
PD-L1 Overexpression, SWI/SNF Complex Deregulation, and Profound Transcriptomic Changes Characterize Cancer-Dependent Exhaustion of Persistently Activated CD4+ T Cells

 , , , , , , add
Show full author list
, , , , , , add
Show full author list
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. CD4+ T Cells Isolation and Stimulation
2.3. Transcriptome Profiling (RNA Sequencing)
2.4. Quantification of mRNA Expression
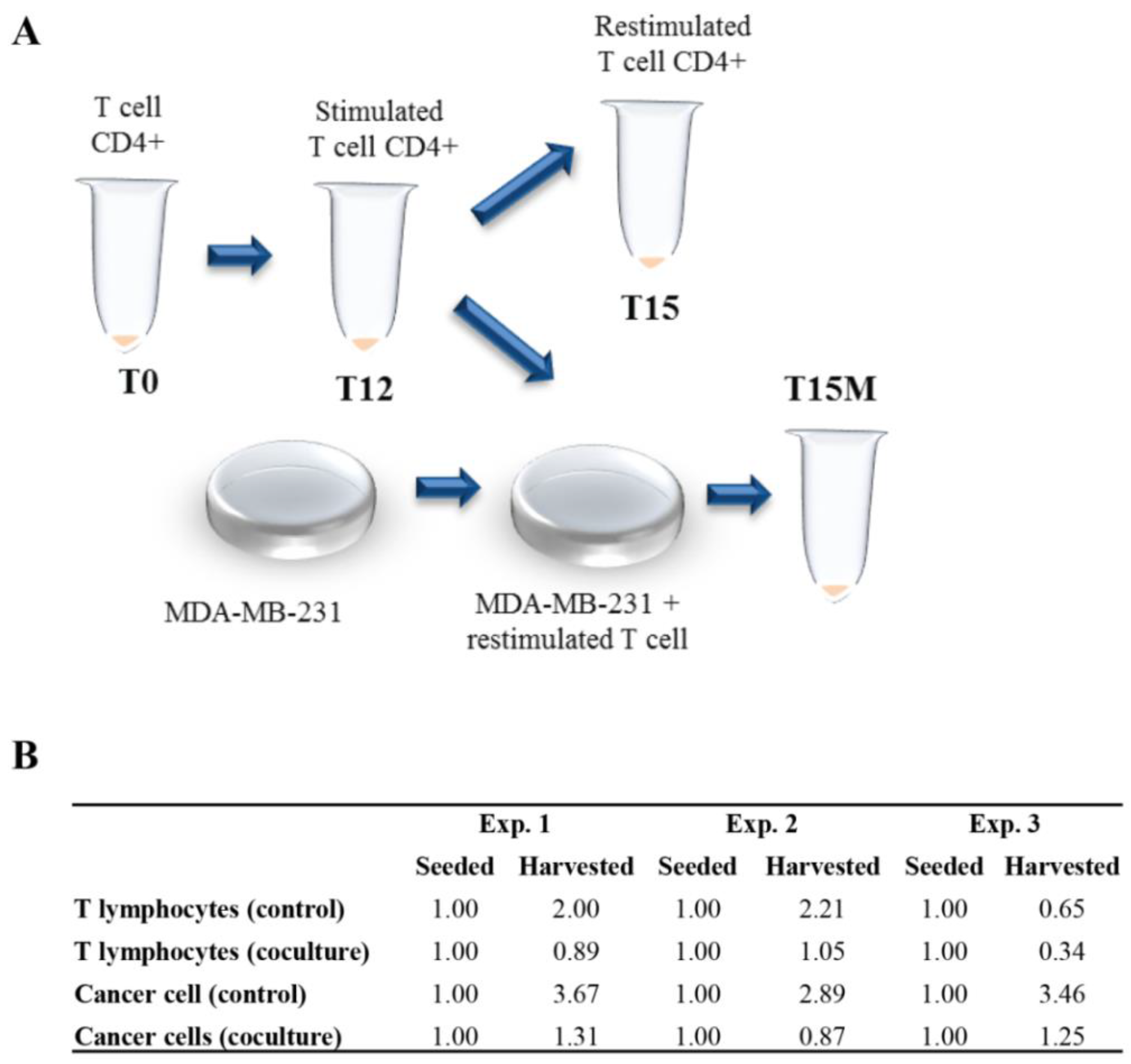
2.5. Co-Culture of Cancer Cells and CD4+ T-Cells
