The initiation and elongation steps of translation are thought to be the most highly regulated and hence have been studied most intensively [13]. Dysregulation of translation in cancer, which occurs because of alterations in the levels or the activity of the components of translational machinery [14], can impact any of these steps.
The major signalling pathways that confer translational control are summarised in Figure 2. A prominent feature of many cancers are mutations that lead to activation of PI3K (phosphatidylinositol-3-kinase)/PKB (protein kinase B) and the Ras (rat sarcoma)/MAPK (mitogen-activated protein kinase) pathways, including loss-of-function mutations of PTEN and gain-of-function mutations of PI3K and genes in the Ras/mitogen-activated protein kinase MAPK pathway [15,16,17,18,19,20]. PTEN (phosphatase and tensin homolog) is a phosphatase which dephosphorylates PtdIns(3,4,5)P3 (PIP3) and thus acts as a tumour suppressor to counteract PI3K activity. One major consequence of these mutations is increased activity of the mechanistic/mammalian target of rapamycin (mTOR) [21,22]. mTOR is a serine/threonine kinase that is part of two distinct complexes, mTOR complex 1 (mTORC1) and complex 2 (mTORC2) [23]. In response to a variety of stimuli including nutrients, oxygen availability, growth factors and hormones, mTORC1 stimulates protein synthesis by phosphorylating substrates such as the ribosomal protein (RP) S6 kinases (S6Ks; S6K 1 and 2 in humans) [24] and the eukaryotic translation initiation factor (eIF) 4E binding proteins (4EBPs; 4EBP1, 2 and 3 in humans). Upon mTORC1-mediated phosphorylation, 4EBPs are released from eIF4E, allowing it to form a heterotrimeric eIF4F complex—with the scaffold protein eIF4G and the RNA helicase eIF4A—that recruits mRNA to the ribosomes [25,26]. In addition, oncogenic activation of the RAS/MAPK pathway is thought to affect translation by stimulating phosphorylation of eIF4E via MAPK-interacting kinases (MNKs), as well as other translation initiation factors (e.g., eIF4B) and ribosomal proteins (e.g., rpS6) [27,28] (Figure 2). Targeting the subunits of the eIF4F complex has attracted substantial interest in BC/PC (e.g., see [29,30]) and will be further discussed below.
2.2. Translation Elongation
Notwithstanding that translation initiation is generally considered the rate limiting step of protein synthesis, more recent evidence shows that aberrant translation elongation may also play a prominent role in cancer [31]. In mammals, translation elongation rates are thought to be primarily regulated by an atypical calcium/calmodulin-dependent protein kinase called the eukaryotic translation elongation factor 2 (eEF2) kinase (eEF2K) [32]. eEF2K reduces elongation rates by phosphorylating and inhibiting eEF2 [33,34,35], a key translation factor that translocates nascent peptide chains from the A-site to the P-site of the ribosome in a GTP-dependent manner [36]. mTORC1 and MAPK suppress eEF2K, thereby increasing elongation rates [37,38] (Figure 2). In contrast, energy-sensing AMP-activating protein kinase (AMPK) phosphorylates and activates eEF2K via mTORC1-dependent and independent mechanisms to suppress translation and reduce energy consumption [39].
2.3. Integrated Stress Response (ISR)
In addition to the mTOR-dependent and -independent inhibitory effects of AMPK on protein synthesis, mTORC1 can be switched off in response to nutrient/energy deprivation or other stressors (e.g., hypoxia, acidosis) via the integrated stress response (ISR) pathway. The function of the ISR pathway is to restore cellular homeostasis or, in situations when its activation is prolonged, induce cell death. Intrinsic cellular stresses such as the unfolded protein response (UPR), which lead to endoplasmic reticulum (EnR) stress, can also trigger the ISR. Three arms of the UPR in mammals regulate distinct but highly orchestrated EnR-stress responses. The Protein Kinase RNA activated (PKR)-like EnR kinase (PERK)/eIF2α arm of the UPR reduces global protein synthesis to relieve EnR overload while inducing translation of a small subset of mRNAs, thus leading to transcriptional reprogramming that enhances EnR folding capacity. In parallel with this, the inositol requiring enzyme 1 (IRE1)/x-box binding protein 1 (XBP1) and the activating transcription factor 6 (ATF6) (and ATF6p50, a cleaved, active form of ATF6) arms of the UPR also contribute to the improvement of EnR folding capacity in 2 ways: (i) by altering transcription, and (ii) by triggering EnR-associated degradation (ERAD), which entails retro-translocation of misfolded proteins from the EnR to the cytosol and their subsequent degradation by the Ub/proteasome system (Figure 2). Additionally, EnR Ca2+ release through the inositol triphosphaste (IP3) receptor (IP3R) evokes cell fate decision events to either allow the cell to trigger or escape from apoptotic death (see [40,41] for reviews).
The ISR arm of the UPR is centered on the phosphorylation of the α-subunit (eIF2α) of heterotrimer eIF2, which is catalysed by PERK [42] or three other kinases (general control nonderepressible 2 (GCN2), PKR and heme-regulated inhibitor (HRI)), that are activated by distinct stresses (amino acid deprivation, viral infection and heme deficiency, respectively) [43,44,45]. Phosphorylation of eIF2α inhibits eIF2B, which exchanges eIF2-GDP for eIF2-GTP and allows recycling of the ternary complex comprising eIF2, GTP and initiator methionyl-tRNA (tRNAiMet). The ensuing limitation in TC levels limits delivery of tRNAiMet and halts global translation while allowing translation of a small subset of mRNAs characterized by inhibitory upstream open reading frames (uORFs) that encode transcription factors involved in stress response (e.g., ATF4, C/EBP homology protein (CHOP)), or are implicated in feedback loops that limit excessive ISR (e.g., GADD34 (growth arrest and DNA damage-inducible gene 34)) (reviewed in [46,47,48], also see Figure 2). Failure to do so ultimately leads to apoptotic cell death, which is exploitable as an anti-cancer strategy.
3. Genomic Alterations to Translation Factors in Breast and Prostate Cancer
Mutation of genes implicated in translation regulation and perturbation of translational programs appears to be a common feature of BC/PC, which provides a strong rationale for targeting translational machinery in these diseases. In particular, mutations in genes encoding proteins upstream of mTORC1, including core components of the PI3K/AKT pathway (i.e., PIK3CA, which encodes the p110α subunit of PI3K, PTEN and AKT1) and the MAPK pathway (MAP3K1, MAP2K4, BRAF and HRAS) are frequent in BC and PC and considered to be drivers of tumourigenesis [49,50,51]. Mutations in the MTOR gene itself are also present in BC (1.85%) and PC (0.4%) patients [52,53,54] (Table 1).
Notably, cancer-related alterations in translational machinery and associated factors do not uniformly affect translation of all cellular mRNAs but rather cause selective reprograming of the translatome to favour synthesis of factors that promote tumorigenesis, tumor progression and drug resistance [23,55]. These differences in translational programs of normal and neoplastic cells could yield a therapeutic window to selectively eradicate cancer cells by targeting translational machinery, while causing minimal toxicity [23]. This concept has spurred recent efforts to design and repurpose translational inhibitors and apply them in oncological indications [14]. Herein, we review recent findings suggesting that dysregulation of the abovementioned mechanisms of translational regulation may play a pivotal role in BC and PC, with a particular focus on the connections between perturbations in mRNA translation, aberrant ER and AR signalling and potential therapeutic applications.
4. Interplay between mRNA Translation and ER Signalling
4.1. ER Selectively Controls Translation Initiation
Early evidence of mRNA translation being regulated by estrogen was provided in the late 1950s and the early 1960s [56,57,58,59,60]. More specifically, estrogen stimulated the incorporation of glycine-2-C14, used as a gross measure of protein synthesis, into rat and chicken cells. However, the effect of estrogen on protein synthesis in BC cells was not rigorously explored until the beginning of the 21st century. For example, an early study found that estrogen facilitated the association of mRNAs with translationally active polyribosomes [61], implying that not all effects of estrogen on BC cell functions were through de novo mRNA synthesis. More recent work is beginning to shed mechanistic insight into how ER regulates mRNA translation. A role for ER in translation appears to relate to its ability to localize to the plasma membrane within lipid rafts (i.e., caveolae)-enriched regions [62], which can be regulated by direct palmitoylation (at C447/C451 in human/mouse respectively) of the ER [63]. There, ER is thought to transactivate tyrosine kinase receptors including epidermal growth factor receptor (EGFR) and insulin-like growth factor 1 (IGF1) receptor (IGF1R) via Src [64,65,66], or to associate with hematopoietic PBX-interacting protein thus facilitating recruitment of Src to the p85 regulatory subunit of PI3K [67]. In this manner, ER is thought to activate PI3K/PKB and MAPK pathways and thus impact translation. Such extra-nuclear functions of nuclear receptors have been proposed to allow more rapid responses than their transcriptional effects [68]. In the context of cap-independent translation, a shorter ER isoform termed ERα46 is translated via an IRES (internal ribosomal entry site)-dependent mechanism in response to EnR stress [69]. However, it is still unclear whether ER has a role in regulating the translation of cancer-associated IRES-dependent mRNAs (reviewed in REF e.g., [70]) such as hypoxia-inducible factor 1α (HIF1α) or the anti-apoptotic protein B-cell lymphoma 2 (BCL2).
Whole-genome/exome sequencing studies revealed that mTORC1 signalling is one of the most upregulated pathways in metastatic BC cells compared to paired primary tumours [71,72,73,74]. The mTOR complexes play a central role in converging upstream signals evoked by ER to control translation initiation. 17β-estradiol (E2, a classic ER agonist) treatment could robustly evoke the phosphorylation of mTORC1 (S6K1 and 4EBP1) and mTORC2 (PKB) downstream targets within 5 min in MCF-7 cells [75]. Analysis of transcriptomic data revealed a strong correlation between ER and mTORC1/2 signalling-regulated genes at the level of gene expression [76]. Indeed, BHPI (3,3-bis(4-hydroxyphenyl)-7-methyl-1,3-dihydro-2H-indol-2-one), a non-competitive ER biomodulator, was able to induce S6K phosphorylation (a readout of mTORC1 activity) and an inhibitor of mTORC1 (rapamycin) reversed the stimulatory effect of E2 on protein synthesis [77]. A recent phosphoproteomics study identified a range of mTORC1 phosphorylation sites from its classic substrates (e.g., S6K, eIF4B, proline-rich Akt substrate of 40 kDa (PRAS40) and La-related protein 1 (LARP1), among others) that could be induced by E2 and are sensitive to rapamycin treatment in MCF-7 cells [78]. Importantly, the authors identified DEPDC6, which encodes for DEPTOR (DEP-domain containing mTOR-interacting protein), as a novel ER-target gene. DEPTOR is a well-established binding partner of mTORC1/2 which negatively regulates the activity of both complexes [79], and thus serves as a tumour suppressor against PC progression [80]. Therefore, the induction of DEPDC6 expression is more likely to act as a negative feedback regulatory mechanism in response to E2-induced mTORC1 activation (Figure 3), but how E2/ER stimulates mTORC1 remains to be elucidated. One obvious possibility is that E2/ER achieves this through the activation of signalling pathways (e.g., PI3K/MAPK) upstream of mTORC1 [81]; alternatively and/or additionally, ER could activate mTORC1 via a non-genomic mechanism as mentioned above. Moreover, ER is a known mTORC1 substrate (see Section 4.2), and therefore phosphorylated/active ER could potentially trigger positive feedback loops to further stimulate mTORC1; such feedback loops are known to be commonly driven by mTORC1 substrates [82,83].
In addition, mTORC1 activation by E2/ER also facilitated the association between the octameric eIF3 and eIF4F, which is required for the formation of 48S preinitiation complex during mammalian translation initiation, as this effect can be reversed by rapamycin [84]. Notably, expression levels of most eIF3 subunits are upregulated in cancer, except for eIF3e and f, which are downregulated in tumour cells and may act as tumour suppressors [85]. eIF3f transcription was greatly reduced upon E2 treatment in MCF-7 cells. However, E2 has minimal effect on eIF3f protein expression, and quite unexpectedly, siRNA-mediated knockdown of ER reduced eIF3f protein levels, implying that the translation of eIF3f was actually enhanced by the presence of ER [85]. How ER can contribute to the synthesis of eIF3f is still unclear; it may involve the activation of mTORC1, since its substrate S6K1 has been shown to interact with and phosphorylate eIF3 [86,87].
Perturbations in the transcription profile may trigger regulatory mechanisms which attempt to compensate for or buffer those changes at the level of mRNA translation, a phenomenon known as translational “offsetting” [88]. Analogous to this, one would expect that altering ER’s transcriptional activity with ER modulators or EDT will induce this adaptive response to maintain translational homeostasis in tumour cells. Analysis of polyribosome-associated mRNAs in BM67 cells (a cell line derived from cPTENfl/fl mice, which was obtained by crossing PB-Cre4 with PTENflox/flox mice) stably expressing shRNAs against ER (shER) revealed that ER depletion triggered translational offsetting [89]. Of importance, mRNAs that were downregulated but translationally offset in shER BM67 frequently contained short 5′-untranslated regions (UTRs) and lacked miRNA target sites at their 3′-UTRs [89]. This is of particular interest since ER is known to either up- or downregulate the expression of several miRNAs in order to fine-tune the translatome as another mechanism to promote BC tumour growth [90,91,92]. In contrast, mRNAs that were induced by ER depletion while being translationally offset are enriched in a specific set of codons decoded by tRNAs bearing a modified uridine (5-methoxycarbonyl-methyl-2-thiouridine) at position 34 (mcm5s2U34) [89], which is formed upon a sequential catalysis by ELP3 (elongator complex protein 3, to generate cm5U), ALKBH8 (AlkB homolog 8, to generate mcm5U) and CTU1/2 (cytoplasmic tRNA 2-thiolation protein 1/2, to generate mcm5s2U). These mcm5s2U34-modifying enzymes, particularly ELP3, have recently been linked to therapy resistance in BRAFV600E driven melanoma [93], BC [94] and colorectal cancer [95]. Importantly, Elp3 mRNA association with polysomes and ELP3 protein expression level was greatly reduced in shER BM67 cells as well as other tested ER-negative cell lines [89], implying that ER may also indirectly impact translation elongation of mRNAs enriched in codons containing mcm5s2U34 tRNAs (Figure 3).
4.2. mTORC1 Directly Controls ER-Mediated Transcription
Direct modification of ER by mTORC1 represents another important link between these factors. ER possesses a highly conserved TOR signalling motif (FPATV, Figure 1), which typically starts with a phenylalanine and is present in most of the mTORC1 substrates [96]. Indeed, mTORC1 can directly phosphorylate ER on Ser104/Ser106, resulting in the enhancement of ER’s transcriptional activity amongst its target genes [97]. This potentially provides a positive feedback link by which ER stimulates a central modulator of protein synthesis to further strengthen its function as a transcription factor (Figure 3). This finding can be substantiated by the fact that, as evidenced by chromatin immunoprecipitation analysis of ER’s binding to the estrogen response element promoter on the pS2 gene, rapamycin was inhibitory to ER’s transcriptional activity in MCF-7 cells [98].
A high level of mTORC1 activity is a predictor of poor progression-free survival (PFS) outcomes in ER+/PR+ BC patients [99]. However, a recent study from Rutkovsky et al. found that 4EBP1, a well-established negative regulator of mTORC1 activity [100], is highly phosphorylated and overexpressed in a range of metastatic ER+ BC cell lines harbouring the 8p11-p12 amplicon, and contributes to BC cell proliferation [101]. DNA amplification of the 8p11-p12 genomic region, which contains the gene encoding 4EBP1 (EIF4EBP1), is frequent in endocrine resistant BC. Knocking down 4EBP1 with lentiviral shRNA reduced the replication of BC cells harbouring the 8p11-p12 amplicon and, unexpectedly, repressed the level of ER protein expression [101]. Hyperphosphorylated 4EBP1 is resistant to ubiquitin-mediated degradation and thus highly stable [102]. Interestingly, knocking down eIF4E can deplete 4E-BP4EBP1 from BC cells [102]; therefore, in addition to amplification of the EIF4EBP1 gene, the observed increase in it therefore suggests it is plausible that increased levels of 4EBP1 in ER+/8p11-p12+ BC cells may also be a result of elevated eIF4E [102]. As further evidence of the relevance of 4EBP1 in breast cancer, genomic profiling studies have identified gene fusions such as TACC1-EIF4EBP1 [103] which are thought to enhance its activity by promoting 4EBP1 autophosphorylation [104,105].
