
Genomic Risk Prediction for Breast Cancer in Older Women

 ,
,  , , , , , add
Show full author list
, , , , , add
Show full author list
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Sample
2.2. Genome-Wide Genotyping and Polygenic Risk Score
2.3. Targeted Gene Panel Sequencing
2.4. Endpoints
2.5. Statistical Analysis
3. Results
3.1. Baseline Characteristics
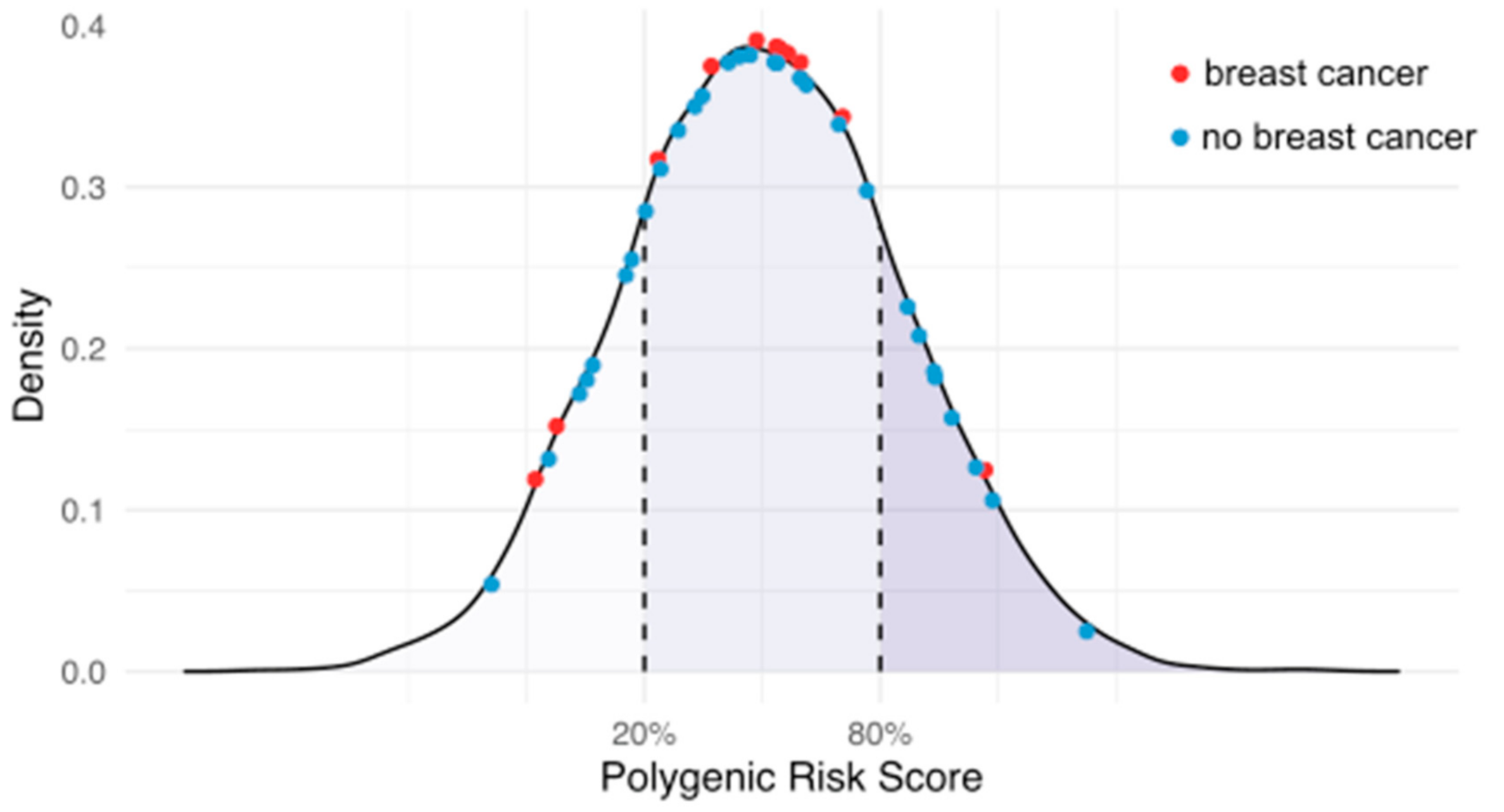
3.2. PRS and Rare Pathogenic Variants
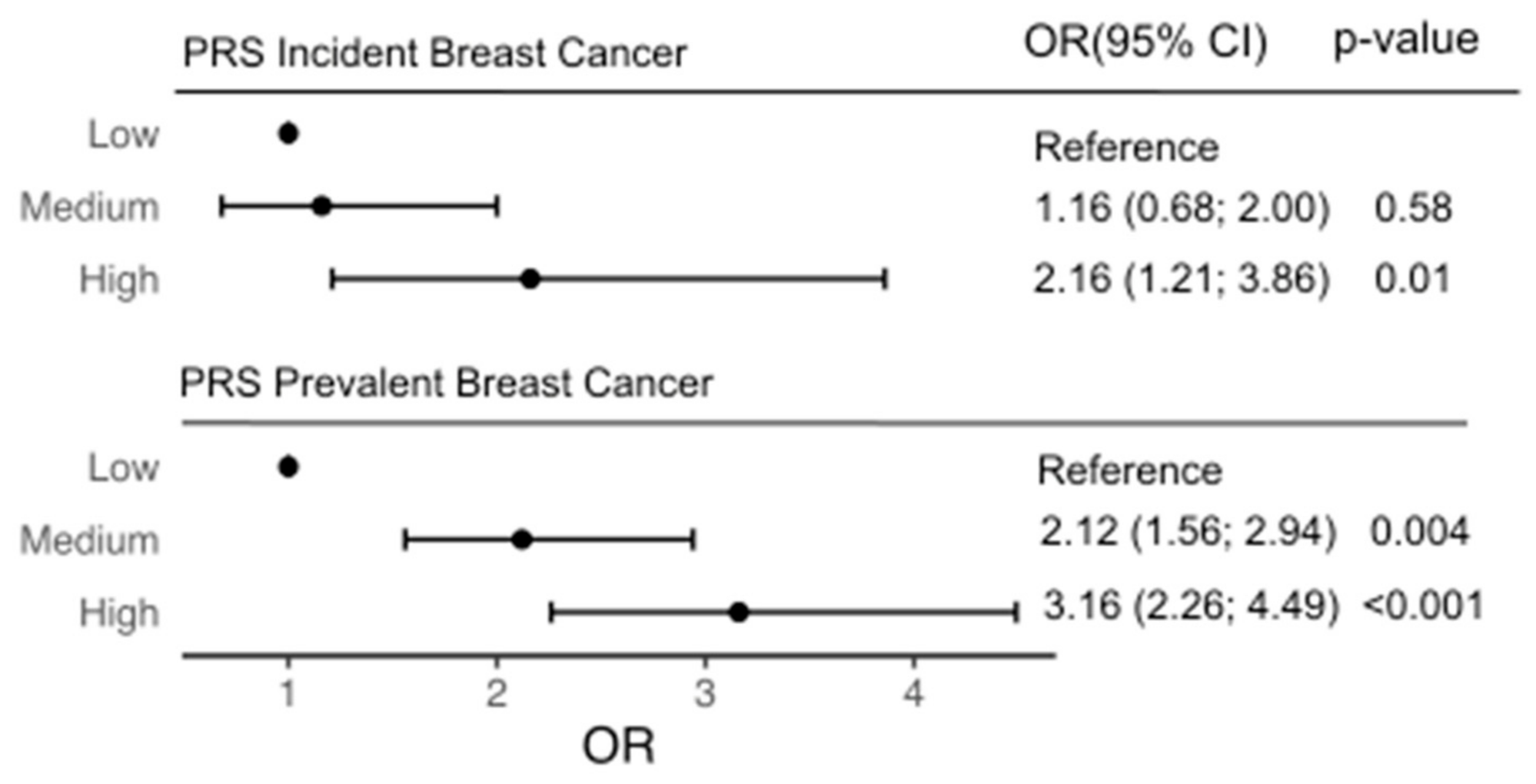
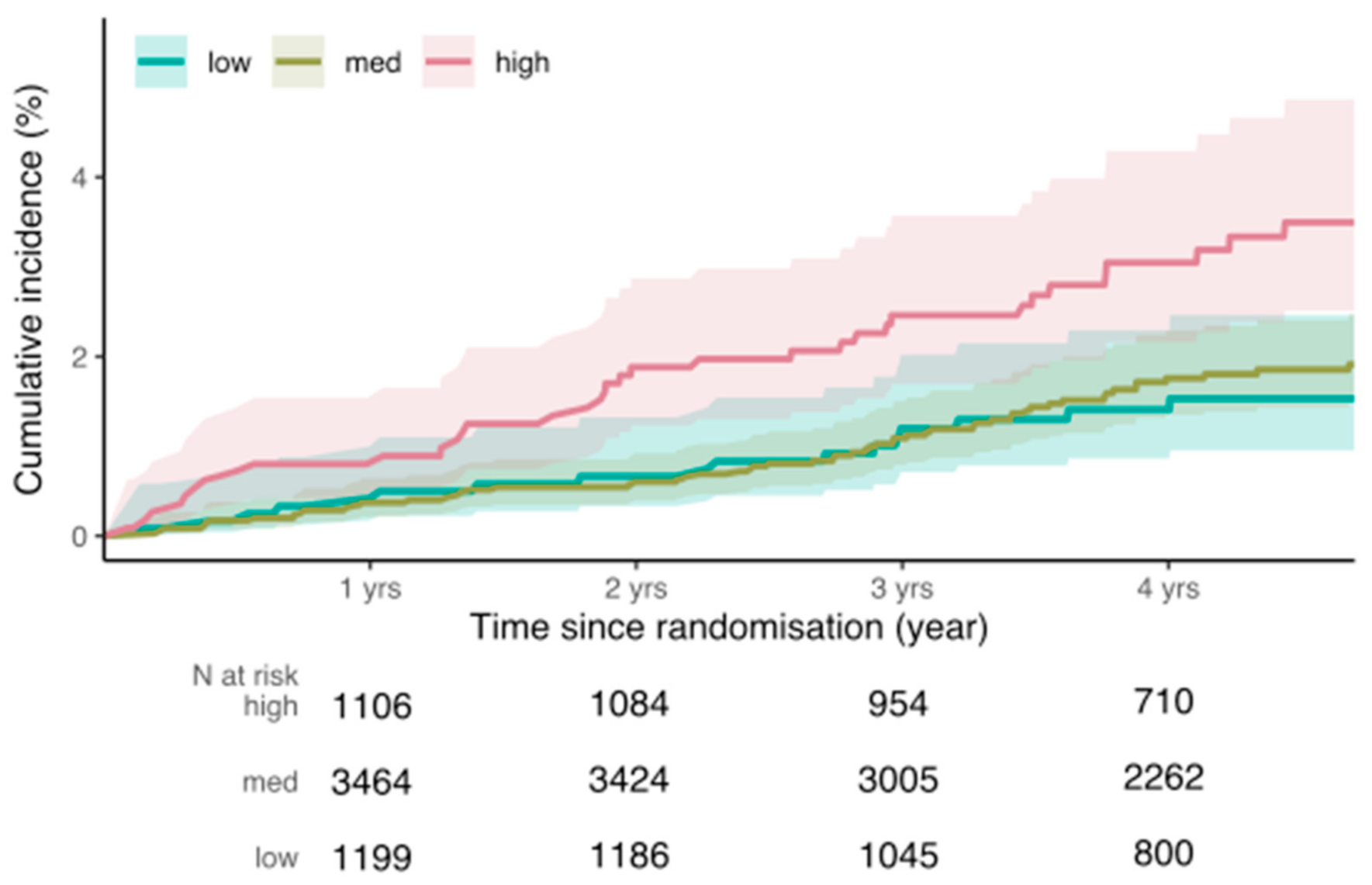
3.3. Incident Breast Cancer Risk
3.4. Prevalent Breast Cancer
3.5. Modification of BC Risk by PRS in Rare PV Carriers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mavaddat, N.; Michailidou, K.; Dennis, J.; Lush, M.; Fachal, L.; Lee, A.; Tyrer, J.P.; Chen, T.H.; Wang, Q.; Bolla, M.K.; et al. Polygenic Risk Scores for Prediction of Breast Cancer and Breast Cancer Subtypes. Am. J. Hum. Genet. 2019, 104, 21–34. [Google Scholar] [CrossRef] [Green Version]
- Lakeman, I.M.M.; Rodriguez-Girondo, M.; Lee, A.; Ruiter, R.; Stricker, B.H.; Wijnant, S.R.A.; Kavousi, M.; Antoniou, A.C.; Schmidt, M.K.; Uitterlinden, A.G.; et al. Validation of the BOADICEA model and a 313-variant polygenic risk score for breast cancer risk prediction in a Dutch prospective cohort. Genet. Med. 2020, 22, 1803–1811. [Google Scholar] [CrossRef]
- Kapoor, P.M.; Mavaddat, N.; Choudhury, P.P.; Wilcox, A.N.; Lindstrom, S.; Behrens, S.; Michailidou, K.; Dennis, J.; Bolla, M.K.; Wang, Q.; et al. Combined associations of a polygenic risk score and classical risk factors with breast cancer risk. J. Natl. Cancer Inst. 2020, 113, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Mars, N.; Koskela, J.T.; Ripatti, P.; Kiiskinen, T.T.J.; Havulinna, A.S.; Lindbohm, J.V.; Ahola-Olli, A.; Kurki, M.; Karjalainen, J.; Palta, P.; et al. Polygenic and clinical risk scores and their impact on age at onset and prediction of cardiometabolic diseases and common cancers. Nat. Med. 2020, 26, 549–557. [Google Scholar] [CrossRef]