2.6. Antibodies and Flow Cytometry
2.7. Protein Analysis Using SDS-PAGE and Western Blot
2.8. Chromatin Immunoprecipitation
2.9. Proteome Identification from Medium Using Mass Spectrometry Method
2.10. Statistical Analysis
3. Results
3.1. The CD4+ T Cells Inhibit Cancer Cell Growth but Concomitantly Cancer Cells Suppress Proliferation of the CD4+ T Cell Population
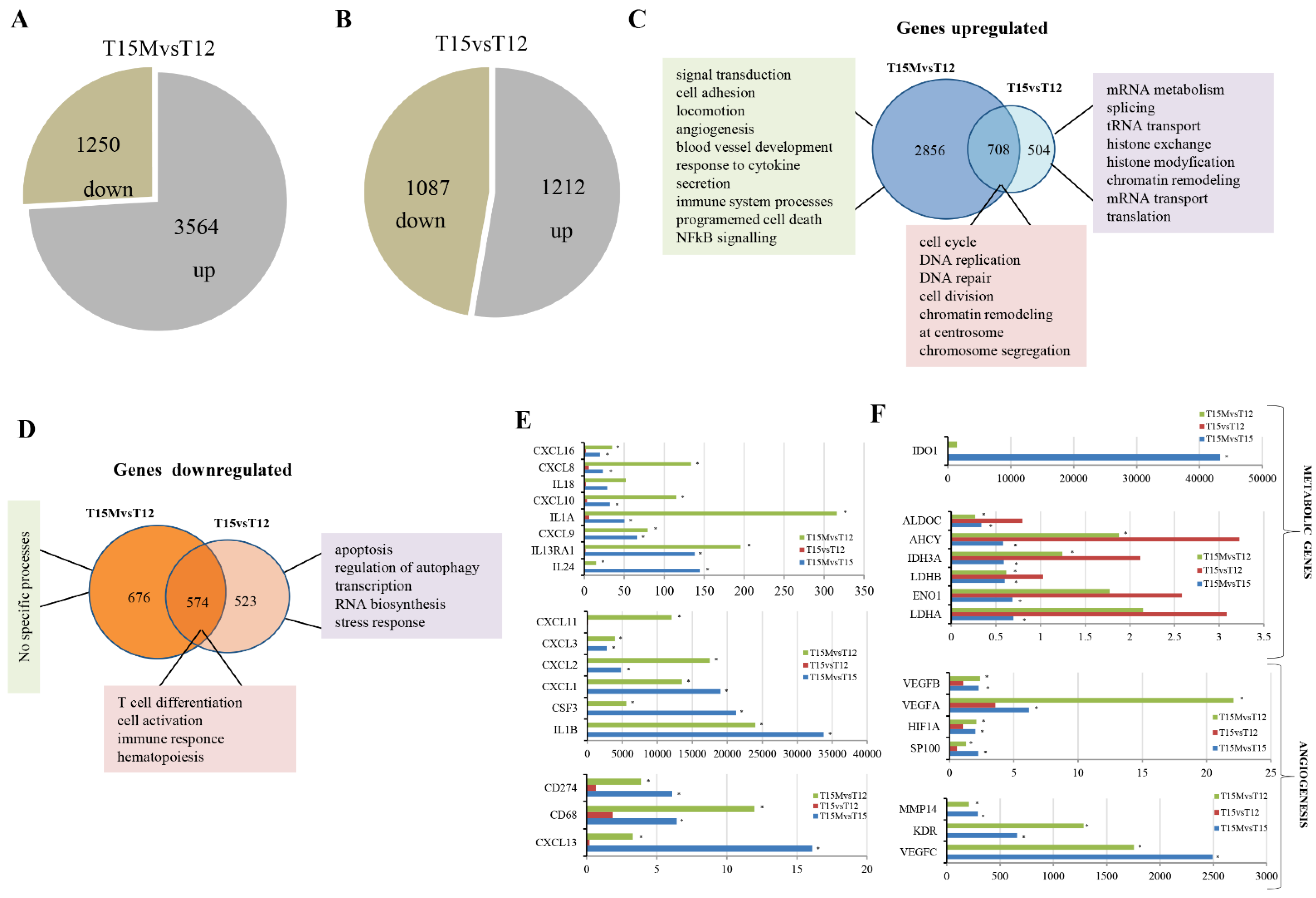
3.2. MDA-MB-231 Cancer Cells Affect the Global Transcriptome in CD4+ T Cells during Co-Culture