4.3. ER Is an Important Player in the UPR
Translation initiation can be activated by anabolic signals from environmental cues favouring cell growth, whereas it is essential for the process to be reversed when cells are facing stressors. In this respect, UPR plays a crucial role in ensuring cell survival by switching off translation initiation in response to stressors. It has been shown that ER can also regulate UPR under catabolic conditions. One of the earliest examples of how ER ligands induce UPR was from Shapiro’s group, who discovered that that the ER inhibitor BHPI rapidly reduced protein synthesis rates in three types of ER+ BC cells (MCF-7, T47D and BG-1/MCF-7), concomitant with an induction of PERK/eIF2α and AMPK/eEF2 phosphorylation, indicative of EnR stress/UPR and the attenuation of translation elongation respectively (Figure 3). Conversely, treating the MCF-7 cells with E2 increased global protein synthesis by approximately 3-fold [77]. The authors further showed that the PERK inhibitor GSK2606414 was able to enhance global protein synthesis to up to 6-fold in BHPI-co-treated MCF-7/T47D cells, whereas knocking down PERK with siRNA strikingly decreased protein synthesis in untreated cells to less than 10% within 24 h, implying that EnR activation was responsible for the reduced protein synthesis in response to BHPI treatment [77]. However, it is still unknown whether the stimulation of the AMPK/eEF2K pathway also contributes to BHPI-mediated protein synthesis inhibition. A more recent study also found that E2 treatment could enhance protein synthesis in MCF-7:5C cells, which are aromatase inhibitor (AI)-resistant MCF-7 cells generated by long-term estrogen deprivation. However, in this model, the effect of E2 was much weaker than was reported in AI-sensitive cell line models [77], implying that estrogen depletion desensitises the cells from ER agonist-induced protein synthesis.
In spite of increasing evidence that ER can modulate the UPR, the mechanism by which it achieves this remains unclear. One possibility is through the regulation of intracellular Ca2+ channels such as IP3R and SERCA, which are Ca2+-ATPases that transport cytoplasmic Ca2+ into the EnR and thus act as secondary messengers for EnR stress/UPR induction (reviewed in [47]). For instance, BHPI can evoke the activation of phospholipase Cγ (PLCγ), which results in elevated IP3 production and subsequently increases intracellular Ca2+ levels by depleting Ca2+ from the EnR reservoir [77] (Figure 3). Whether ER’s ability to activate PLCγ is due to direct regulation of its gene, similar to other UPR-related genes [92], is unknown.
5. New Roles for AR Signalling in mRNA Translation
5.1. AR Indirectly Regulates Translation Initiation
Similar to estrogenic control of protein synthesis [56,57,58,59,60], a central role for androgens in stimulation of protein synthesis in the prostate was also proposed over half a century ago [106]. Initial observations suggested that androgen-dependent induction of protein synthesis in prostate is rapid [107,108,109,110] and thus unlikely to stem only from the effects of AR on transcription. However, the mechanisms underlying AR translational reprogramming in PC are incompletely understood. Highlighting this issue, a recent study found that de novo protein synthesis was strikingly enhanced in castrated cPTENfl/fl mice, which apparently contradicts early historical findings [30]. Enhanced protein synthesis in this model was correlated with a decrease in 4EBP1 levels and concomitant increase in eIF4F complex assembly [30]. Low levels or complete loss of AR also coincided with reduced 4EBP1 expression in AR-low/null human AR program-independent prostate cancer cell line and metastatic castration-resistant PC (CRPC) LuCaP patient-derived xenografts (PDXs). Consistently, AR-low PC cells exhibited higher sensitivity to 4EBP1 over-expression as compared to corresponding models with high levels of AR [30]. As such, these findings suggest that the absence of AR can also promote protein synthesis, and that the effects of AR on translation may be mediated via regulation of 4EBP1/eIF4F. It has also been shown that AR inhibition by bicalutamide in metastatic CRPC PDXs promoted eIF4E phosphorylation, and sensitized the PC cells to MNK inhibition [111]. Taken together, these reports support the notion that the components of eIF4F may represent a druggable target for AR-low PC.
In addition to affecting 4EBP1 levels, AR may also regulate 4EBP1 activity via modulating mTORC1 signaling. For instance, androgen treatment induced expression of L-type amino acid transporters 3 (LAT3) in LNCaP (PC cell line carrying a AR-FLT877A mutation) cells, whereas bicalutamide reversed these effects [112]. LAT3 is highly expressed in primary prostate tumours [113,114,115,116] and depleting LAT3 effectively diminished prostate cancer cell proliferation in vitro [112]. AR can also regulate mTORC1 indirectly via a signalling cascade involving kallikrein related peptidase 4 (KLK4), promyelocytic leukemia zinc finger (PLZF) and regulated in development and DNA damage responses 1 (REDD1) [117] (Figure 4). Association of the AR-regulated factor KLK4 [118] leads to destabilization of PLZF and REDD1, relieving mTORC1 from REDD1-mediated inhibition. In other words, one would expect that the ablation of AR inhibits mTORC1, but this is seemingly contradictory to the fact that AR-low PC cells have a high protein synthesis rate [30]. One explanation can be that low levels of 4EBP1 expression in AR-low/null cells render eIF4E insensitive to mTORC1 inhibition [30]; hence the eIF4F complex becomes “constitutively active”.
The effect of AR signalling on mTORC1 activity appears to be context-dependent. In contrast to the findings described above, Zhang et al. recently demonstrated that knockdown of AR can trigger mTORC1 stimulation in hepatocellular carcinoma (HCC) cells MHCC-97L, as a result of downregulation of the classic AR target, FK506 binding protein 5 (FKBP5), which acts as an AKT/mTORC1 upstream inhibitor [119]. In addition, although rapamycin treatment is known to enhance AR’s transcriptional activity in PC cells [120], rapamycin blunted the transcriptional activity of AR in HCC (SNU423 and MHCC-97L) cells. Therefore, it appears that either the stimulation (PC) or the depletion (HCC) of AR can activate mTORC1; similarly, mTOR inhibition can either stimulate (PC) or repress (HCC) transcriptional activity of the AR. This apparent paradox may be reconciled as a cell type-specific phenomenon but warrants further investigation. One possibility is that long-term rapamycin treatment can suppress mTORC1 and mTORC2 in some cell lines (e.g., LNCaP and PC3 [120,121]), but not others [121], and therefore the effect of mTOR inhibition may depend on its effect on mTORC2. The use of an ATP-competitive mTOR inhibitor (mTORi) that strongly suppresses the activation of both mTORC1 and 2 ([122], also see Section 6 below), or mTOR complex-specific genetic ablation, may help to dissect the precise role of the regulation of AR by mTOR.
Beyond control of translation factors, other mechanisms of interplay between AR and mTOR have been documented. For example, it was found that AR and mTOR can interact on chromatin, and co-regulate the transcription of genes implicated in metabolic rewiring in PC cells [123,124,125]. Additionally, AR/mTOR can also promote the cleavage and hence the nuclear translocation and activation of sterol regulatory element-binding transcription factor 1 to induce the expression of crucial lipogenic genes such as FASN and SCD1 [125]. As such, it is tempting to speculate that by targeting both AR and mTOR one may even “kill two birds with one stone”—simultaneously modulating oncogenic translational and transcriptional activities within a PC cell.
In comparison to cap-dependent translation, how AR regulates IRES-dependent/cap-independent translation is less well understood. A dramatic increase in IRES signals was observed in the testis of mice treated with testosterone and siRNA-based silencing of AR reduced IRES signals in vivo [126], suggesting that AR activation promotes IRES-dependent translation. However, in another study, AR inhibition with bicalutamide was also shown to promote IRES-dependent translation in CRPC cells [111]. In short, more work is required to precisely elucidate the role of AR in IRES-dependent/cap-independent translation.
5.2. AR and UPR
Multiple lines of evidence link the AR and UPR pathways in PC. Early work found that AR expression is positively correlated with several UPR-related genes in prostate tumours [127], which may relate to the fact that transcription of some UPR-related genes (e.g., NDRG1 and HERPUD1) is responsive to androgen treatment in PC cells [128]. This work suggested that AR stimulates UPR, which was confirmed by the finding that the synthetic androgen R1881 induces the IRE1/XBP1 arm of UPR in LNCaP cells (Figure 4) [129]. Conversely, silencing AR in LNCaP cells reversed the induction of ERN1 transcription and reduced the levels of spliced XBP1 [127,130]. Importantly, XBP1 was found to be highly expressed in PC patient tumours compared to benign tissues [127]. The synthetic androgen also induces ERAD in several PC cell lines, and high expression of ERAD-related genes (gp78, Hrd1 and SVIP) has been observed in PC patient tissues [131]. Additionally, Yang et al. found that DHT-induced AR protein expression in murine embryonic stem cells (mESCs) correlated with induction of all three arms of UPR, evidenced by increased XBP1 mRNA splicing, elevated phosphorylation of PERK, upregulated levels of mRNA encoding CHOP mRNA and ATF6p50 proteins [132]. Sheng et al., however, reported only modest increases in mRNA levels of ATF6/ATF6p50 in response to R1881, implying a weaker activation of the ATF6 arm of UPR in comparison to the IRE1α/XBP1 arm. ATF4 and CHOP levels were also shown to be elevated in this study, although surprisingly the levels of phosphorylated PERK/eIF2α and total PERK were markedly decreased [127]. These apparent discrepancies between the studies indicate that the extent to which AR-mediated regulation of eIF4F complex assembly [30] and PERK/eIF2α phosphorylation/ISR [127] contributes to translational perturbations in prostate cancer remains to be established. Nevertheless, the overall body of evidence demonstrates that AR (activation) induces UPR, which can lead to apoptotic death in some non-PC cells (e.g., mESCs and ovarian cells) [132,133]. The reader is also directed to an excellent recent review [134] which further covers this topic.
6. Targeting Pathways That Regulate mRNA Translation in BC and PC
6.1. Targeting Pathways That Regulate mRNA Translation
6.1.1. PI3K
Most of the PI3KCA mutants in BC are oncogenic (promote growth, proliferation and survival of BC cells) and enhance the activity of the PI3K pathway, particularly in response to ligands such as insulin and IGFs [135,136]. Accordingly, depletion of PI3KCA and PI3KCB is synergistic with estrogen deprivation in ER+ BC cells with PI3KCA mutations and/or PI3KCB amplification [137]. Dual PI3K/mTORis (BKM120, BYL719, RAD001 and BEZ235) also exert strong anti-proliferative effects on a range of ER+/human epidermal growth factor receptor 2 (HER2)+ BC cell lines carrying PI3KCA mutations both in vitro and in vivo [138]. Paradoxically, PI3KCA mutations are favourable prognostic markers of PFS [139,140] for ER+/HER2+ BC patients, and despite high levels of PI3K/AKT signalling, they appear to be associated with reduced mTORC1 activity [140]. ER+/HER2+ BC patients harbouring PI3KCA mutations are also more responsive to tamoxifen (a selective ER modulator) monotherapy [140]. This can be explained in part by elevated levels of genes that negatively regulate mTORC1 such as PP2A and PML [140], feedback mechanisms in response to PI3K activation (see [82,83]) and/or mutant-specific signalling patterns. Moreover, a recent study found that PDXs from the luminal AR+ subtype (LAR+) of ER−/PR−/HER2− BC (TNBC), which accounts for up to 9% of all TNBCs [141] and is characterized by AR activation, are highly enriched in PI3KCA and AKT1 mutations (100% in LAR+ TNBC vs. 7.5% in other subtypes of TNBCs) [142]. Enzalutamide-resistant LAR+ TNBC PDXs are remarkedly sensitive to PI3K, mTOR or PI3K-mTOR dual inhibitors [142]. PI3K and MAPK pathway inhibitors have been extensively studied as anti-BC/PC agents and interested readers are referred to several excellent reviews on this topic [143,144,145,146].
6.1.2. MNK
MNKs are promising therapeutic targets within the translational control signalling network. Targeting MNKs may be a safer option since they are unessential to normal cell physiology, as evidenced by the fact that MNK1/2 double knockout mice are viable and fertile [50]. Interestingly, Njar’s group discovered that a group of compounds, the C-4 heteroaryl retinamides, could simultaneously provoke the degradation of AR-FL, AR-V7 (AR splice variant 7, a variant that lacks the ligand-binding domain and hence can signal in the absence of androgen) and MNK1/2 [147,148]. Retinamides exhibited substantial anti-proliferative, anti-migratory/invasive (in vitro) and anti-growth (xenograft) properties in a range of AR-positve and -negative PC cell lines [147,148], suggesting that simultaneously inhibiting the AR and the MNKs can potentially be a new strategy against AR+ PC; this also implies that the MNK inhibitors (MNKis) may be useful in an AR-negative context. In support of this notion, it has been demonstrated that AR serves as a suppressor of eIF4E phosphorylation [111], which is catalysed only by MNKs [50], and thus loss of AR would evoke MNK activation. On the other hand, mTORis may also reactivate MNK2 by alleviating it from inhibitory post-translational modifications (phosphorylation at Ser74 and Ser437) catalysed by mTORC1 [149,150]. Indeed, in a PC patient tissue microarray analysis, MNK2Ser74 was less phosphorylated in tumour specimens with a high Gleason score [149]. In support of these observations, the MNKi CGP57380 can overcome the resistance to mTORis in PC cells [111]. A monotherapy regimen with a more specific MNKi, eFT508 [151], completed a CRPC-related phase II clinical trial (NCT03690141) in April 2021.
Similarly, tamoxifen-resistant ER+ BC cells also exhibited elevated mTORC1 activity, concomitant with high levels of total and phosphorylated eIF4E; suppression of mTORC1/MNK1 activity using everolimus/CGP57380, or reducing eIF4E levels via si/shRNA, resensitized cells to tamoxifen [152]. CGP57380 exerts some off-target effects on other kinases (CK1, BRSK2 and MKK1) [153], a cautionary note when interpreting results with this agent.
6.1.3. eIF4E
Because inhibition of global translation will unavoidably disrupt physiological functions of the cell, one can expect that future therapeutic strategies aiming at mRNA translation will tend to be more selective towards specific sets of targets. Most of the factors implicated in translation regulation are essential for normal cell function, which is a major hurdle for any treatment strategy targeting the translation machinery. However, eIF4E may serve as a key translation target for anti-cancer treatment. A whole body haplo-insufficient eIF4E+/− genetically engineered mouse model (GEMM) revealed that a “full dosage” of eIF4E is not required for normal development, yet it aids the translation of specific subsets of mRNAs, particularly those that regulate the production of reactive oxygen species, to promote tumour progression [154]. In addition, genetic ablation of the MNKs, the only in vivo eIF4E kinases, did not affect murine viability or fertility [27]. Importantly, eIF4E is highly phosphorylated in advanced PC patient biopsies [149,155]. Knocking-in mutation of eIF4ESer209 (MNKs’ phosphorylation site) to alanine (eIF4ES209A/S209A) in mouse embryonic fibroblasts conferred them resistance to oncogenic transformation [155]. Using the cPTENfl/fl GEMM, it has also been shown that cPTENfl/fl × eIF4ES209A/S209A knock-in mice are resistant to the loss of PTEN-induced PC development [155]. Moreover, Hsieh et al. [156] reported that INK128, an ATP-competitive mTORi (see Section 6 below) that blocks the activities of both mTORC1 and 2, exerted strong cytotoxicity in and effectively prevented the metastasis of PC cells in cPTENfl/fl mice [156]. Collectively, these studies serve as a basis for future research on the potential of targeting eIF4E in PC.
6.1.4. eIF4A
An alternative therapeutic strategy to targeting upstream signalling pathways is direct interference with the components of the eIF4F complex. A study from Modelska et al. [38] has revealed that high levels of eIF4A1, eIF4B and eIF4E are all linked to unfavourable clinical outcomes for ER− BC patients, whereas the tumour suppressor programmed cell death 4 (PDCD4), which binds to and inhibits eIF4A [157], is positively correlated with favourable outcomes in ER+ BC patients. Knock-down of eIF4A or eIF4B using siRNA or over-expressing PDCD4 effectively diminished MCF-7 cell (an ER+/PR+ BC cell line) proliferation in vitro [38]. Investigation into drugs that specifically target the eIFs is still at an early stage, although some have showed promising results in preclinical studies in BC models [158,159]. For instance, eIF4A inhibitors (eIF4Ais), which are derivatives of a class of natural products called the flavaglines, were shown to have strong anti-tumour actions against BC/PC cells both in vitro and in vivo [158,159,160,161,162,163,164,165]. Most of the studies related to eIF4Ais are still at a preclinical stage; however, a newly synthesized flavagline derivative (eFT226 [166]) has just entered phase I/II clinical trials (NCT04092673) against selected advanced solid tumours. Interestingly, targeting eIF4A influenced the BC transcriptome and translatome in a manner distinct from mTORi-mediated eIF4E inhibition [167], indicating that eIF4A and eIF4E may affect different subsets of messages in cancer cells and suggesting that concomitant targeting of eIF4A and eIF4E is a plausible combinatorial therapeutic strategy.