- Michailidou, K.; Lindstrom, S.; Dennis, J.; Beesley, J.; Hui, S.; Kar, S.; Lemacon, A.; Soucy, P.; Glubb, D.; Rostamianfar, A.; et al. Association analysis identifies 65 new breast cancer risk loci. Nature 2017, 551, 92–94. [Google Scholar] [CrossRef] [Green Version]
- Anglian Breast Cancer Study Group. Prevalence and penetrance of BRCA1 and BRCA2 mutations in a population-based series of breast cancer cases. Br. J. Cancer 2000, 83, 1301–1308. [Google Scholar] [CrossRef] [PubMed]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.A.; Mooij, T.M.; Roos-Blom, M.J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mavaddat, N.; Peock, S.; Frost, D.; Ellis, S.; Platte, R.; Fineberg, E.; Evans, D.G.; Izatt, L.; Eeles, R.A.; Adlard, J.; et al. Cancer risks for BRCA1 and BRCA2 mutation carriers: Results from prospective analysis of EMBRACE. J. Natl. Cancer Inst. 2013, 105, 812–822. [Google Scholar] [CrossRef] [Green Version]
- Barnes, D.R.; Rookus, M.A.; McGuffog, L.; Leslie, G.; Mooij, T.M.; Dennis, J.; Mavaddat, N.; Adlard, J.; Ahmed, M.; Aittomaki, K.; et al. Polygenic risk scores and breast and epithelial ovarian cancer risks for carriers of BRCA1 and BRCA2 pathogenic variants. Genet. Med. 2020, 22, 1653–1666. [Google Scholar] [CrossRef]
- Kuchenbaecker, K.B.; McGuffog, L.; Barrowdale, D.; Lee, A.; Soucy, P.; Dennis, J.; Domchek, S.M.; Robson, M.; Spurdle, A.B.; Ramus, S.J.; et al. Evaluation of Polygenic Risk Scores for Breast and Ovarian Cancer Risk Prediction in BRCA1 and BRCA2 Mutation Carriers. J. Natl. Cancer Inst. 2017, 109, djw302. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.J.; Cunningham, A.P.; Tischkowitz, M.; Simard, J.; Pharoah, P.D.; Easton, D.F.; Antoniou, A.C. Incorporating truncating variants in PALB2, CHEK2, and ATM into the BOADICEA breast cancer risk model. Genet. Med. 2016, 18, 1190–1198. [Google Scholar] [CrossRef] [Green Version]