6.2. Targeting Ribosome Biogenesis
Eukaryotic ribosomes, comprised of ~80 RPs and 4 ribosomal RNAs, are macromolecular complexes that function to synthesize proteins from mRNAs. Using a genome-wide CRISPR screen (over 70,000 single guide RNAs) coupled with patient-derived metastatic BC cells, Ebright et al. [168] recently discovered that ribosomal genes, especially eL15, uL29 and eL13 (encoding RPL15, RPL35 and RPL13, respectively) were highly enriched in circulating tumour cells (CTCs). In particular, over-expression of RPL15 in CTCs promoted global protein synthesis and metastasis in the lung in mice. In addition, patient CTCs with the highest expression levels of RPs were associated with the worst clinical outcomes. Other factors that were highly correlated with poor patient survival rates included genes that encode for the eIFs and mTOR. A combinational therapy using omacetaxine, an FDA-approved translational elongation inhibitor, and palbociclib, a CDK4/6 inhibitor approved for BC treatment, effectively prevented the metastasis of RPL15-enriched CTCs [168]. Similar to these findings in BC, Rebello et al. [169] found that the combination of two drugs (CX-5461 and CX-6258), two orally available agents which inhibit RNA polymerase I transcription and the PIM kinase respectively, exerted strong anti-proliferative and pro-apoptotic effects on both high c-Myc and PTEN-null murine prostate tumour models. A later study demonstrated that the CX-5461/CX-6258 combination therapy effectively inhibited growth, increased DNA damage responses and suppressed mTOR signalling in four distinct PC PDX models obtained from CRPC patients resistant to second-generation ADT agents, including abiraterone and/or enzalutamide [170]. Taken together, these studies highlighted a promising future of targeting the translation machinery, especially the ribosomes, for BC/PC treatments.
6.3. Targeting the UPR
To cope with the abnormally high protein synthesis demand under a nutrient deprived, hypoxic and acidotic microenvironment, it is critical for cancer cells to maintain energetic balance and proteostasis, for example, by triggering ISR or activating AMPK. Nguyen et al. recently discovered that, unexpectedly, the co-existence of PTEN deletion and high c-Myc expression (cPTENfl/fl × MycTg) in murine prostates resulted in dampening of the increase in global protein synthesis (compared to PTEN-null or high c-Myc alone), which occurred concomitantly with an induction of phosphorylated PERK, one of the four eIF2α kinases [42]. In this study, the ablation of PERK rescued the reduction in protein synthesis in cPTENfl/fl × MycTg mice [171]. As such, one can expect that ISR inhibition may lead to devastating consequences for PC cells but have a limited toxic effect on normal cells. Indeed, the PERK inhibitor ISRIB [172] induced apoptosis in PC cells, reduced tumour volume, and prolonged the survival rates of cPTENfl/fl × MycTg mice, as well as of mice bearing metastatic CRPC PDXs [171]. However, although alleviation of ISR exerted strong anti-tumour effects in the cPTENfl/fl × MycTg GEMM [171], it is not clear whether the dampening of protein synthesis rates in those mice reflects the induction of ISR.
An alternative therapeutic strategy for PC would be to target the IRE1/XBP1-axis of UPR. Toyocamycin, an IRE1α inhibitor, effectively diminished LNCaP or VCaP xenograft growth in vivo [127]. Similarly, MKC8866, another specific inhibitor to IRE1α RNase activities, potentiated the activity of conventional AR-targeted therapies (enzalutamide, abiraterone and cabazitaxel) in mice bearing VCaP xenografts [129]. Taken together, these studies [127,129] thus provide a molecular basis for targeting the IRE1/XBP1 arm of UPR for PC treatment.
7. Drawbacks and New Hopes in Treatments Targeting mTOR in BC and PC
7.1. Rationale for Application of mTORis as an Anti-BC/PC Treatment
As described above, there are various potentially druggable targets within the translation machinery that can be exploited for anti-BC/PC therapies. Among them, mTORis are one of the most promising strategies. This is mainly due to a suite of unique features associated with mTOR. Firstly, mTOR is a protein kinase initially discovered to be the sole target of a highly specific natural compound, rapamycin, whereas most of the early identified kinase inhibitors possess severe off-target effects [153,173,174]. Secondly, targeting upstream signalling components such as PI3K, AKT, Raf, MEK, etc., will unavoidably trigger stronger side effects since they also regulate other essential cell processes, whereas mTOR acts downstream of these signalling pathways and its best established role so far is the homeostatic control of protein synthesis. It has been shown that the ratio of eIF4E to 4EBPs correlates with response to mTORC active site kinase inhibitors [175]. Lastly, mTORis, especially rapamycin analogues (rapalogs), have been in development for >20 years; indeed, some have been approved by the FDA for clinical use against certain types of cancer (e.g., kidney, renal and pancreatic) since the early 2000s.
7.2. Early Trials Using Rapalogs in BC/PC
Rapalogs were initially successful in early clinical trials against BC [176,177,178], as evidenced by reasonable anti-tumour activities, well tolerated toxicity and prolonged PFS in patients (Table 2, also see [179] for an early review). In 2012, a milestone phase III study (BOLERO-2) performed by Baselga et al. proved that everolimus (a rapalog, 10 mg daily) combined with exemestane (an AI, 25 mg daily) could improve PFS in ER+ BC compared to exemestane treatment alone [180]. Accordingly, the FDA approved the use of this combinational therapy for ER+ BC treatment. However, a follow-up study of BOLERO-2 failed to demonstrate that everolimus plus exemestane improved PFS [181]. Another similar combination, temsirolimus (rapalog) plus letrozole (AI), also failed to provide therapeutic benefits to BC patients [182]. Although a recent study showed that the addition of everolimus to fulvestrant (a selective ER degrader) treatment prolonged PFS in HR+/EGFR2− patients, this was associated with an increase in the occurrence of adverse events [183]. Accordingly, interest in rapalogs as a treatment for BC patients has waned considerably and they are no longer being tested in clinical trials.
Trials of rapalogs in PC, primarily in the CRPC setting, have also suffered setbacks recently (Table 2). Despite modest improvement of PFS in PTEN-null CRPC patients with everolimus treatment alone [187], most of the rapalog monotherapy regimens failed to show therapeutic benefits in PC patients [186,191,192], perhaps due to the fact that rapamycin can stimulate AR’s transcriptional activity [120]. A combination of everolimus and the AR antagonist bicalutamide in a phase II trial was unable to reach the primary endpoint due to low anti-tumour activity [185]. Similarly, temsirolimus and bevacizumab (vascular endothelial growth factor A inhibitor) treatment together not only failed to provide any improvement in terms of clinical outcome but also produced severe side effects in patients [192]. Moreover, two trials involving the combination of dual PI3K/mTOR inhibitor BEZ235 with abiraterone were also withdrawn during the early stages due to high levels of toxicity [188,189]. It has been well known that rapamycin does not inhibit mTORC1 completely [193] and may not affect mTORC2 [121], whereas the ATP-competitive mTORis can simultaneously and completely blunt the activation of wild-type mTOR-associated mTORC1 and 2 [194]. INK128, an ATP-competitive mTORi, was initially identified as a strong candidate for PC treatment due to prominent anti-tumour activity against PC progression in the cPTENfl/fl GEMM [156]. Unfortunately, a recent study found that INK128 exerted strong side-effects (grade 3 dyspnea and maculopapular rash) in CRPC patients, and thus the trial was also forced to be discontinued [190].
Some recent studies have unravelled mechanisms behind the resistance of BC/PC to mTOR inhibition. Reactivation of the PI3K/AKT pathway is a classic feedback mechanism driven by the rapalogs [83,122]. Yang et al. discovered that the induction of PI3K/AKT stimulation by everolimus in ER+ BC cells was mediated by the ER, which also required the transactivation of insulin receptor/IGF1R and insulin receptor substrates [195]. Additionally, screening of secreted proteins from the ER+ BC tumour microenvironment has revealed that the induction of fibroblast growth factor 2 was primarily responsible for conferring resistance to the BC cells against PI3K/mTORis [196].
In short, lessons from clinical trials of mTORis in BC and PC and associated mechanistic studies have established a challenging paradigm: the first generation of mTORis usually do not provide further clinical benefits on top of existing anti-BC/PC therapies, likely because they are relatively “weak” inhibitors against the mTORCs [193]; whereas “stronger” drugs such as the dual PI3K/mTOR or the ATP-competitive inhibitors are often associated with severe adverse effects on patients, since they also affect physiological functions of non-malignant cells [197] and may trigger feedback mechanisms to activate other oncogenic pathways [83]. The question of how to leverage mTORis such that significant anti-tumour activity is achieved with tolerable side effects remains outstanding.
7.3. The Future of mTOR Inhibition as a Therapy for Breast and Prostate Cancer
Despite some unfavourable outcomes from those mTORi-related trials, more recent studies provide new hope. For example, fulvestrant-resistant ER+/HER2 kinase domain mutant (i.e., G309A, L755S and V777L) cells possess high level of PI3K/mTORC1 activity, as evidenced by increased levels of phosphorylated AKT and RP S6, respectively, and Everolimus was able to resensitize those cells to fulvestrant or estrogen deprivation [198]. In addition, an alternative approach would be to test other mTORis, especially beyond the rapalogs. GDC-0084, a dual PI3K/mTOR inhibitor designed to cross the blood-brain barrier [199], was recently shown to exert growth inhibitory effects on BC brain metastatic cell lines bearing PIK3CA mutations both in vitro and in vivo [200]. Earlier this year, La Manna et al. also reported the first attempt of using RapaLink-1 against PC; exposure of two bone-metastatic PC PDX models to RapaLink-1 strongly reduced their cancer stem cell features and prevented tumour growth in vivo [201]. However, although results from these preclinical studies appear promising, patients receiving these agents are more likely to experience stronger side effects than those who were administered with the rapalogs (see Section 7.2). Therefore, when it comes to future clinical trials, it is of utmost importance to ensure the safety aspect of these drugs does not compromise their potential as novel treatments.
8. Conclusions, Future Perspectives and Outstanding Questions
While ER and AR are clear drivers of neoplastic growth, our understanding of their involvement in reshaping the translatome during cancer progression remains incomplete. We propose that the most pressing outstanding questions for basic science and for clinical translation are:
- -
- How is translation modulated during adaption to anti-estrogen/androgen therapies and in response to direct alterations to AR/ER (e.g., mutations and truncations)?
- -
- Why do different ER/AR ligands exert distinct downstream effects on protein synthesis?
- -
- How do cancer cells balance the need for increased protein synthesis while avoiding UPR?
- -
- Will single-cell technologies address key mechanistic questions related to the interplay between AR/ER signalling and protein synthesis?
- -
- Does the cytotoxicity of drugs that target the translation modulators (e.g., mTORis) primarily rely on their effect on protein synthesis?
- -
- How do we identify BC/PC patients who are likely to benefit from protein synthesis-targeted therapies? Can we use translation-related genetic profiles to tailor personalized treatment regimens? For example, patients harbouring PTEN/PI3K/mTOR mutations would be expected to gain the most benefit from PI3K/mTOR inhibitors.
Funding
E.P.K. and L.F. are supported by Victorian Cancer Agency Fellowships awarded by the Department of Health and Human Services (ECRF20018 and MCRF16007). LF is also supported by a grant from the National Health and Medical Research Council, Australia (APP1141339) and the Victorian Medical Acceleration Fund (VMRAF). L.A.S. is supported by: a Principal Cancer Research Fellowship awarded by Cancer Council’s Beat Cancer project on behalf of its donors, the South Australian Government through the Department of Health, and the Australian Government through the Medical Research Future Fund; and grants from the Cancer Council of South Australia (Beat Cancer Project Grant 1185012), the Movember Foundation and the Prostate Cancer Foundation of Australia (Movember Revolutionary Team Award MRTA-3) and The Hospital Research Foundation (C-PJ-126-Exper-2019).