- Breast Cancer Association Consortium; Dorling, L.; Carvalho, S.; Allen, J.; Gonzalez-Neira, A.; Luccarini, C.; Wahlstrom, C.; Pooley, K.A.; Parsons, M.T.; Fortuno, C.; et al. Breast Cancer Risk Genes—Association Analysis in More than 113,000 Women. N. Engl. J. Med. 2021, 384, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Hart, S.N.; Gnanaolivu, R.; Huang, H.; Lee, K.Y.; Na, J.; Gao, C.; Lilyquist, J.; Yadav, S.; Boddicker, N.J.; et al. A Population-Based Study of Genes Previously Implicated in Breast Cancer. N. Engl. J. Med. 2021, 384, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Bahcall, O. Common variation and heritability estimates for breast, ovarian and prostate cancers. Nat. Genet. 2013, 10, 304. [Google Scholar] [CrossRef]
- McNeil, J.J.; Wolfe, R.; Woods, R.L.; Tonkin, A.M.; Donnan, G.A.; Nelson, M.R.; Reid, C.M.; Lockery, J.E.; Kirpach, B.; Storey, E.; et al. Effect of Aspirin on Cardiovascular Events and Bleeding in the Healthy Elderly. N. Engl. J. Med. 2018, 379, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- McNeil, J.J.; Woods, R.L.; Nelson, M.R.; Reid, C.M.; Kirpach, B.; Wolfe, R.; Storey, E.; Shah, R.C.; Lockery, J.E.; Tonkin, A.M.; et al. Effect of Aspirin on Disability-free Survival in the Healthy Elderly. N. Engl. J. Med. 2018, 379, 1499–1508. [Google Scholar] [CrossRef] [PubMed]
- McNeil, J.J.; Nelson, M.R.; Woods, R.L.; Lockery, J.E.; Wolfe, R.; Reid, C.M.; Kirpach, B.; Shah, R.C.; Ives, D.G.; Storey, E.; et al. Effect of Aspirin on All-Cause Mortality in the Healthy Elderly. N. Engl. J. Med. 2018, 379, 1519–1528. [Google Scholar] [CrossRef]
- ASPREE Investigator Group. Study design of ASPirin in Reducing Events in the Elderly (ASPREE): A randomized, controlled trial. Contemp. Clin. Trials 2013, 36, 555–564. [Google Scholar] [CrossRef] [Green Version]
- Nelson, M.R.; Reid, C.M.; Ames, D.A.; Beilin, L.J.; Donnan, G.A.; Gibbs, P.; Johnston, C.I.; Krum, H.; Storey, E.; Tonkin, A.; et al. Feasibility of conducting a primary prevention trial of low-dose aspirin for major adverse cardiovascular events in older people in Australia: Results from the ASPirin in Reducing Events in the Elderly (ASPREE) pilot study. Med. J. Aust. 2008, 189, 105–109. [Google Scholar] [CrossRef]
- Lockery, J.E.; Collyer, T.A.; Abhayaratna, W.P.; Fitzgerald, S.M.; McNeil, J.J.; Nelson, M.R.; Orchard, S.G.; Reid, C.; Stocks, N.P.; Trevaks, R.E.; et al. Recruiting general practice patients for large clinical trials: Lessons from the Aspirin in Reducing Events in the Elderly (ASPREE) study. Med. J. Aust. 2019, 210, 168–173. [Google Scholar] [CrossRef] [Green Version]
- McNeil, J.J.; Woods, R.L.; Nelson, M.R.; Murray, A.M.; Reid, C.M.; Kirpach, B.; Storey, E.; Shah, R.C.; Wolfe, R.S.; Tonkin, A.M.; et al. Baseline Characteristics of Participants in the ASPREE (ASPirin in Reducing Events in the Elderly) Study. J Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 1586–1593. [Google Scholar] [CrossRef] [Green Version]
- Genomes Project Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar]
- Taliun, D.; Harris, D.N.; Kessler, M.D.; Carlson, J.; Szpiech, Z.A.; Torres, R.; Taliun, S.A.G.; Corvelo, A.; Gogarten, S.M.; Kang, H.M.; et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 2021, 590, 290–299. [Google Scholar] [CrossRef]
- Lacaze, P.; Sebra, R.; Riaz, M.; Tiller, J.; Revote, J.; Phung, J.; Parker, E.J.; Orchard, S.G.; Lockery, J.E.; Wolfe, R.; et al. Medically actionable pathogenic variants in a population of 13,131 healthy elderly individuals. Genet. Med. 2020, 22, 1883–1886. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Orchard, S.G.; Lockery, J.E.; Gibbs, P.; Polekhina, G.; Wolfe, R.; Zalcberg, J.; Haydon, A.; McNeil, J.J.; Nelson, M.R.; Reid, C.M.; et al. Cancer history and risk factors in healthy older people enrolling in the ASPREE clinical trial. Contemp. Clin. Trials 2020, 96, 106095. [Google Scholar] [CrossRef]