Acknowledgments
The authors would like to thank Ivan Topisirovic for critical reading of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Risbridger, G.P.; Davis, I.D.; Birrell, S.N.; Tilley, W.D. Breast and prostate cancer: More similar than different. Nat. Rev. Cancer 2010, 10, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Manavathi, B.; Samanthapudi, V.S.; Gajulapalli, V.N. Estrogen receptor coregulators and pioneer factors: The orchestrators of mammary gland cell fate and development. Front. Cell Dev. Biol. 2014, 2, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obinata, D.; Takayama, K.; Takahashi, S.; Inoue, S. Crosstalk of the Androgen Receptor with Transcriptional Collaborators: Potential Therapeutic Targets for Castration-Resistant Prostate Cancer. Cancers 2017, 9, 22. [Google Scholar] [CrossRef] [Green Version]
- Karamouzis, M.V.; Papavassiliou, K.A.; Adamopoulos, C.; Papavassiliou, A.G. Targeting Androgen/Estrogen Receptors Crosstalk in Cancer. Trends Cancer 2016, 2, 35–48. [Google Scholar] [CrossRef]
- Pomerantz, M.M.; Qiu, X.; Zhu, Y.; Takeda, D.Y.; Pan, W.; Baca, S.C.; Gusev, A.; Korthauer, K.D.; Severson, T.M.; Ha, G.; et al. Prostate cancer reactivates developmental epigenomic programs during metastatic progression. Nat. Genet. 2020, 52, 790–799. [Google Scholar] [CrossRef]
- Kovacs, T.; Szabo-Meleg, E.; Abraham, I.M. Estradiol-Induced Epigenetically Mediated Mechanisms and Regulation of Gene Expression. Int. J. Mol. Sci. 2020, 21, 3177. [Google Scholar] [CrossRef]
- Coutinho, I.; Day, T.K.; Tilley, W.D.; Selth, L.A. Androgen receptor signaling in castration-resistant prostate cancer: A lesson in persistence. Endocr. Relat. Cancer 2016, 23, T179–T197. [Google Scholar] [CrossRef]
- Belachew, E.B.; Sewasew, D.T. Molecular Mechanisms of Endocrine Resistance in Estrogen-Positive Breast Cancer. Front. Endocrinol. 2021, 12, 599586. [Google Scholar] [CrossRef]
- Dobrzycka, K.M.; Townson, S.M.; Jiang, S.; Oesterreich, S. Estrogen receptor corepressors—A role in human breast cancer? Endocr. Relat. Cancer 2003, 10, 517–536. [Google Scholar] [CrossRef] [Green Version]
- Baniahmad, A. Nuclear hormone receptor co-repressors. J. Steroid Biochem. Mol. Biol. 2005, 93, 89–97. [Google Scholar] [CrossRef]
- Groner, A.C.; Brown, M. Role of steroid receptor and coregulator mutations in hormone-dependent cancers. J. Clin. Investig. 2017, 127, 1126–1135. [Google Scholar] [CrossRef] [Green Version]
- Schuller, A.P.; Green, R. Roadblocks and resolutions in eukaryotic translation. Nat. Rev. Mol. Cell Biol. 2018, 19, 526–541. [Google Scholar] [CrossRef]
- Kong, J.; Lasko, P. Translational control in cellular and developmental processes. Nat. Rev. Genet. 2012, 13, 383–394. [Google Scholar] [CrossRef]
- Truitt, M.L.; Ruggero, D. New frontiers in translational control of the cancer genome. Nat. Rev. Cancer 2016, 16, 288–304. [Google Scholar] [CrossRef] [Green Version]
- Tahmasebi, S.; Khoutorsky, A.; Mathews, M.B.; Sonenberg, N. Translation deregulation in human disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 791–807. [Google Scholar] [CrossRef]
- Potter, C.J.; Pedraza, L.G.; Xu, T. Akt regulates growth by directly phosphorylating Tsc2. Nat. Cell Biol. 2002, 4, 658–665. [Google Scholar] [CrossRef]
- Manning, B.D.; Tee, A.R.; Logsdon, M.N.; Blenis, J.; Cantley, L.C. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol. Cell 2002, 10, 151–162. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef]
- Ma, L.; Chen, Z.; Erdjument-Bromage, H.; Tempst, P.; Pandolfi, P.P. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 2005, 121, 179–193. [Google Scholar] [CrossRef] [Green Version]
- Roux, P.P.; Ballif, B.A.; Anjum, R.; Gygi, S.P.; Blenis, J. Tumor-Promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc. Natl. Acad. Sci. USA 2004, 101, 13489–13494. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Mossmann, D.; Park, S.; Hall, M.N. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 2018, 18, 744–757. [Google Scholar] [CrossRef] [PubMed]
- Bhat, M.; Robichaud, N.; Hulea, L.; Sonenberg, N.; Pelletier, J.; Topisirovic, I. Targeting the translation machinery in cancer. Nat. Rev. Drug Discov. 2015, 14, 261–278. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Kuo, C.J.; Crabtree, G.R.; Blenis, J. Rapamycin-FKBP specifically blocks growth-dependent activation of and signaling by the 70 kd S6 protein kinases. Cell 1992, 69, 1227–1236. [Google Scholar] [CrossRef]
- Pause, A.; Belsham, G.J.; Gingras, A.C.; Donze, O.; Lin, T.A.; Lawrence, J.C., Jr.; Sonenberg, N. Insulin-Dependent stimulation of protein synthesis by phosphorylation of a regulator of 5′-cap function. Nature 1994, 371, 762–767. [Google Scholar] [CrossRef]
- Pelletier, J.; Sonenberg, N. The Organizing Principles of Eukaryotic Ribosome Recruitment. Annu. Rev. Biochem. 2019, 88, 307–335. [Google Scholar] [CrossRef]
- Ueda, T.; Watanabe-Fukunaga, R.; Fukuyama, H.; Nagata, S.; Fukunaga, R. Mnk2 and Mnk1 are essential for constitutive and inducible phosphorylation of eukaryotic initiation factor 4E but not for cell growth or development. Mol. Cell Biol. 2004, 24, 6539–6549. [Google Scholar] [CrossRef] [Green Version]
- Roux, P.P.; Topisirovic, I. Signaling Pathways Involved in the Regulation of mRNA Translation. Mol. Cell Biol. 2018, 38. [Google Scholar] [CrossRef] [Green Version]
- Zindy, P.; Berge, Y.; Allal, B.; Filleron, T.; Pierredon, S.; Cammas, A.; Beck, S.; Mhamdi, L.; Fan, L.; Favre, G.; et al. Formation of the eIF4F translation-initiation complex determines sensitivity to anticancer drugs targeting the EGFR and HER2 receptors. Cancer Res. 2011, 71, 4068–4073. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Horn, J.L.; Banda, K.; Goodman, A.Z.; Lim, Y.; Jana, S.; Arora, S.; Germanos, A.A.; Wen, L.; Hardin, W.R.; et al. The androgen receptor regulates a druggable translational regulon in advanced prostate cancer. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Knight, J.R.P.; Garland, G.; Poyry, T.; Mead, E.; Vlahov, N.; Sfakianos, A.; Grosso, S.; De-Lima-Hedayioglu, F.; Mallucci, G.R.; von der Haar, T.; et al. Control of translation elongation in health and disease. Dis. Model. Mech. 2020, 13. [Google Scholar] [CrossRef] [Green Version]
- Proud, C.G. Regulation and roles of elongation factor 2 kinase. Biochem. Soc. Trans. 2015, 43, 328–332. [Google Scholar] [CrossRef]
- Carlberg, U.; Nilsson, A.; Nygard, O. Functional properties of phosphorylated elongation factor 2. Eur. J. Biochem. 1990, 191, 639–645. [Google Scholar] [CrossRef]
- Ryazanov, A.G.; Shestakova, E.A.; Natapov, P.G. Phosphorylation of elongation factor 2 by EF-2 kinase affects rate of translation. Nature 1988, 334, 170–173. [Google Scholar] [CrossRef]
- Ryazanov, A.G.; Natapov, P.G.; Shestakova, E.A.; Severin, F.F.; Spirin, A.S. Phosphorylation of the elongation factor 2: The fifth Ca2+/calmodulin-dependent system of protein phosphorylation. Biochimie 1988, 70, 619–626. [Google Scholar] [CrossRef]
- Moazed, D.; Noller, H.F. Intermediate states in the movement of transfer RNA in the ribosome. Nature 1989, 342, 142–148. [Google Scholar] [CrossRef]
- Wang, X.; Regufe da Mota, S.; Liu, R.; Moore, C.E.; Xie, J.; Lanucara, F.; Agarwala, U.; Pyr Dit Ruys, S.; Vertommen, D.; Rider, M.H.; et al. Eukaryotic elongation factor 2 kinase activity is controlled by multiple inputs from oncogenic signaling. Mol. Cell Biol. 2014, 34, 4088–4103. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, W.; Williams, M.; Terada, N.; Alessi, D.R.; Proud, C.G. Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J. 2001, 20, 4370–4379. [Google Scholar] [CrossRef]
- Horman, S.; Beauloye, C.; Vertommen, D.; Vanoverschelde, J.L.; Hue, L.; Rider, M.H. Myocardial ischemia and increased heart work modulate the phosphorylation state of eukaryotic elongation factor-2. J. Biol. Chem. 2003, 278, 41970–41976. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- Wang, M.; Kaufman, R.J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 2014, 14, 581–597. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Dever, T.E.; Feng, L.; Wek, R.C.; Cigan, A.M.; Donahue, T.F.; Hinnebusch, A.G. Phosphorylation of initiation factor 2 alpha by protein kinase GCN2 mediates gene-specific translational control of GCN4 in yeast. Cell 1992, 68, 585–596. [Google Scholar] [CrossRef]
- Ranu, R.S.; London, I.M. Regulation of protein synthesis in rabbit reticulocyte lysates: Additional initiation factor required for formation of ternary complex (eIF-2.GTP.Met-tRNAf) and demonstration of inhibitory effect of heme-regulated protein kinase. Proc. Natl. Acad. Sci. USA 1979, 76, 1079–1083. [Google Scholar] [CrossRef] [Green Version]
- Meurs, E.F.; Watanabe, Y.; Kadereit, S.; Barber, G.N.; Katze, M.G.; Chong, K.; Williams, B.R.; Hovanessian, A.G. Constitutive expression of human double-stranded RNA-activated p68 kinase in murine cells mediates phosphorylation of eukaryotic initiation factor 2 and partial resistance to encephalomyocarditis virus growth. J. Virol. 1992, 66, 5805–5814. [Google Scholar] [CrossRef] [Green Version]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Costa-Mattioli, M.; Walter, P. The integrated stress response: From mechanism to disease. Science 2020, 368. [Google Scholar] [CrossRef]
- Martincorena, I.; Raine, K.M.; Gerstung, M.; Dawson, K.J.; Haase, K.; Van Loo, P.; Davies, H.; Stratton, M.R.; Campbell, P.J. Universal Patterns of Selection in Cancer and Somatic Tissues. Cell 2017, 171, 1029–1041.e21. [Google Scholar] [CrossRef]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385.e18. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Warrell, J.; Li, S.; McGillivray, P.D.; Meyerson, W.; Salichos, L.; Harmanci, A.; Martinez-Fundichely, A.; Chan, C.W.Y.; Nielsen, M.M.; et al. Passenger Mutations in More Than 2500 Cancer Genomes: Overall Molecular Functional Impact and Consequences. Cell 2020, 180, 915–927.e16. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forbes, S.A.; Beare, D.; Boutselakis, H.; Bamford, S.; Bindal, N.; Tate, J.; Cole, C.G.; Ward, S.; Dawson, E.; Ponting, L.; et al. COSMIC: Somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017, 45, D777–D783. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Kusnadi, E.P.; Trigos, A.S.; Cullinane, C.; Goode, D.L.; Larsson, O.; Devlin, J.R.; Chan, K.T.; De Souza, D.P.; McConville, M.J.; McArthur, G.A.; et al. Reprogrammed mRNA translation drives resistance to therapeutic targeting of ribosome biogenesis. EMBO J. 2020, 39, e105111. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, T.H. Isotopic studies on estrogen-induced accelerations of ribonucleic acid and protein synthesis. Proc. Natl. Acad. Sci. USA 1963, 49, 373–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noteboom, W.D.; Gorski, J. An Early Effect of Estrogen on Protein Synthesis. Proc. Natl. Acad. Sci. USA 1963, 50, 250–255. [Google Scholar] [CrossRef] [Green Version]
- Mueller, G.C.; Gorski, J.; Aizawa, Y. The role of protein synthesis in early estrogen action. Proc. Natl. Acad. Sci. USA 1961, 47, 164–169. [Google Scholar] [CrossRef] [Green Version]
- Schjeide, O.A.; Binz, S.; Ragan, N. Estrogen-Induced serum protein synthesis in the liver of the chicken embryo. Growth 1960, 24, 401–410. [Google Scholar]
- Schjeide, O.A.; Simons, S.; Ragan, N. Effect of x-irradiation on estrogen-induced protein synthesis in the chick. Growth 1959, 23, 273–290. [Google Scholar]
- Pennequin, P.; Robins, D.M.; Schimke, R.T. Regulation of translation of ovalbumin messenger RNA by estrogens and progesterone in oviduct of withdrawn chicks. Eur. J. Biochem. 1978, 90, 51–58. [Google Scholar] [CrossRef]
- Lu, Q.; Pallas, D.C.; Surks, H.K.; Baur, W.E.; Mendelsohn, M.E.; Karas, R.H. Striatin assembles a membrane signaling complex necessary for rapid, nongenomic activation of endothelial NO synthase by estrogen receptor alpha. Proc. Natl. Acad. Sci. USA 2004, 101, 17126–17131. [Google Scholar] [CrossRef] [Green Version]
- Adlanmerini, M.; Solinhac, R.; Abot, A.; Fabre, A.; Raymond-Letron, I.; Guihot, A.L.; Boudou, F.; Sautier, L.; Vessieres, E.; Kim, S.H.; et al. Mutation of the palmitoylation site of estrogen receptor alpha in vivo reveals tissue-specific roles for membrane versus nuclear actions. Proc. Natl. Acad. Sci. USA 2014, 111, E283–E290. [Google Scholar] [CrossRef] [Green Version]
- Razandi, M.; Pedram, A.; Park, S.T.; Levin, E.R. Proximal events in signaling by plasma membrane estrogen receptors. J. Biol. Chem. 2003, 278, 2701–2712. [Google Scholar] [CrossRef] [Green Version]
- Song, R.X.; Zhang, Z.; Chen, Y.; Bao, Y.; Santen, R.J. Estrogen signaling via a linear pathway involving insulin-like growth factor I receptor, matrix metalloproteinases, and epidermal growth factor receptor to activate mitogen-activated protein kinase in MCF-7 breast cancer cells. Endocrinology 2007, 148, 4091–4101. [Google Scholar] [CrossRef] [Green Version]
- Bartucci, M.; Morelli, C.; Mauro, L.; Ando, S.; Surmacz, E. Differential insulin-like growth factor I receptor signaling and function in estrogen receptor (ER)-positive MCF-7 and ER-negative MDA-MB-231 breast cancer cells. Cancer Res. 2001, 61, 6747–6754. [Google Scholar]
- Manavathi, B.; Acconcia, F.; Rayala, S.K.; Kumar, R. An inherent role of microtubule network in the action of nuclear receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 15981–15986. [Google Scholar] [CrossRef] [Green Version]
- Hammes, S.R.; Levin, E.R. Extranuclear steroid receptors: Nature and actions. Endocr. Rev. 2007, 28, 726–741. [Google Scholar] [CrossRef]
- Chantalat, E.; Boudou, F.; Laurell, H.; Palierne, G.; Houtman, R.; Melchers, D.; Rochaix, P.; Filleron, T.; Stella, A.; Burlet-Schiltz, O.; et al. The AF-1-deficient estrogen receptor ERalpha46 isoform is frequently expressed in human breast tumors. Breast Cancer Res. 2016, 18, 123. [Google Scholar] [CrossRef] [Green Version]