- Song, M.; Kraft, P.; Joshi, A.D.; Barrdahl, M.; Chatterjee, N. Testing calibration of risk models at extremes of disease risk. Biostatistics 2015, 16, 143–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeLong, E.R.; DeLong, D.M.; Clarke-Pearson, D.L. Comparing the areas under two or more correlated receiver operating characteristic curves: A nonparametric approach. Biometrics 1988, 44, 837–845. [Google Scholar] [CrossRef]
- Sun, X.; Xu, W. Fast Implementation of DeLong’s Algorithm for Comparing the Areas under Correlated Receiver Operating Characteristic Curves. IEEE Signal Process. Lett. 2014, 21, 1389–1393. [Google Scholar] [CrossRef]
- Fahed, A.C.; Wang, M.; Homburger, J.R.; Patel, A.P.; Bick, A.G.; Neben, C.L.; Lai, C.; Brockman, D.; Philippakis, A.; Ellinor, P.T.; et al. Polygenic background modifies penetrance of monogenic variants for tier 1 genomic conditions. Nat. Commun. 2020, 11, 3635. [Google Scholar] [CrossRef]
- Sudlow, C.; Gallacher, J.; Allen, N.; Beral, V.; Burton, P.; Danesh, J.; Downey, P.; Elliott, P.; Green, J.; Landray, M.; et al. UK biobank: An open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015, 12, e1001779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stovitz, S.D.; Banack, H.R.; Kaufman, J.S. “Depletion of the susceptibles” taught through a story, a table and basic arithmetic. BMJ Evid. Based Med. 2018, 23, 199. [Google Scholar] [CrossRef] [PubMed]
- Khera, A.V.; Chaffin, M.; Aragam, K.G.; Haas, M.E.; Roselli, C.; Choi, S.H.; Natarajan, P.; Lander, E.S.; Lubitz, S.A.; Ellinor, P.T.; et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 2018, 50, 1219–1224. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Mavaddat, N.; Wilcox, A.N.; Cunningham, A.P.; Carver, T.; Hartley, S.; Babb de Villiers, C.; Izquierdo, A.; Simard, J.; Schmidt, M.K.; et al. BOADICEA: A comprehensive breast cancer risk prediction model incorporating genetic and nongenetic risk factors. Genet. Med. 2019, 21, 1708–1718. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Characteristics | Total | Low-Risk PRS (Q1) | Moderate-Risk PRS (Q2–4) | High-Risk PRS (Q5) |
|---|---|---|---|---|
| Participants | n = 6339 | n = 1268 | n = 3803 | n = 1268 |
| Sex = Female (%) | 6339 (100) | 1268 (100) | 3803 (100) | 1268 (100) |
| Mean Age at Enrolment, Years | 75.1 | 75.1 | 75.1 | 75.2 |
| Age Group, Years (%) | ||||
| 70–74 | 3825 (60.3) | 778 (61.4) | 2287 (60.1) | 760 (59.9) |
| 75–79 | 1599 (25.5) | 304 (24.0) | 975 (25.6) | 320 (25.2) |
| 80–84 | 706 (11.1) | 136 (10.7) | 429 (11.3) | 141 (11.1) |
| 85+ | 211 (3.3) | 50 (3.9) | 112 (2.9) | 47 (3.7) |
| Current or Former Smoker (%) | 1974 (31.1) | 391 (30.8) | 1195 (31.4) | 388 (30.6) |
| Diabetes (%) | 497 (7.8) | 97 (7.6) | 289 (7.6) | 111 (8.8) |
| Randomized to Aspirin (%) | 3170 (50.0) | 630 (49.7) | 1898 (49.9) | 642 (50.6) |
| Body Mass Index kg/m2 ((mean) (SD)) | 28.03 (5.09) | 28.02 (5.02) | 28.05 (5.10) | 28.00 (5.13) |
| Current Alcohol Consumption (%) | 4730 (74.6) | 941 (74.2) | 2850 (74.9) | 939 (74.1) |
| Hormone Replacement Therapy * | 533 (8.4) | 103 (8.1) | 321 (8.4) | 109 (8.6) |
| Progesterone-Only HRT | 6 (0.2) | 1 (0.1) | 3 (0.1) | 2 (0.2) |
| Family History of Breast Cancer (%) ‡ | 850 (13.4) | 135 (10.6) | 500 (13.1) | 215 (17.0) |
| Prevalent Breast Cancer | ||||
| Cases | 475 | 47 | 288 | 140 |
| Diagnosed <49 Years | 60 | 6 | 39 | 15 |
| Diagnosed 50+ Years | 415 | 41 | 249 | 125 |
| Incident Breast Cancer ¶ | ||||
| Cases | 129 | 21 | 66 | 42 |
| Polygenic Risk Score (mean (SD)) | 0.1 (0.53) | −0.93 (0.26) | −0.13 (0.27) | 0.69 (0.28) |
| Variable | PRS as Continuous Variable | PRS as Categorical Variable | ||||
|---|---|---|---|---|---|---|
| Hazard Ratio | 95% CI | p-Value | Hazard Ratio | 95% CI | p-Value | |
| Polygenic Score (per standard deviation) | 1.43 | (1.18; 1.73) | <0.001 | |||
| Low PRS (Q1) | Reference | |||||
| Moderate PRS (Q2,3,4) | 1.16 | (0.68; 2.00) | 0.58 | |||
| High PRS (Q5) | 2.16 | (1.21; 3.86) | 0.009 | |||
| Pathogenic Variants (n = 41 carriers) | No incident events | No incident events | ||||
| Family History of Breast Cancer * (Y/N) | 1.81 | (1.15; 2.85) | 0.01 | 1.83 | (1.16; 2.88) | 0.009 |
| Age at Enrolment | 0.97 | (0.92; 1.02) | 0.21 | 0.97 | (0.92; 1.02) | 0.22 |
| Treatment (Aspirin) | 1.16 | (0.80; 1.69) | 0.44 | 1.15 | (0.79; 1.68) | 0.45 |
| Number of Children | 0.81 | (0.66; 0.99) | 0.04 | 0.81 | (0.66; 0.99) | 0.04 |
| Body Mass Index (kg/m2 (mean) SD) | 1.14 | (0.95; 1.37) | 0.17 | 1.14 | (0.95; 1.37) | 0.15 |
| Alcohol (None) | Reference | Reference | ||||
| Alcohol (Low) | 1.16 | (0.68; 1.97) | 0.59 | 1.16 | (0.68; 1.98) | 0.58 |
| Alcohol (High) | 1.70 | (1.01; 2.85) | 0.04 | 1.70 | (1.02; 2.86) | 0.04 |
| HRT ‡ (Y/N) | 1.54 | (0.88; 2.71) | 0.13 | 1.51 | (0.86; 2.65) | 0.15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lacaze, P.; Bakshi, A.; Riaz, M.; Orchard, S.G.; Tiller, J.; Neumann, J.T.; Carr, P.R.; Joshi, A.D.; Cao, Y.; Warner, E.T.; et al. Genomic Risk Prediction for Breast Cancer in Older Women. Cancers 2021, 13, 3533. https://doi.org/10.3390/cancers13143533
Lacaze P, Bakshi A, Riaz M, Orchard SG, Tiller J, Neumann JT, Carr PR, Joshi AD, Cao Y, Warner ET, et al. Genomic Risk Prediction for Breast Cancer in Older Women. Cancers. 2021; 13(14):3533. https://doi.org/10.3390/cancers13143533
Chicago/Turabian StyleLacaze, Paul, Andrew Bakshi, Moeen Riaz, Suzanne G. Orchard, Jane Tiller, Johannes T. Neumann, Prudence R. Carr, Amit D. Joshi, Yin Cao, Erica T. Warner, and et al. 2021. "Genomic Risk Prediction for Breast Cancer in Older Women" Cancers 13, no. 14: 3533. https://doi.org/10.3390/cancers13143533