- Silvera, D.; Formenti, S.C.; Schneider, R.J. Translational control in cancer. Nat. Rev. Cancer 2010, 10, 254–266. [Google Scholar] [CrossRef]
- Paul, M.R.; Pan, T.C.; Pant, D.K.; Shih, N.N.; Chen, Y.; Harvey, K.L.; Solomon, A.; Lieberman, D.; Morrissette, J.J.; Soucier-Ernst, D.; et al. Genomic landscape of metastatic breast cancer identifies preferentially dysregulated pathways and targets. J. Clin. Investig. 2020, 130, 4252–4265. [Google Scholar] [CrossRef] [PubMed]
- Adamo, B.; Deal, A.M.; Burrows, E.; Geradts, J.; Hamilton, E.; Blackwell, K.L.; Livasy, C.; Fritchie, K.; Prat, A.; Harrell, J.C.; et al. Phosphatidylinositol 3-kinase pathway activation in breast cancer brain metastases. Breast Cancer Res. 2011, 13, R125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brastianos, P.K.; Carter, S.L.; Santagata, S.; Cahill, D.P.; Taylor-Weiner, A.; Jones, R.T.; Van Allen, E.M.; Lawrence, M.S.; Horowitz, P.M.; Cibulskis, K.; et al. Genomic Characterization of Brain Metastases Reveals Branched Evolution and Potential Therapeutic Targets. Cancer Discov. 2015, 5, 1164–1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunus, J.M.; Quinn, M.C.; Patch, A.M.; Pearson, J.V.; Bailey, P.J.; Nones, K.; McCart Reed, A.E.; Miller, D.; Wilson, P.J.; Al-Ejeh, F.; et al. Integrated genomic and transcriptomic analysis of human brain metastases identifies alterations of potential clinical significance. J. Pathol. 2015, 237, 363–378. [Google Scholar] [CrossRef]
- Yu, J.; Henske, E.P. Estrogen-Induced activation of mammalian target of rapamycin is mediated via tuberin and the small GTPase Ras homologue enriched in brain. Cancer Res. 2006, 66, 9461–9466. [Google Scholar] [CrossRef] [Green Version]
- Martin, E.C.; Rhodes, L.V.; Elliott, S.; Krebs, A.E.; Nephew, K.P.; Flemington, E.K.; Collins-Burow, B.M.; Burow, M.E. microRNA regulation of mammalian target of rapamycin expression and activity controls estrogen receptor function and RAD001 sensitivity. Mol. Cancer 2014, 13, 229. [Google Scholar] [CrossRef] [Green Version]
- Andruska, N.D.; Zheng, X.; Yang, X.; Mao, C.; Cherian, M.M.; Mahapatra, L.; Helferich, W.G.; Shapiro, D.J. Estrogen receptor alpha inhibitor activates the unfolded protein response, blocks protein synthesis, and induces tumor regression. Proc. Natl. Acad. Sci. USA 2015, 112, 4737–4742. [Google Scholar] [CrossRef] [Green Version]
- Cuesta, R.; Gritsenko, M.A.; Petyuk, V.A.; Shukla, A.K.; Tsai, C.F.; Liu, T.; McDermott, J.E.; Holz, M.K. Phosphoproteome Analysis Reveals Estrogen-ER Pathway as a Modulator of mTOR Activity Via DEPTOR. Mol. Cell. Proteom. 2019, 18, 1607–1618. [Google Scholar] [CrossRef]
- Peterson, T.R.; Laplante, M.; Thoreen, C.C.; Sancak, Y.; Kang, S.A.; Kuehl, W.M.; Gray, N.S.; Sabatini, D.M. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 2009, 137, 873–886. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Xiong, X.; Cui, D.; Yang, F.; Wei, D.; Li, H.; Shu, J.; Bi, Y.; Dai, X.; Gong, L.; et al. DEPTOR is an in vivo tumor suppressor that inhibits prostate tumorigenesis via the inactivation of mTORC1/2 signals. Oncogene 2020, 39, 1557–1571. [Google Scholar] [CrossRef] [Green Version]
- Takizawa, I.; Lawrence, M.G.; Balanathan, P.; Rebello, R.; Pearson, H.B.; Garg, E.; Pedersen, J.; Pouliot, N.; Nadon, R.; Watt, M.J.; et al. Estrogen receptor alpha drives proliferation in PTEN-deficient prostate carcinoma by stimulating survival signaling, MYC expression and altering glucose sensitivity. Oncotarget 2015, 6, 604–616. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Proud, C.G. Crosstalk between mTOR complexes. Nat. Cell Biol. 2013, 15, 1263–1265. [Google Scholar] [CrossRef]
- Xie, J.; Proud, C.G. Signaling crosstalk between the mTOR complexes. Translation 2014, 2, e28174. [Google Scholar] [CrossRef]
- Cuesta, R.; Berman, A.Y.; Alayev, A.; Holz, M.K. Estrogen receptor alpha promotes protein synthesis by fine-tuning the expression of the eukaryotic translation initiation factor 3 subunit f (eIF3f). J. Biol. Chem. 2019, 294, 2267–2278. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Pan, X.; Hershey, J.W. Individual overexpression of five subunits of human translation initiation factor eIF3 promotes malignant transformation of immortal fibroblast cells. J. Biol. Chem. 2007, 282, 5790–5800. [Google Scholar] [CrossRef] [Green Version]
- Martineau, Y.; Wang, X.; Alain, T.; Petroulakis, E.; Shahbazian, D.; Fabre, B.; Bousquet-Dubouch, M.P.; Monsarrat, B.; Pyronnet, S.; Sonenberg, N. Control of Paip1-eukayrotic translation initiation factor 3 interaction by amino acids through S6 kinase. Mol. Cell. Biol. 2014, 34, 1046–1053. [Google Scholar] [CrossRef] [Green Version]
- Holz, M.K.; Ballif, B.A.; Gygi, S.P.; Blenis, J. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell 2005, 123, 569–580. [Google Scholar] [CrossRef] [Green Version]
- Cenik, C.; Cenik, E.S.; Byeon, G.W.; Grubert, F.; Candille, S.I.; Spacek, D.; Alsallakh, B.; Tilgner, H.; Araya, C.L.; Tang, H.; et al. Integrative analysis of RNA, translation, and protein levels reveals distinct regulatory variation across humans. Genome Res. 2015, 25, 1610–1621. [Google Scholar] [CrossRef] [Green Version]
- Lorent, J.; Kusnadi, E.P.; van Hoef, V.; Rebello, R.J.; Leibovitch, M.; Ristau, J.; Chen, S.; Lawrence, M.G.; Szkop, K.J.; Samreen, B.; et al. Translational offsetting as a mode of estrogen receptor alpha-dependent regulation of gene expression. EMBO J. 2019, 38, e101323. [Google Scholar] [CrossRef] [PubMed]
- Castellano, L.; Giamas, G.; Jacob, J.; Coombes, R.C.; Lucchesi, W.; Thiruchelvam, P.; Barton, G.; Jiao, L.R.; Wait, R.; Waxman, J.; et al. The estrogen receptor-alpha-induced microRNA signature regulates itself and its transcriptional response. Proc. Natl. Acad. Sci. USA 2009, 106, 15732–15737. [Google Scholar] [CrossRef] [Green Version]
- Maillot, G.; Lacroix-Triki, M.; Pierredon, S.; Gratadou, L.; Schmidt, S.; Benes, V.; Roche, H.; Dalenc, F.; Auboeuf, D.; Millevoi, S.; et al. Widespread estrogen-dependent repression of micrornas involved in breast tumor cell growth. Cancer Res. 2009, 69, 8332–8340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, S.T.; Westerling, T.; Brown, M. Loss of estrogen-regulated microRNA expression increases HER2 signaling and is prognostic of poor outcome in luminal breast cancer. Cancer Res. 2015, 75, 436–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapino, F.; Delaunay, S.; Rambow, F.; Zhou, Z.; Tharun, L.; De Tullio, P.; Sin, O.; Shostak, K.; Schmitz, S.; Piepers, J.; et al. Codon-specific translation reprogramming promotes resistance to targeted therapy. Nature 2018, 558, 605–609. [Google Scholar] [CrossRef] [PubMed]
- Delaunay, S.; Rapino, F.; Tharun, L.; Zhou, Z.; Heukamp, L.; Termathe, M.; Shostak, K.; Klevernic, I.; Florin, A.; Desmecht, H.; et al. Elp3 links tRNA modification to IRES-dependent translation of LEF1 to sustain metastasis in breast cancer. J. Exp. Med. 2016, 213, 2503–2523. [Google Scholar] [CrossRef] [Green Version]
- Ladang, A.; Rapino, F.; Heukamp, L.C.; Tharun, L.; Shostak, K.; Hermand, D.; Delaunay, S.; Klevernic, I.; Jiang, Z.; Jacques, N.; et al. Elp3 drives Wnt-dependent tumor initiation and regeneration in the intestine. J. Exp. Med. 2015, 212, 2057–2075. [Google Scholar] [CrossRef] [Green Version]
- Schalm, S.S.; Blenis, J. Identification of a conserved motif required for mTOR signaling. Curr. Biol. 2002, 12, 632–639. [Google Scholar] [CrossRef] [Green Version]
- Alayev, A.; Salamon, R.S.; Berger, S.M.; Schwartz, N.S.; Cuesta, R.; Snyder, R.B.; Holz, M.K. mTORC1 directly phosphorylates and activates ERalpha upon estrogen stimulation. Oncogene 2016, 35, 3535–3543. [Google Scholar] [CrossRef]
- Becker, M.A.; Ibrahim, Y.H.; Cui, X.; Lee, A.V.; Yee, D. The IGF pathway regulates ERalpha through a S6K1-dependent mechanism in breast cancer cells. Mol. Endocrinol. 2011, 25, 516–528. [Google Scholar] [CrossRef] [Green Version]
- Meric-Bernstam, F.; Chen, H.; Akcakanat, A.; Do, K.A.; Lluch, A.; Hennessy, B.T.; Hortobagyi, G.N.; Mills, G.B.; Gonzalez-Angulo, A. Aberrations in translational regulation are associated with poor prognosis in hormone receptor-positive breast cancer. Breast Cancer Res. 2012, 14, R138. [Google Scholar] [CrossRef] [Green Version]
- Brunn, G.J.; Hudson, C.C.; Sekulic, A.; Williams, J.M.; Hosoi, H.; Houghton, P.J.; Lawrence, J.C., Jr.; Abraham, R.T. Phosphorylation of the translational repressor PHAS-I by the mammalian target of rapamycin. Science 1997, 277, 99–101. [Google Scholar] [CrossRef]
- Rutkovsky, A.C.; Yeh, E.S.; Guest, S.T.; Findlay, V.J.; Muise-Helmericks, R.C.; Armeson, K.; Ethier, S.P. Eukaryotic initiation factor 4E-binding protein as an oncogene in breast cancer. BMC Cancer 2019, 19, 491. [Google Scholar] [CrossRef]
- Yanagiya, A.; Suyama, E.; Adachi, H.; Svitkin, Y.V.; Aza-Blanc, P.; Imataka, H.; Mikami, S.; Martineau, Y.; Ronai, Z.A.; Sonenberg, N. Translational homeostasis via the mRNA cap-binding protein, eIF4E. Mol. Cell 2012, 46, 847–858. [Google Scholar] [CrossRef] [Green Version]
- Parris, T.Z.; Ronnerman, E.W.; Engqvist, H.; Biermann, J.; Truve, K.; Nemes, S.; Forssell-Aronsson, E.; Solinas, G.; Kovacs, A.; Karlsson, P.; et al. Genome-Wide multi-omics profiling of the 8p11-p12 amplicon in breast carcinoma. Oncotarget 2018, 9, 24140–24154. [Google Scholar] [CrossRef] [Green Version]
- Lasorella, A.; Sanson, M.; Iavarone, A. FGFR-TACC gene fusions in human glioma. Neuro Oncol. 2017, 19, 475–483. [Google Scholar] [CrossRef] [Green Version]
- Brennan, C. FGFR-TACC approaches the first turn in the race for targetable GBM mutations. Neuro Oncol. 2017, 19, 461–462. [Google Scholar] [CrossRef] [Green Version]
- Williams-Ashman, H.G. Androgenic control of nucleic acid and protein synthesis in male accessory genital organs. J. Cell. Physiol. 1965, 66 (Suppl. 1), 111–124. [Google Scholar] [CrossRef]
- Liang, T.; Liao, S. A very rapid effect of androgen on initiation of protein synthesis in prostate. Proc. Natl. Acad. Sci. USA 1975, 72, 706–709. [Google Scholar] [CrossRef] [Green Version]
- Kochakian, C.D. Androgen Regulation of Nucleic Acid and Protein Biosynthesis in the Prostate. Natl. Cancer Inst. Monogr. 1963, 12, 263–274. [Google Scholar]
- Kochakian, C.D.; Hill, J.; Aonuma, S. Regulation of protein biosynthesis in mouse kidney by androgens. Endocrinology 1963, 72, 354–363. [Google Scholar] [CrossRef]
- Bernelli-Zazzera, A.; Bassi, M.; Comolli, R.; Lucchelli, P. Action of testosterone propionate and 4-chlorotestosterone acetate on protein synthesis in vitro. Nature 1958, 182, 663. [Google Scholar] [CrossRef]
- D’Abronzo, L.S.; Bose, S.; Crapuchettes, M.E.; Beggs, R.E.; Vinall, R.L.; Tepper, C.G.; Siddiqui, S.; Mudryj, M.; Melgoza, F.U.; Durbin-Johnson, B.P.; et al. The androgen receptor is a negative regulator of eIF4E phosphorylation at S209: Implications for the use of mTOR inhibitors in advanced prostate cancer. Oncogene 2017, 36, 6359–6373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Bailey, C.G.; Ng, C.; Tiffen, J.; Thoeng, A.; Minhas, V.; Lehman, M.L.; Hendy, S.C.; Buchanan, G.; Nelson, C.C.; et al. Androgen receptor and nutrient signaling pathways coordinate the demand for increased amino acid transport during prostate cancer progression. Cancer Res. 2011, 71, 7525–7536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holzbeierlein, J.; Lal, P.; LaTulippe, E.; Smith, A.; Satagopan, J.; Zhang, L.; Ryan, C.; Smith, S.; Scher, H.; Scardino, P.; et al. Gene expression analysis of human prostate carcinoma during hormonal therapy identifies androgen-responsive genes and mechanisms of therapy resistance. Am. J. Pathol. 2004, 164, 217–227. [Google Scholar] [CrossRef] [Green Version]
- Chandran, U.R.; Ma, C.; Dhir, R.; Bisceglia, M.; Lyons-Weiler, M.; Liang, W.; Michalopoulos, G.; Becich, M.; Monzon, F.A. Gene expression profiles of prostate cancer reveal involvement of multiple molecular pathways in the metastatic process. BMC Cancer 2007, 7, 64. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.P.; Landsittel, D.; Jing, L.; Nelson, J.; Ren, B.; Liu, L.; McDonald, C.; Thomas, R.; Dhir, R.; Finkelstein, S.; et al. Gene expression alterations in prostate cancer predicting tumor aggression and preceding development of malignancy. J. Clin. Oncol. 2004, 22, 2790–2799. [Google Scholar] [CrossRef]
- Varambally, S.; Yu, J.; Laxman, B.; Rhodes, D.R.; Mehra, R.; Tomlins, S.A.; Shah, R.B.; Chandran, U.; Monzon, F.A.; Becich, M.J.; et al. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell 2005, 8, 393–406. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Qu, S.; Tesikova, M.; Wang, L.; Kristian, A.; Maelandsmo, G.M.; Kong, H.; Zhang, T.; Jeronimo, C.; Teixeira, M.R.; et al. Molecular circuit involving KLK4 integrates androgen and mTOR signaling in prostate cancer. Proc. Natl. Acad. Sci. USA 2013, 110, E2572–E2581. [Google Scholar] [CrossRef] [Green Version]
- Velasco, A.M.; Gillis, K.A.; Li, Y.; Brown, E.L.; Sadler, T.M.; Achilleos, M.; Greenberger, L.M.; Frost, P.; Bai, W.; Zhang, Y. Identification and validation of novel androgen-regulated genes in prostate cancer. Endocrinology 2004, 145, 3913–3924. [Google Scholar] [CrossRef] [Green Version]
- Wang, L. FKBP51 regulation of AKT/protein kinase B phosphorylation. Curr. Opin. Pharmacol. 2011, 11, 360–364. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Mikhailova, M.; Bose, S.; Pan, C.X.; deVere White, R.W.; Ghosh, P.M. Regulation of androgen receptor transcriptional activity by rapamycin in prostate cancer cell proliferation and survival. Oncogene 2008, 27, 7106–7117. [Google Scholar] [CrossRef] [Green Version]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef]
- Xie, J.; Wang, X.; Proud, C.G. mTOR inhibitors in cancer therapy. F1000Research 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Audet-Walsh, E.; Dufour, C.R.; Yee, T.; Zouanat, F.Z.; Yan, M.; Kalloghlian, G.; Vernier, M.; Caron, M.; Bourque, G.; Scarlata, E.; et al. Nuclear mTOR acts as a transcriptional integrator of the androgen signaling pathway in prostate cancer. Genes Dev. 2017, 31, 1228–1242. [Google Scholar] [CrossRef] [Green Version]
- Giguere, V. DNA-PK, Nuclear mTOR, and the Androgen Pathway in Prostate Cancer. Trends Cancer 2020, 6, 337–347. [Google Scholar] [CrossRef]
- Audet-Walsh, E.; Vernier, M.; Yee, T.; Laflamme, C.; Li, S.; Chen, Y.; Giguere, V. SREBF1 Activity Is Regulated by an AR/mTOR Nuclear Axis in Prostate Cancer. Mol. Cancer Res. 2018, 16, 1396–1405. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Herrera, I.G.; Prado-Lourenco, L.; Pileur, F.; Conte, C.; Morin, A.; Cabon, F.; Prats, H.; Vagner, S.; Bayard, F.; Audigier, S.; et al. Testosterone regulates FGF-2 expression during testis maturation by an IRES-dependent translational mechanism. FASEB J. 2006, 20, 476–478. [Google Scholar] [CrossRef] [Green Version]
- Sheng, X.; Arnoldussen, Y.J.; Storm, M.; Tesikova, M.; Nenseth, H.Z.; Zhao, S.; Fazli, L.; Rennie, P.; Risberg, B.; Waehre, H.; et al. Divergent androgen regulation of unfolded protein response pathways drives prostate cancer. EMBO Mol. Med. 2015, 7, 788–801. [Google Scholar] [CrossRef]
- Segawa, T.; Nau, M.E.; Xu, L.L.; Chilukuri, R.N.; Makarem, M.; Zhang, W.; Petrovics, G.; Sesterhenn, I.A.; McLeod, D.G.; Moul, J.W.; et al. Androgen-Induced expression of endoplasmic reticulum (ER) stress response genes in prostate cancer cells. Oncogene 2002, 21, 8749–8758. [Google Scholar] [CrossRef] [Green Version]
- Sheng, X.; Nenseth, H.Z.; Qu, S.; Kuzu, O.F.; Frahnow, T.; Simon, L.; Greene, S.; Zeng, Q.; Fazli, L.; Rennie, P.S.; et al. IRE1alpha-XBP1s pathway promotes prostate cancer by activating c-MYC signaling. Nat. Commun. 2019, 10, 323. [Google Scholar] [CrossRef] [Green Version]
- Stelloo, S.; Linder, S.; Nevedomskaya, E.; Valle-Encinas, E.; de Rink, I.; Wessels, L.F.A.; van der Poel, H.; Bergman, A.M.; Zwart, W. Androgen modulation of XBP1 is functionally driving part of the AR transcriptional program. Endocr. Relat. Cancer 2020, 27, 67–79. [Google Scholar] [CrossRef]
- Erzurumlu, Y.; Ballar, P. Androgen Mediated Regulation of Endoplasmic Reticulum-Associated Degradation and its Effects on Prostate Cancer. Sci. Rep. 2017, 7, 40719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.C.; Fu, H.C.; Hsiao, B.L.; Sobue, G.; Adachi, H.; Huang, F.J.; Hsuuw, Y.D.; Wei, K.T.; Chang, C.; Huang, K.E.; et al. Androgen receptor inclusions acquire GRP78/BiP to ameliorate androgen-induced protein misfolding stress in embryonic stem cells. Cell Death Dis. 2013, 4, e607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azhary, J.M.K.; Harada, M.; Takahashi, N.; Nose, E.; Kunitomi, C.; Koike, H.; Hirata, T.; Hirota, Y.; Koga, K.; Wada-Hiraike, O.; et al. Endoplasmic Reticulum Stress Activated by Androgen Enhances Apoptosis of Granulosa Cells via Induction of Death Receptor 5 in PCOS. Endocrinology 2019, 160, 119–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doultsinos, D.; Mills, I. The role of the androgen receptor as a driver and mitigator of cellular stress. J. Mol. Endocrinol. 2020, 65, R19–R33. [Google Scholar] [CrossRef] [PubMed]
- Isakoff, S.J.; Engelman, J.A.; Irie, H.Y.; Luo, J.; Brachmann, S.M.; Pearline, R.V.; Cantley, L.C.; Brugge, J.S. Breast cancer-associated PIK3CA mutations are oncogenic in mammary epithelial cells. Cancer Res. 2005, 65, 10992–11000. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Liu, G.; Dziubinski, M.; Yang, Z.; Ethier, S.P.; Wu, G. Comprehensive analysis of oncogenic effects of PIK3CA mutations in human mammary epithelial cells. Breast Cancer Res. Treat. 2008, 112, 217–227. [Google Scholar] [CrossRef]
- Crowder, R.J.; Phommaly, C.; Tao, Y.; Hoog, J.; Luo, J.; Perou, C.M.; Parker, J.S.; Miller, M.A.; Huntsman, D.G.; Lin, L.; et al. PIK3CA and PIK3CB inhibition produce synthetic lethality when combined with estrogen deprivation in estrogen receptor-positive breast cancer. Cancer Res. 2009, 69, 3955–3962. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, N.A.; McDonald, K.; Tong, L.; von Euw, E.; Kalous, O.; Conklin, D.; Hurvitz, S.A.; di Tomaso, E.; Schnell, C.; Linnartz, R.; et al. Targeting PI3K/mTOR overcomes resistance to HER2-targeted therapy independent of feedback activation of AKT. Clin. Cancer Res. 2014, 20, 3507–3520. [Google Scholar] [CrossRef] [Green Version]
- Kalinsky, K.; Jacks, L.M.; Heguy, A.; Patil, S.; Drobnjak, M.; Bhanot, U.K.; Hedvat, C.V.; Traina, T.A.; Solit, D.; Gerald, W.; et al. PIK3CA mutation associates with improved outcome in breast cancer. Clin. Cancer Res. 2009, 15, 5049–5059. [Google Scholar] [CrossRef] [Green Version]
- Loi, S.; Haibe-Kains, B.; Majjaj, S.; Lallemand, F.; Durbecq, V.; Larsimont, D.; Gonzalez-Angulo, A.M.; Pusztai, L.; Symmans, W.F.; Bardelli, A.; et al. PIK3CA mutations associated with gene signature of low mTORC1 signaling and better outcomes in estrogen receptor-positive breast cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 10208–10213. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Coussy, F.; Lavigne, M.; de Koning, L.; Botty, R.E.; Nemati, F.; Naguez, A.; Bataillon, G.; Ouine, B.; Dahmani, A.; Montaudon, E.; et al. Response to mTOR and PI3K inhibitors in enzalutamide-resistant luminal androgen receptor triple-negative breast cancer patient-derived xenografts. Theranostics 2020, 10, 1531–1543. [Google Scholar] [CrossRef]
- Verret, B.; Cortes, J.; Bachelot, T.; Andre, F.; Arnedos, M. Efficacy of PI3K inhibitors in advanced breast cancer. Ann. Oncol. 2019, 30 (Suppl. 10), x12–x20. [Google Scholar] [CrossRef]
- Shorning, B.Y.; Dass, M.S.; Smalley, M.J.; Pearson, H.B. The PI3K-AKT-mTOR Pathway and Prostate Cancer: At the Crossroads of AR, MAPK, and WNT Signaling. Int. J. Mol. Sci. 2020, 21, 4507. [Google Scholar] [CrossRef]
- Braglia, L.; Zavatti, M.; Vinceti, M.; Martelli, A.M.; Marmiroli, S. Deregulated PTEN/PI3K/AKT/mTOR signaling in prostate cancer: Still a potential druggable target? Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118731. [Google Scholar] [CrossRef]
- Park, S.; Kim, Y.S.; Kim, D.Y.; So, I.; Jeon, J.H. PI3K pathway in prostate cancer: All resistant roads lead to PI3K. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 198–206. [Google Scholar] [CrossRef]
- Mbatia, H.W.; Ramalingam, S.; Ramamurthy, V.P.; Martin, M.S.; Kwegyir-Afful, A.K.; Njar, V.C. Novel C-4 heteroaryl 13-cis-retinamide Mnk/AR degrading agents inhibit cell proliferation and migration and induce apoptosis in human breast and prostate cancer cells and suppress growth of MDA-MB-231 human breast and CWR22Rv1 human prostate tumor xenografts in mice. J. Med. Chem. 2015, 58, 1900–1914. [Google Scholar] [CrossRef]
- Ramamurthy, V.P.; Ramalingam, S.; Gediya, L.; Kwegyir-Afful, A.K.; Njar, V.C. Simultaneous targeting of androgen receptor (AR) and MAPK-interacting kinases (MNKs) by novel retinamides inhibits growth of human prostate cancer cell lines. Oncotarget 2015, 6, 3195–3210. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Shen, K.; Jones, A.T.; Yang, J.; Tee, A.R.; Shen, M.H.; Yu, M.; Irani, S.; Wong, D.; Merrett, J.E.; et al. Reciprocal signaling between mTORC1 and MNK2 controls cell growth and oncogenesis. Cell. Mol. Life Sci. 2020, 249–270. [Google Scholar] [CrossRef]
- Stead, R.L.; Proud, C.G. Rapamycin enhances eIF4E phosphorylation by activating MAP kinase-interacting kinase 2a (Mnk2a). FEBS Lett. 2013, 587, 2623–2628. [Google Scholar] [CrossRef] [Green Version]
- Reich, S.H.; Sprengeler, P.A.; Chiang, G.G.; Appleman, J.R.; Chen, J.; Clarine, J.; Eam, B.; Ernst, J.T.; Han, Q.; Goel, V.K.; et al. Structure-based Design of Pyridone-Aminal eFT508 Targeting Dysregulated Translation by Selective Mitogen-activated Protein Kinase Interacting Kinases 1 and 2 (MNK1/2) Inhibition. J. Med. Chem. 2018, 61, 3516–3540. [Google Scholar] [CrossRef] [PubMed]
- Geter, P.A.; Ernlund, A.W.; Bakogianni, S.; Alard, A.; Arju, R.; Giashuddin, S.; Gadi, A.; Bromberg, J.; Schneider, R.J. Hyperactive mTOR and MNK1 phosphorylation of eIF4E confer tamoxifen resistance and estrogen independence through selective mRNA translation reprogramming. Genes Dev. 2017, 31, 2235–2249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bain, J.; Plater, L.; Elliott, M.; Shpiro, N.; Hastie, C.J.; McLauchlan, H.; Klevernic, I.; Arthur, J.S.; Alessi, D.R.; Cohen, P. The selectivity of protein kinase inhibitors: A further update. Biochem. J. 2007, 408, 297–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truitt, M.L.; Conn, C.S.; Shi, Z.; Pang, X.; Tokuyasu, T.; Coady, A.M.; Seo, Y.; Barna, M.; Ruggero, D. Differential Requirements for eIF4E Dose in Normal Development and Cancer. Cell 2015, 162, 59–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furic, L.; Rong, L.; Larsson, O.; Koumakpayi, I.H.; Yoshida, K.; Brueschke, A.; Petroulakis, E.; Robichaud, N.; Pollak, M.; Gaboury, L.A.; et al. eIF4E phosphorylation promotes tumorigenesis and is associated with prostate cancer progression. Proc. Natl. Acad. Sci. USA 2010, 107, 14134–14139. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, A.C.; Liu, Y.; Edlind, M.P.; Ingolia, N.T.; Janes, M.R.; Sher, A.; Shi, E.Y.; Stumpf, C.R.; Christensen, C.; Bonham, M.J.; et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 2012, 485, 55–61. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.S.; Jansen, A.P.; Komar, A.A.; Zheng, X.; Merrick, W.C.; Costes, S.; Lockett, S.J.; Sonenberg, N.; Colburn, N.H. The transformation suppressor Pdcd4 is a novel eukaryotic translation initiation factor 4A binding protein that inhibits translation. Mol. Cell. Biol. 2003, 23, 26–37. [Google Scholar] [CrossRef] [Green Version]
- Howard, C.M.; Estrada, M.; Terrero, D.; Tiwari, A.K.; Raman, D. Identification of Cardiac Glycosides as Novel Inhibitors of eIF4A1-Mediated Translation in Triple-Negative Breast Cancer Cells. Cancers 2020, 12, 2169. [Google Scholar] [CrossRef]
- Sridharan, S.; Robeson, M.; Bastihalli-Tukaramrao, D.; Howard, C.M.; Subramaniyan, B.; Tilley, A.M.C.; Tiwari, A.K.; Raman, D. Targeting of the Eukaryotic Translation Initiation Factor 4A Against Breast Cancer Stemness. Front. Oncol. 2019, 9, 1311. [Google Scholar] [CrossRef] [Green Version]
- Cencic, R.; Carrier, M.; Galicia-Vazquez, G.; Bordeleau, M.E.; Sukarieh, R.; Bourdeau, A.; Brem, B.; Teodoro, J.G.; Greger, H.; Tremblay, M.L.; et al. Antitumor activity and mechanism of action of the cyclopenta[b]benzofuran, silvestrol. PLoS ONE 2009, 4, e5223. [Google Scholar] [CrossRef] [Green Version]
- Garrido, M.F.; Martin, N.J.; Bertrand, M.; Gaudin, C.; Commo, F.; El Kalaany, N.; Al Nakouzi, N.; Fazli, L.; Del Nery, E.; Camonis, J.; et al. Regulation of eIF4F Translation Initiation Complex by the Peptidyl Prolyl Isomerase FKBP7 in Taxane-resistant Prostate Cancer. Clin. Cancer Res. 2019, 25, 710–723. [Google Scholar] [CrossRef] [Green Version]
- Modelska, A.; Turro, E.; Russell, R.; Beaton, J.; Sbarrato, T.; Spriggs, K.; Miller, J.; Graf, S.; Provenzano, E.; Blows, F.; et al. The malignant phenotype in breast cancer is driven by eIF4A1-mediated changes in the translational landscape. Cell Death Dis. 2015, 6, e1603. [Google Scholar] [CrossRef] [Green Version]
- Nasr, Z.; Robert, F.; Porco, J.A., Jr.; Muller, W.J.; Pelletier, J. eIF4F suppression in breast cancer affects maintenance and progression. Oncogene 2013, 32, 861–871. [Google Scholar] [CrossRef] [Green Version]
- Jin, C.; Rajabi, H.; Rodrigo, C.M.; Porco, J.A., Jr.; Kufe, D. Targeting the eIF4A RNA helicase blocks translation of the MUC1-C oncoprotein. Oncogene 2013, 32, 2179–2188. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.M.; Kabotyanski, E.B.; Dou, Y.; Reineke, L.C.; Zhang, P.; Zhang, X.H.; Malovannaya, A.; Jung, S.Y.; Mo, Q.; Roarty, K.P.; et al. FGFR1-Activated Translation of WNT Pathway Components with Structured 5′ UTRs Is Vulnerable to Inhibition of EIF4A-Dependent Translation Initiation. Cancer Res. 2018, 78, 4229–4240. [Google Scholar] [CrossRef] [Green Version]
- Ernst, J.T.; Thompson, P.A.; Nilewski, C.; Sprengeler, P.A.; Sperry, S.; Packard, G.; Michels, T.; Xiang, A.; Tran, C.; Wegerski, C.J.; et al. Design of Development Candidate eFT226, a First in Class Inhibitor of Eukaryotic Initiation Factor 4A RNA Helicase. J. Med. Chem. 2020, 63, 5879–5955. [Google Scholar] [CrossRef]
- Rubio, C.A.; Weisburd, B.; Holderfield, M.; Arias, C.; Fang, E.; DeRisi, J.L.; Fanidi, A. Transcriptome-Wide characterization of the eIF4A signature highlights plasticity in translation regulation. Genome Biol. 2014, 15, 476. [Google Scholar] [CrossRef]
- Ebright, R.Y.; Lee, S.; Wittner, B.S.; Niederhoffer, K.L.; Nicholson, B.T.; Bardia, A.; Truesdell, S.; Wiley, D.F.; Wesley, B.; Li, S.; et al. Deregulation of ribosomal protein expression and translation promotes breast cancer metastasis. Science 2020, 367, 1468–1473. [Google Scholar] [CrossRef]
- Rebello, R.J.; Kusnadi, E.; Cameron, D.P.; Pearson, H.B.; Lesmana, A.; Devlin, J.R.; Drygin, D.; Clark, A.K.; Porter, L.; Pedersen, J.; et al. The Dual Inhibition of RNA Pol I Transcription and PIM Kinase as a New Therapeutic Approach to Treat Advanced Prostate Cancer. Clin. Cancer Res. 2016, 22, 5539–5552. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.G.; Obinata, D.; Sandhu, S.; Selth, L.A.; Wong, S.Q.; Porter, L.H.; Lister, N.; Pook, D.; Pezaro, C.J.; Goode, D.L.; et al. Patient-Derived Models of Abiraterone- and Enzalutamide-resistant Prostate Cancer Reveal Sensitivity to Ribosome-directed Therapy. Eur. Urol. 2018, 74, 562–572. [Google Scholar] [CrossRef]
- Nguyen, H.G.; Conn, C.S.; Kye, Y.; Xue, L.; Forester, C.M.; Cowan, J.E.; Hsieh, A.C.; Cunningham, J.T.; Truillet, C.; Tameire, F.; et al. Development of a stress response therapy targeting aggressive prostate cancer. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidrauski, C.; Acosta-Alvear, D.; Khoutorsky, A.; Vedantham, P.; Hearn, B.R.; Li, H.; Gamache, K.; Gallagher, C.M.; Ang, K.K.; Wilson, C.; et al. Pharmacological brake-release of mRNA translation enhances cognitive memory. eLife 2013, 2, e00498. [Google Scholar] [CrossRef] [PubMed]
- Bain, J.; McLauchlan, H.; Elliott, M.; Cohen, P. The specificities of protein kinase inhibitors: An update. Biochem. J. 2003, 371, 199–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, S.P.; Reddy, H.; Caivano, M.; Cohen, P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 2000, 351, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Alain, T.; Morita, M.; Fonseca, B.D.; Yanagiya, A.; Siddiqui, N.; Bhat, M.; Zammit, D.; Marcus, V.; Metrakos, P.; Voyer, L.A.; et al. eIF4E/4E-BP ratio predicts the efficacy of mTOR targeted therapies. Cancer Res. 2012, 72, 6468–6476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, S.; Scheulen, M.E.; Johnston, S.; Mross, K.; Cardoso, F.; Dittrich, C.; Eiermann, W.; Hess, D.; Morant, R.; Semiglazov, V.; et al. Phase II study of temsirolimus (CCI-779), a novel inhibitor of mTOR, in heavily pretreated patients with locally advanced or metastatic breast cancer. J. Clin. Oncol. 2005, 23, 5314–5322. [Google Scholar] [CrossRef]
- Awada, A.; Cardoso, F.; Fontaine, C.; Dirix, L.; De Greve, J.; Sotiriou, C.; Steinseifer, J.; Wouters, C.; Tanaka, C.; Zoellner, U.; et al. The oral mTOR inhibitor RAD001 (everolimus) in combination with letrozole in patients with advanced breast cancer: Results of a phase I study with pharmacokinetics. Eur. J. Cancer 2008, 44, 84–91. [Google Scholar] [CrossRef]
- Ellard, S.L.; Clemons, M.; Gelmon, K.A.; Norris, B.; Kennecke, H.; Chia, S.; Pritchard, K.; Eisen, A.; Vandenberg, T.; Taylor, M.; et al. Randomized phase II study comparing two schedules of everolimus in patients with recurrent/metastatic breast cancer: NCIC Clinical Trials Group IND.163. J. Clin. Oncol. 2009, 27, 4536–4541. [Google Scholar] [CrossRef]
- Zagouri, F.; Sergentanis, T.N.; Chrysikos, D.; Filipits, M.; Bartsch, R. mTOR inhibitors in breast cancer: A systematic review. Gynecol. Oncol. 2012, 127, 662–672. [Google Scholar] [CrossRef]
- Baselga, J.; Campone, M.; Piccart, M.; Burris, H.A., 3rd; Rugo, H.S.; Sahmoud, T.; Noguchi, S.; Gnant, M.; Pritchard, K.I.; Lebrun, F.; et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N. Engl. J. Med. 2012, 366, 520–529. [Google Scholar] [CrossRef] [Green Version]
- Piccart, M.; Hortobagyi, G.N.; Campone, M.; Pritchard, K.I.; Lebrun, F.; Ito, Y.; Noguchi, S.; Perez, A.; Rugo, H.S.; Deleu, I.; et al. Everolimus plus exemestane for hormone-receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: Overall survival results from BOLERO-2dagger. Ann. Oncol. 2014, 25, 2357–2362. [Google Scholar] [CrossRef]
- Wolff, A.C.; Lazar, A.A.; Bondarenko, I.; Garin, A.M.; Brincat, S.; Chow, L.; Sun, Y.; Neskovic-Konstantinovic, Z.; Guimaraes, R.C.; Fumoleau, P.; et al. Randomized phase III placebo-controlled trial of letrozole plus oral temsirolimus as first-line endocrine therapy in postmenopausal women with locally advanced or metastatic breast cancer. J. Clin. Oncol. 2013, 31, 195–202. [Google Scholar] [CrossRef] [Green Version]
- Kornblum, N.; Zhao, F.; Manola, J.; Klein, P.; Ramaswamy, B.; Brufsky, A.; Stella, P.J.; Burnette, B.; Telli, M.; Makower, D.F.; et al. Randomized Phase II Trial of Fulvestrant Plus Everolimus or Placebo in Postmenopausal Women with Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Metastatic Breast Cancer Resistant to Aromatase Inhibitor Therapy: Results of PrE0102. J. Clin. Oncol. 2018, 36, 1556–1563. [Google Scholar] [CrossRef]
- Armstrong, A.J.; Netto, G.J.; Rudek, M.A.; Halabi, S.; Wood, D.P.; Creel, P.A.; Mundy, K.; Davis, S.L.; Wang, T.; Albadine, R.; et al. A pharmacodynamic study of rapamycin in men with intermediate-to high-risk localized prostate cancer. Clin. Cancer Res. 2010, 16, 3057–3066. [Google Scholar] [CrossRef] [Green Version]
- Nakabayashi, M.; Werner, L.; Courtney, K.D.; Buckle, G.; Oh, W.K.; Bubley, G.J.; Hayes, J.H.; Weckstein, D.; Elfiky, A.; Sims, D.M.; et al. Phase II trial of RAD001 and bicalutamide for castration-resistant prostate cancer. BJU Int. 2012, 110, 1729–1735. [Google Scholar] [CrossRef]
- Kruczek, K.; Ratterman, M.; Tolzien, K.; Sulo, S.; Lestingi, T.M.; Nabhan, C. A phase II study evaluating the toxicity and efficacy of single-agent temsirolimus in chemotherapy-naive castration-resistant prostate cancer. Br. J. Cancer 2013, 109, 1711–1716. [Google Scholar] [CrossRef] [Green Version]
- Templeton, A.J.; Dutoit, V.; Cathomas, R.; Rothermundt, C.; Bartschi, D.; Droge, C.; Gautschi, O.; Borner, M.; Fechter, E.; Stenner, F.; et al. Phase 2 trial of single-agent everolimus in chemotherapy-naive patients with castration-resistant prostate cancer (SAKK 08/08). Eur. Urol. 2013, 64, 150–158. [Google Scholar] [CrossRef]
- Wei, X.X.; Hsieh, A.C.; Kim, W.; Friedlander, T.; Lin, A.M.; Louttit, M.; Ryan, C.J. A Phase I Study of Abiraterone Acetate Combined with BEZ235, a Dual PI3K/mTOR Inhibitor, in Metastatic Castration Resistant Prostate Cancer. Oncologist 2017, 22, e503–e543. [Google Scholar] [CrossRef] [Green Version]
- Massard, C.; Chi, K.N.; Castellano, D.; de Bono, J.; Gravis, G.; Dirix, L.; Machiels, J.P.; Mita, A.; Mellado, B.; Turri, S.; et al. Phase Ib dose-finding study of abiraterone acetate plus buparlisib (BKM120) or dactolisib (BEZ235) in patients with castration-resistant prostate cancer. Eur. J. Cancer 2017, 76, 36–44. [Google Scholar] [CrossRef]
- Graham, L.; Banda, K.; Torres, A.; Carver, B.S.; Chen, Y.; Pisano, K.; Shelkey, G.; Curley, T.; Scher, H.I.; Lotan, T.L.; et al. A phase II study of the dual mTOR inhibitor MLN0128 in patients with metastatic castration resistant prostate cancer. Investig. New Drugs 2018, 36, 458–467. [Google Scholar] [CrossRef]
- Koshkin, V.S.; Mir, M.C.; Barata, P.; Gul, A.; Gupta, R.; Stephenson, A.J.; Kaouk, J.; Berglund, R.; Magi-Galluzzi, C.; Klein, E.A.; et al. Randomized phase II trial of neoadjuvant everolimus in patients with high-risk localized prostate cancer. Investig. New Drugs 2019, 37, 559–566. [Google Scholar] [CrossRef]
- Barata, P.C.; Cooney, M.; Mendiratta, P.; Gupta, R.; Dreicer, R.; Garcia, J.A. Phase I/II study evaluating the safety and clinical efficacy of temsirolimus and bevacizumab in patients with chemotherapy refractory metastatic castration-resistant prostate cancer. Investig. New Drugs 2019, 37, 331–337. [Google Scholar] [CrossRef]
- Thoreen, C.C.; Sabatini, D.M. Rapamycin inhibits mTORC1, but not completely. Autophagy 2009, 5, 725–726. [Google Scholar] [CrossRef] [Green Version]
- Thoreen, C.C.; Kang, S.A.; Chang, J.W.; Liu, Q.; Zhang, J.; Gao, Y.; Reichling, L.J.; Sim, T.; Sabatini, D.M.; Gray, N.S. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J. Biol. Chem. 2009, 284, 8023–8032. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Schwartz, G.N.; Marotti, J.D.; Chen, V.; Traphagen, N.A.; Gui, J.; Miller, T.W. Estrogen receptor alpha drives mTORC1 inhibitor-induced feedback activation of PI3K/AKT in ER+ breast cancer. Oncotarget 2018, 9, 8810–8822. [Google Scholar] [CrossRef] [Green Version]
- Shee, K.; Yang, W.; Hinds, J.W.; Hampsch, R.A.; Varn, F.S.; Traphagen, N.A.; Patel, K.; Cheng, C.; Jenkins, N.P.; Kettenbach, A.N.; et al. Therapeutically targeting tumor microenvironment-mediated drug resistance in estrogen receptor-positive breast cancer. J. Exp. Med. 2018, 215, 895–910. [Google Scholar] [CrossRef] [Green Version]
- Barlow, A.D.; Xie, J.; Moore, C.E.; Campbell, S.C.; Shaw, J.A.; Nicholson, M.L.; Herbert, T.P. Rapamycin toxicity in MIN6 cells and rat and human islets is mediated by the inhibition of mTOR complex 2 (mTORC2). Diabetologia 2012, 55, 1355–1365. [Google Scholar] [CrossRef] [Green Version]
- Croessmann, S.; Formisano, L.; Kinch, L.N.; Gonzalez-Ericsson, P.I.; Sudhan, D.R.; Nagy, R.J.; Mathew, A.; Bernicker, E.H.; Cristofanilli, M.; He, J.; et al. Combined Blockade of Activating ERBB2 Mutations and ER Results in Synthetic Lethality of ER+/HER2 Mutant Breast Cancer. Clin. Cancer Res. 2019, 25, 277–289. [Google Scholar] [CrossRef] [Green Version]
- Heffron, T.P.; Ndubaku, C.O.; Salphati, L.; Alicke, B.; Cheong, J.; Drobnick, J.; Edgar, K.; Gould, S.E.; Lee, L.B.; Lesnick, J.D.; et al. Discovery of Clinical Development Candidate GDC-0084, a Brain Penetrant Inhibitor of PI3K and mTOR. ACS Med. Chem. Lett. 2016, 7, 351–356. [Google Scholar] [CrossRef] [Green Version]
- Ippen, F.M.; Alvarez-Breckenridge, C.A.; Kuter, B.M.; Fink, A.L.; Bihun, I.V.; Lastrapes, M.; Penson, T.; Schmidt, S.P.; Wojtkiewicz, G.R.; Ning, J.; et al. The Dual PI3K/mTOR Pathway Inhibitor GDC-0084 Achieves Antitumor Activity in PIK3CA-Mutant Breast Cancer Brain Metastases. Clin. Cancer Res. 2019, 25, 3374–3383. [Google Scholar] [CrossRef] [Green Version]
- La Manna, F.; De Menna, M.; Patel, N.; Karkampouna, S.; De Filippo, M.; Klima, I.; Kloen, P.; Beimers, L.; Thalmann, G.N.; Pelger, R.C.M.; et al. Dual-mTOR Inhibitor Rapalink-1 Reduces Prostate Cancer Patient-Derived Xenograft Growth and Alters Tumor Heterogeneity. Front. Oncol. 2020, 10, 1012. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Structural domains of human ER and AR proteins. Both ER and AR belong to the nuclear receptor superfamily and share the same functional domains including the N-terminal domain (NTD), a DNA-binding domain (DBD), a hinge region and a ligand-binding domain (LBD). Activation function (AF) domains (AF-1, AF-2 and AF-5, the latter present only in AR) are shown.
Figure 1.
Structural domains of human ER and AR proteins. Both ER and AR belong to the nuclear receptor superfamily and share the same functional domains including the N-terminal domain (NTD), a DNA-binding domain (DBD), a hinge region and a ligand-binding domain (LBD). Activation function (AF) domains (AF-1, AF-2 and AF-5, the latter present only in AR) are shown.

Figure 2.
Signalling pathways that control translation. In response to extracellular stimuli, oncogenic signalling pathways, such as Ras/MAPK/MNK and PI3K/PKB, are activated and subsequently switch on/off downstream signalling events including mTORC1/S6K/eEF2K, mTORC1/4EBP/eIF4E and MNK/eIF4E pathways that drive/prevent translation initiation of elongation. In contrast, stress signals primarily act through three UPR pathways (PERK/eIF2α/ATF4; IRE1-XBP1; ATF6/ATF6p50) to restore protein homeostasis, which is achieved by blocking translation initiation (PERK/eIF2α), inducing the transcription of EnR folding-related genes (all three arms) and ERAD (through IRE1-XBP1 and ATF6/ATF6p50).
Figure 2.
Signalling pathways that control translation. In response to extracellular stimuli, oncogenic signalling pathways, such as Ras/MAPK/MNK and PI3K/PKB, are activated and subsequently switch on/off downstream signalling events including mTORC1/S6K/eEF2K, mTORC1/4EBP/eIF4E and MNK/eIF4E pathways that drive/prevent translation initiation of elongation. In contrast, stress signals primarily act through three UPR pathways (PERK/eIF2α/ATF4; IRE1-XBP1; ATF6/ATF6p50) to restore protein homeostasis, which is achieved by blocking translation initiation (PERK/eIF2α), inducing the transcription of EnR folding-related genes (all three arms) and ERAD (through IRE1-XBP1 and ATF6/ATF6p50).

Figure 3.
Schematic presentation summarizes reported major ER-driven translational control. ER can modulate translation initiation via stimulation of mTORC1 and eIF2α. It can also affect UPR via regulating the release of EnR Ca2+. ER inhibition triggers the AMPK/eEF2K pathway, and subsequently alleviates translation elongation. ER may also indirectly impact on translation elongation of mRNAs enriched in codons containing mcm5s2U34-modified tRNAs.
Figure 3.
Schematic presentation summarizes reported major ER-driven translational control. ER can modulate translation initiation via stimulation of mTORC1 and eIF2α. It can also affect UPR via regulating the release of EnR Ca2+. ER inhibition triggers the AMPK/eEF2K pathway, and subsequently alleviates translation elongation. ER may also indirectly impact on translation elongation of mRNAs enriched in codons containing mcm5s2U34-modified tRNAs.

Figure 4.
Signalling pathways implicated in AR-driven translational control. AR has been shown to modulate translation initiation through signalling cues (MNK and mTORC1, the latter via LAT3 and KLK4) that converge on eIF4E, a translation initiation factor that promotes the translation of mRNAs with certain patterns (e.g., guanine-rich translational element (GRTE)-containing messages). AR can also act as a regulator of the UPR by directly inducing IRE1, which leads to upregulation of ERAD-related genes.
Figure 4.
Signalling pathways implicated in AR-driven translational control. AR has been shown to modulate translation initiation through signalling cues (MNK and mTORC1, the latter via LAT3 and KLK4) that converge on eIF4E, a translation initiation factor that promotes the translation of mRNAs with certain patterns (e.g., guanine-rich translational element (GRTE)-containing messages). AR can also act as a regulator of the UPR by directly inducing IRE1, which leads to upregulation of ERAD-related genes.


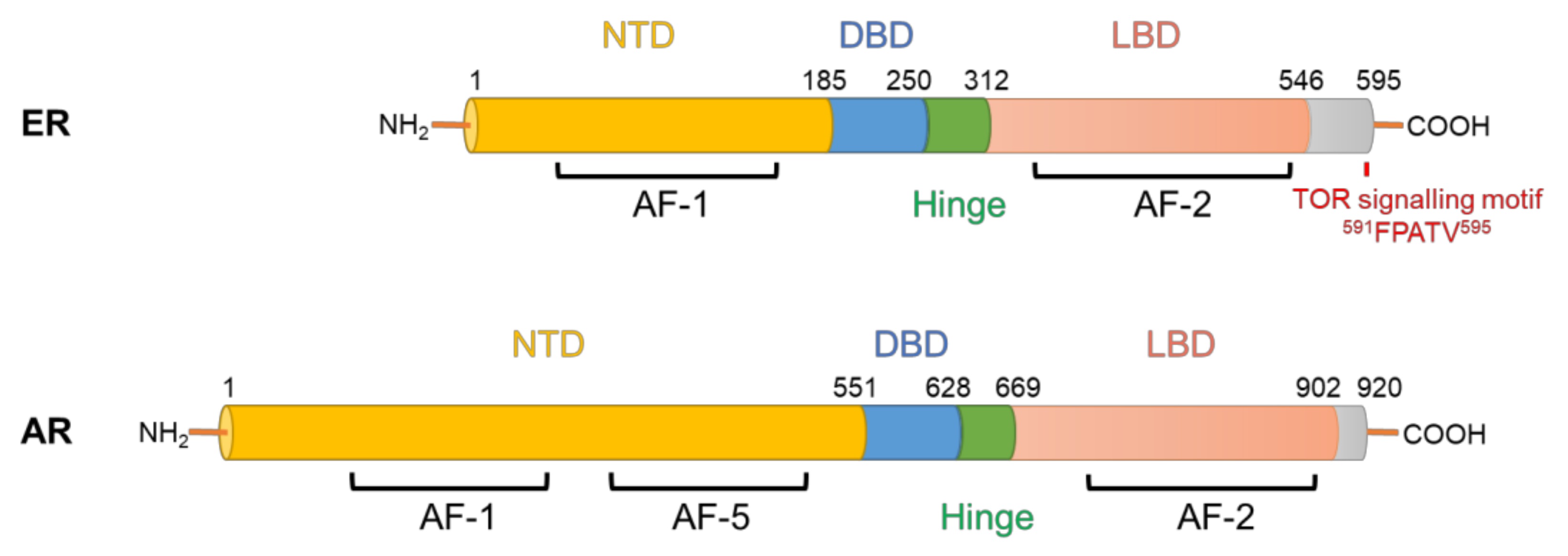{kind=link}
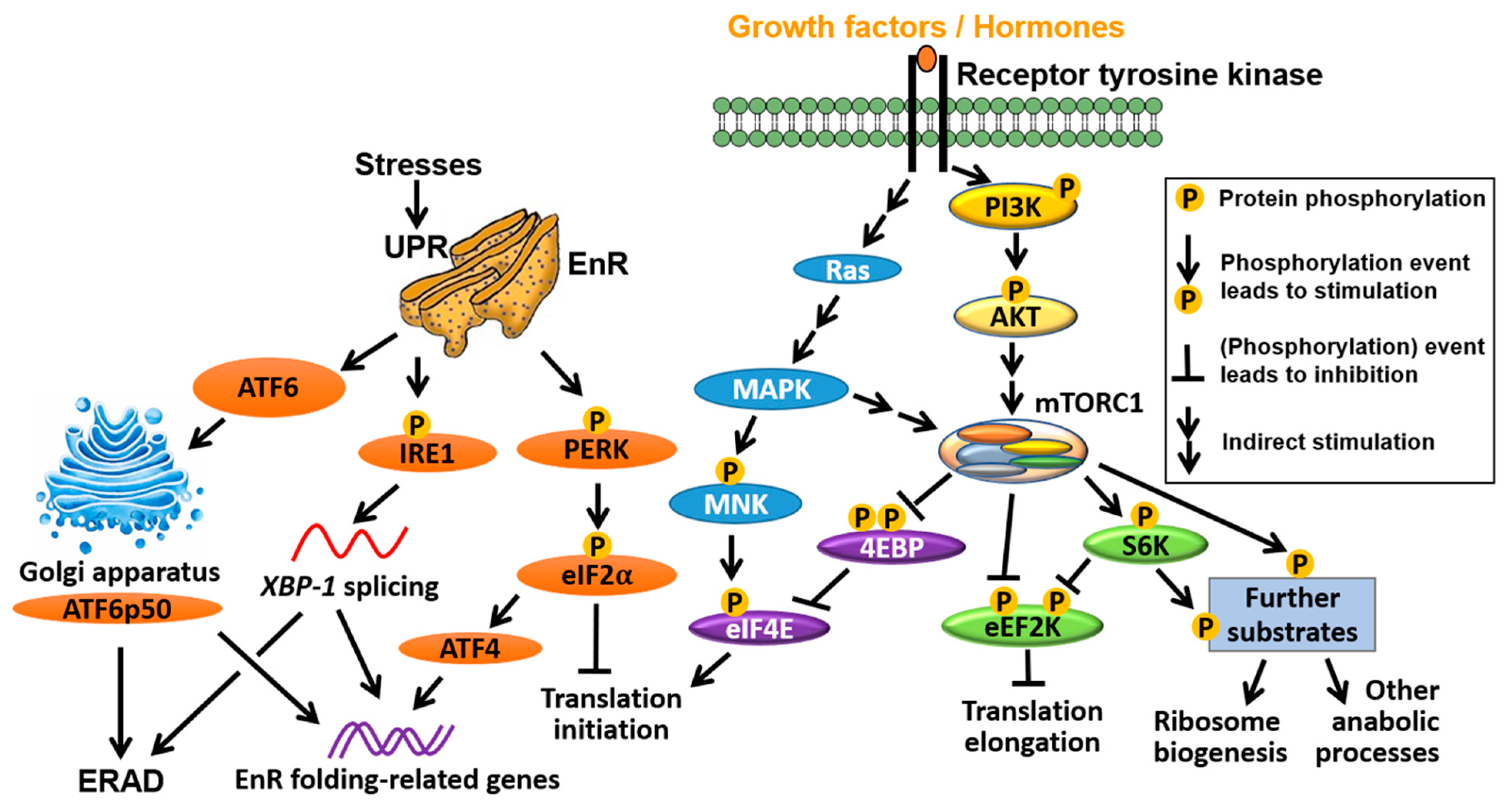{kind=link}
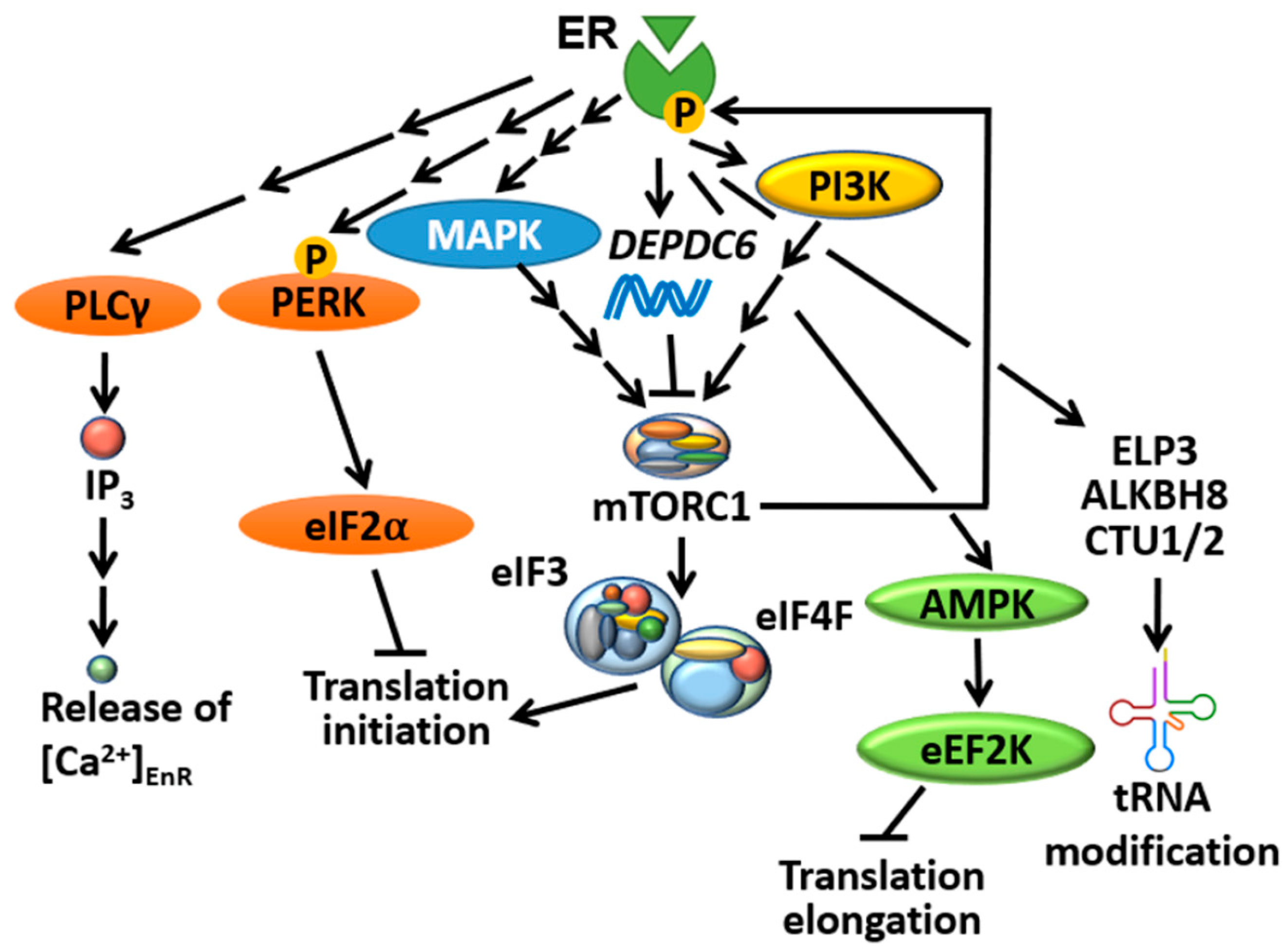{kind=link}
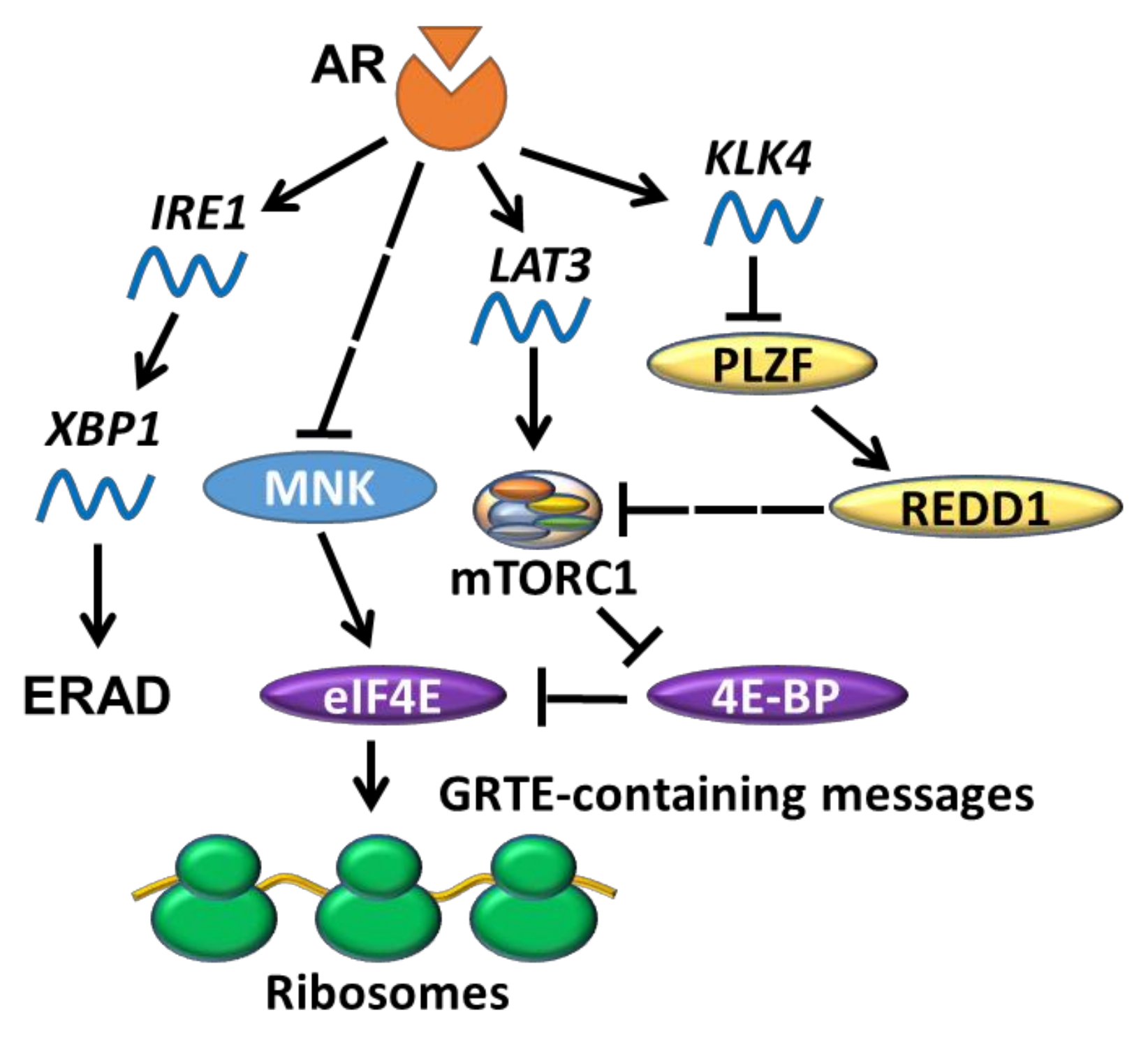{kind=link}
Table 1.
Frequency of mutations in selected translation-related genes encoding proteins upstream of mTORC1 in BC/PC according to cBioPortal (TCGA PanCan 2018; cbioportal.org).
Table 1.
Frequency of mutations in selected translation-related genes encoding proteins upstream of mTORC1 in BC/PC according to cBioPortal (TCGA PanCan 2018; cbioportal.org).
| Gene | BC (Invasive Carcinoma, 1084 Samples) | PC (494 Samples) | ||
|---|---|---|---|---|
| No. of Mutated Samples | % of Mutated Samples | No. of Mutated Samples | % of Mutated Samples | |
| AKT1 | 27 | 2.49% | 2 | 0.40% |
| AKT2 | 4 | 0.37% | 0 | 0.00% |
| AKT3 | 8 | 0.74% | 1 | 0.20% |
| BRAF | 7 | 0.65% | 7 | 1.42% |
| CRAF | 7 | 0.65% | 0 | 0.00% |
| HRAS | 5 | 0.46% | 4 | 0.81% |
| KRAS | 6 | 0.55% | 2 | 0.40% |
| MAP2K4 | 7 | 0.65% | 1 | 0.20% |
| MAP3K1 | 7 | 0.65% | 1 | 0.20% |
| MTOR | 20 | 1.85% | 2 | 0.40% |
| PIK3CA | 333 | 30.72% | 10 | 2.02% |
| PIK3CB | 10 | 0.92% | 3 | 0.61% |
| PIK3CD | 11 | 1.01% | 2 | 0.40% |
| PTEN | 56 | 5.17% | 13 | 2.63% |
Table 2.
A selection of reported clinical trials related to mTORis and their respective outcomes in BC/PC (chronological order). Abbreviations not defined in the main text: DI: dual PI3K/mTOR inhibitor; FA: FDA approval; Gen.: generation/class of mTORi (first listed under the column of “Drug(s)”); Ref.: reference.
Table 2.
A selection of reported clinical trials related to mTORis and their respective outcomes in BC/PC (chronological order). Abbreviations not defined in the main text: DI: dual PI3K/mTOR inhibitor; FA: FDA approval; Gen.: generation/class of mTORi (first listed under the column of “Drug(s)”); Ref.: reference.
| Year | Cancer Type | Drug(s) | Gen. | Phase | Outcome Summary | Ref. |
|---|---|---|---|---|---|---|
| 2005 | Localized or metastatic BC | Temsirolimus (CCI-779) only | 1st | II | Patients treated with temsirolimus showed anti-tumour activity and well tolerated toxicity. | [176] |
| 2008 | Advanced BC | Everolimus (RAD001) and letrozole | 1st | I | The combinational therapy showed anti-tumour activity, toxicity also well tolerated. | [177] |
| 2009 | Recurrent or metastatic BC | Everolimus only | 1st | II | Continuous daily dosing but not weekly dosing had anti-tumour activity. The drug was well tolerated but some patients developed pneumonitis. | [178] |
| 2010 | Localized PC | Sirolimus (rapamycin) only | 1st | I | Daily dosing had no effect on tumour proliferation/apoptosis despite suppresion of RP S6 phsophorylation. | [184] |
| 2012 | CRPC | Everolimus and bicalutamide | 1st | II | Combinational therapy was well tolerated despite 56% cases of grade 1/2 mucositis, but it had low activity and did not achieve the primary endpoint. | [185] |
| 2012 | HR+ BC | Everolimus and exemestane | 1st | III, FA | Combinational therapy improved PFS of patients previously treated with non-steroidal AIs. | [180] |
| 2013 | Locally advanced or metastatic BC | Temsirolimus and letrozole | 1st | III | In comparison to lotrozole monotherapy, daily and orally administrated temsirolimus failed to confer added PFS benefit to aromatase inhibitor-resistant ER+ patients. | [182] |
| 2013 | CRPC | Temsirolimus only | 1st | II | Weekly dosing of the drug had minimal therapeutic activity, and the study was put on halt at an early stage. | [186] |
| 2013 | CRPC | Everolimus only | 1st | II | The monotherapy regime modestly improved PFS, especially in PTEN−/− patients. Toxicity was also manageable. | [187] |
| 2014 | Advanced BC | Everolimus and exemestane | 1st | III | Addition of everolimus to exemestane treatment did not provide further improvement to HR+ BC patients at the secondary endpoint. | [181] |
| 2017 | CRPC | BEZ235 with abiraterone/prednisone | DI | I | The combinational therapy was poorly tolerated; adverse effects included mucositis, hypotension, dyspnea and pneumonitis. | [188] |
| 2017 | CRPC | BEZ235/BKM120 with abiraterone | DI | Ib | The trial was discontinued due to high levels of toxicity and poor pharmacokinetics among the participants. | [189] |
| 2018 | CRPC | MLN0128 (INK128) | 2nd | II | MLN0128 exhibited high levels of toxicity in patients; dyspnea and maculopapular rash were the main grade 3 adverse events. | [190] |
| 2018 | ER+/EGFR2−BC | Everolimus and fulvestrant | 1st | II | Despite increased cases of adverse events, improved PFS was observed with combinational therapy-treated patients. | [183] |
| 2019 | High-risk localized PC | Everolimus | 1st | II | Everolimus showed limited clinical activity. | [191] |
| 2019 | CRPC | Temsirolimus and bevacizumab | 1st | I/II | The combinational therapy did not improve the clinical outcome of the participants, and also induced severe adverse events. | [192] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Xie, J.; Kusnadi, E.P.; Furic, L.; Selth, L.A. Regulation of mRNA Translation by Hormone Receptors in Breast and Prostate Cancer. Cancers 2021, 13, 3254. https://doi.org/10.3390/cancers13133254
AMA Style
Xie J, Kusnadi EP, Furic L, Selth LA. Regulation of mRNA Translation by Hormone Receptors in Breast and Prostate Cancer. Cancers. 2021; 13(13):3254. https://doi.org/10.3390/cancers13133254
Chicago/Turabian StyleXie, Jianling, Eric P. Kusnadi, Luc Furic, and Luke A. Selth. 2021. "Regulation of mRNA Translation by Hormone Receptors in Breast and Prostate Cancer" Cancers 13, no. 13: 3254. https://doi.org/10.3390/cancers13133254
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.