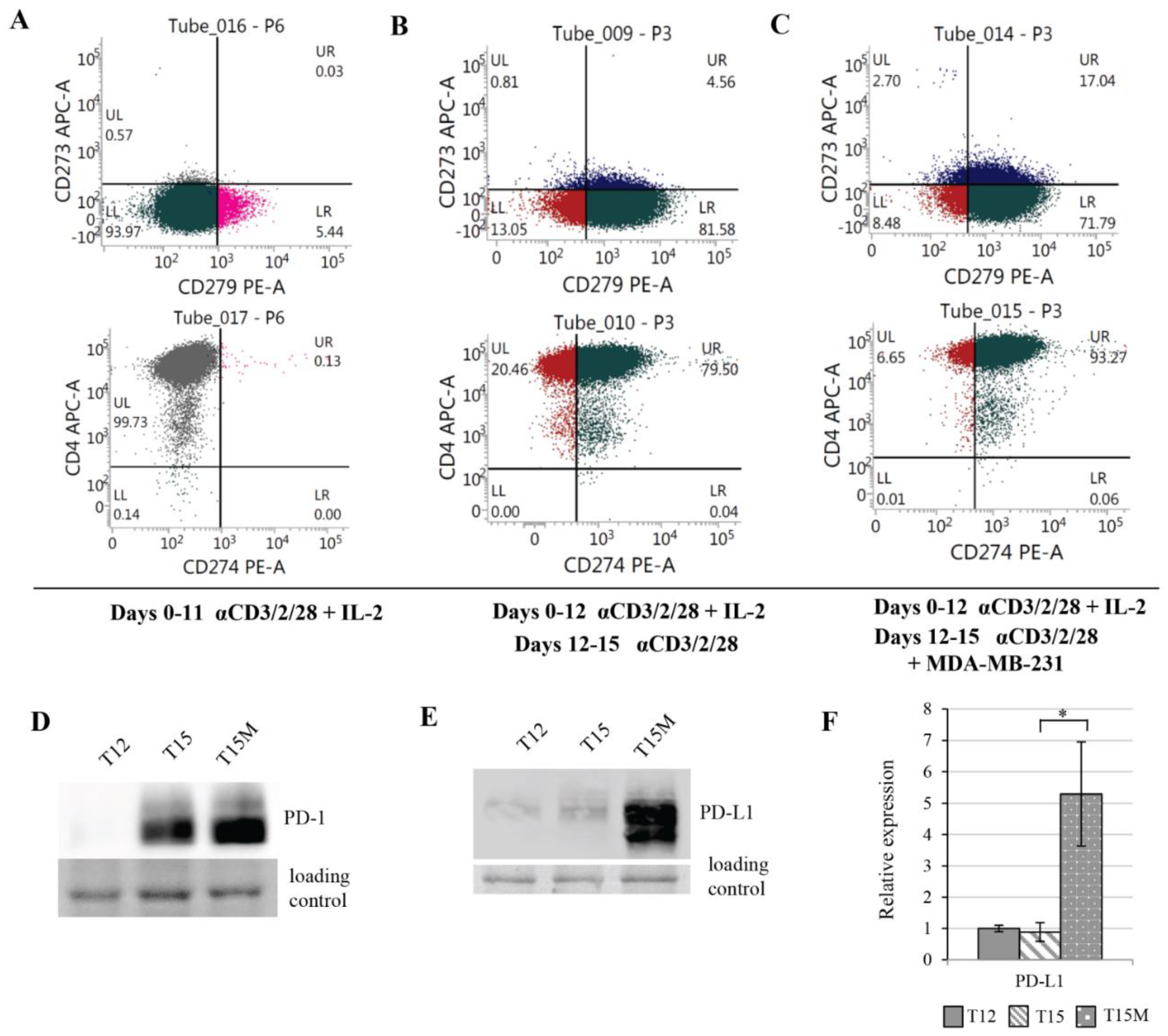
3.3. MDA-MB-231 Cancer Cells Promote Upregulation of Genes Encoding PD-1 and PD-1-Ligands in Persistently Activated CD4+ T Cells
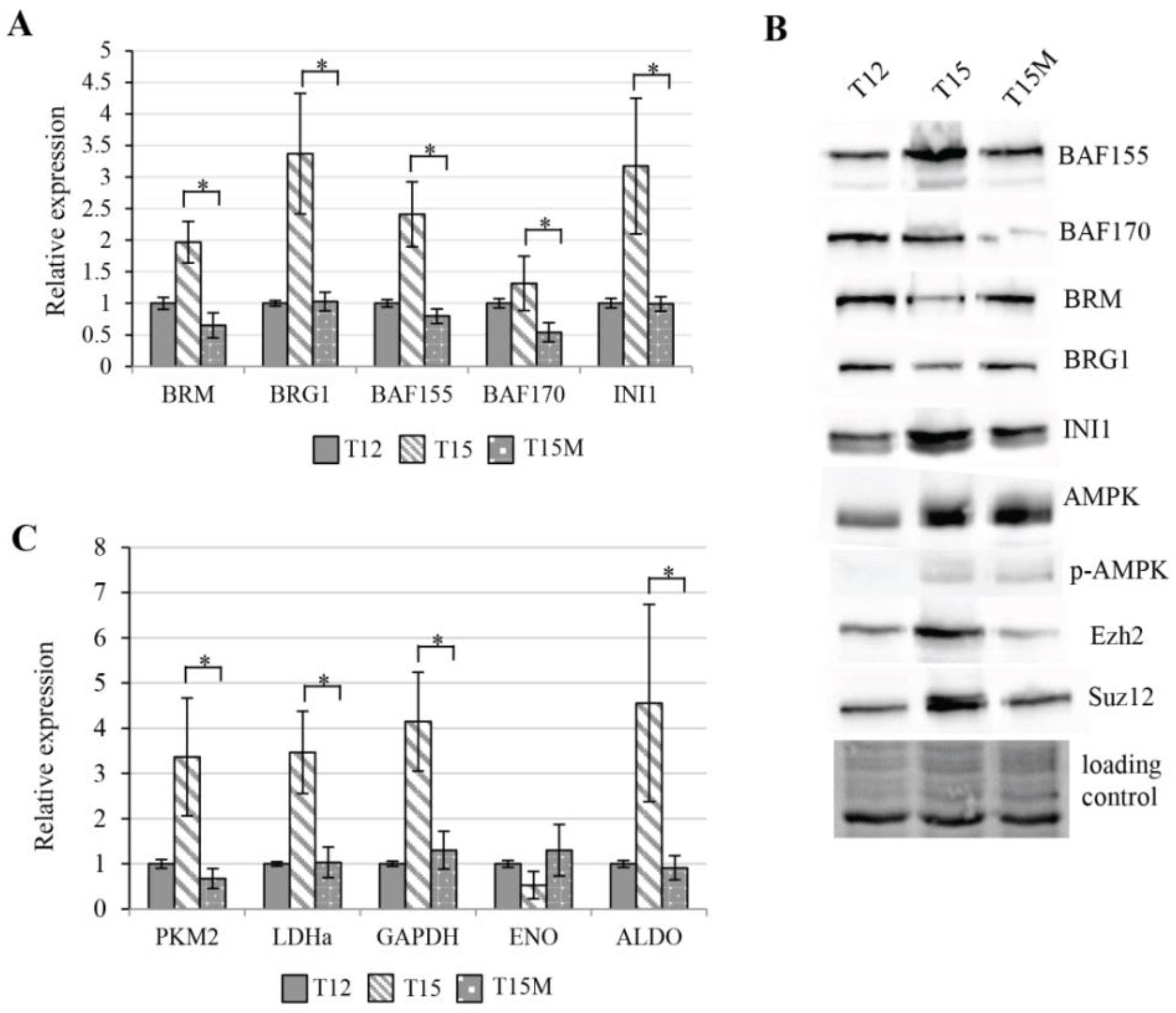
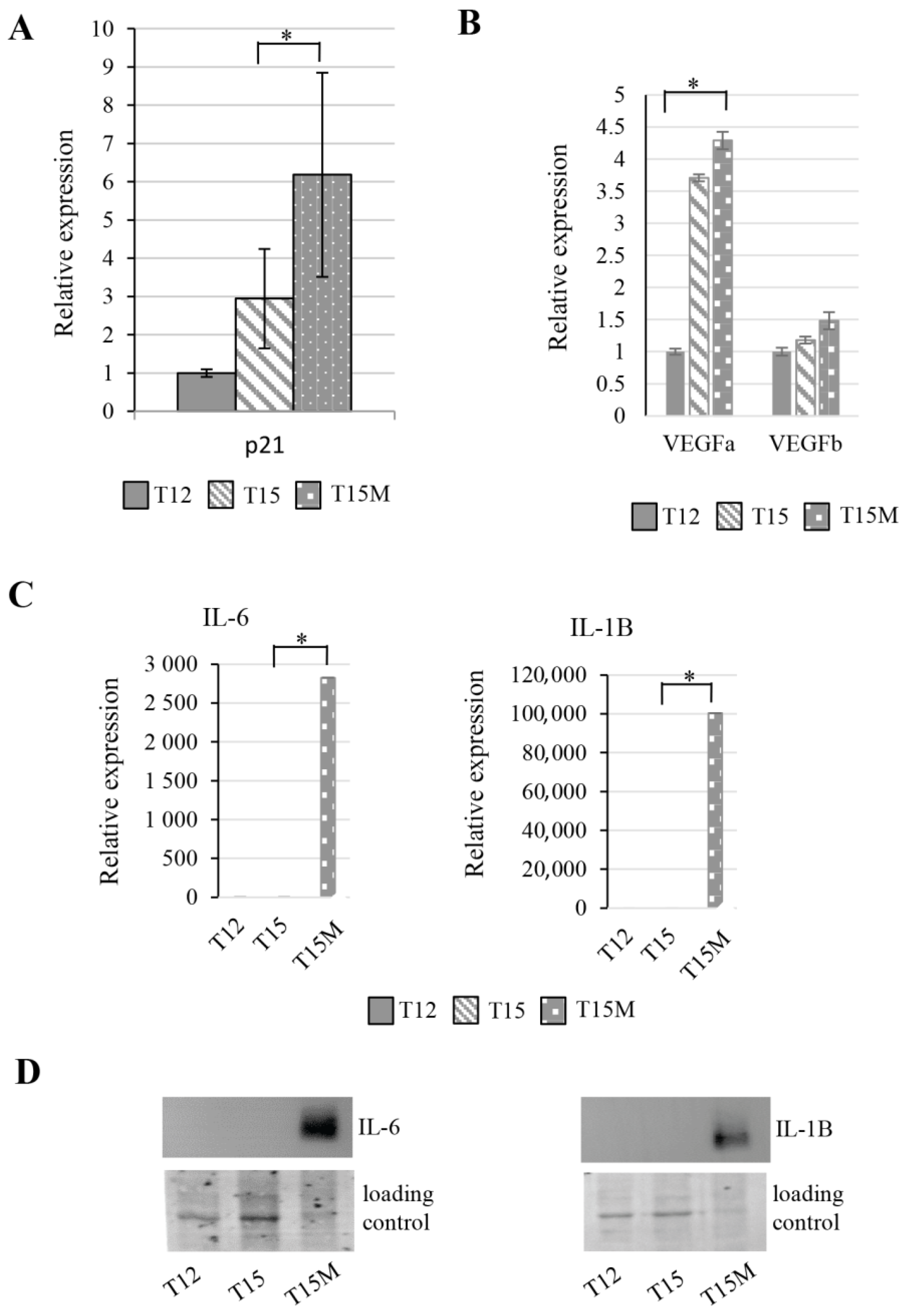
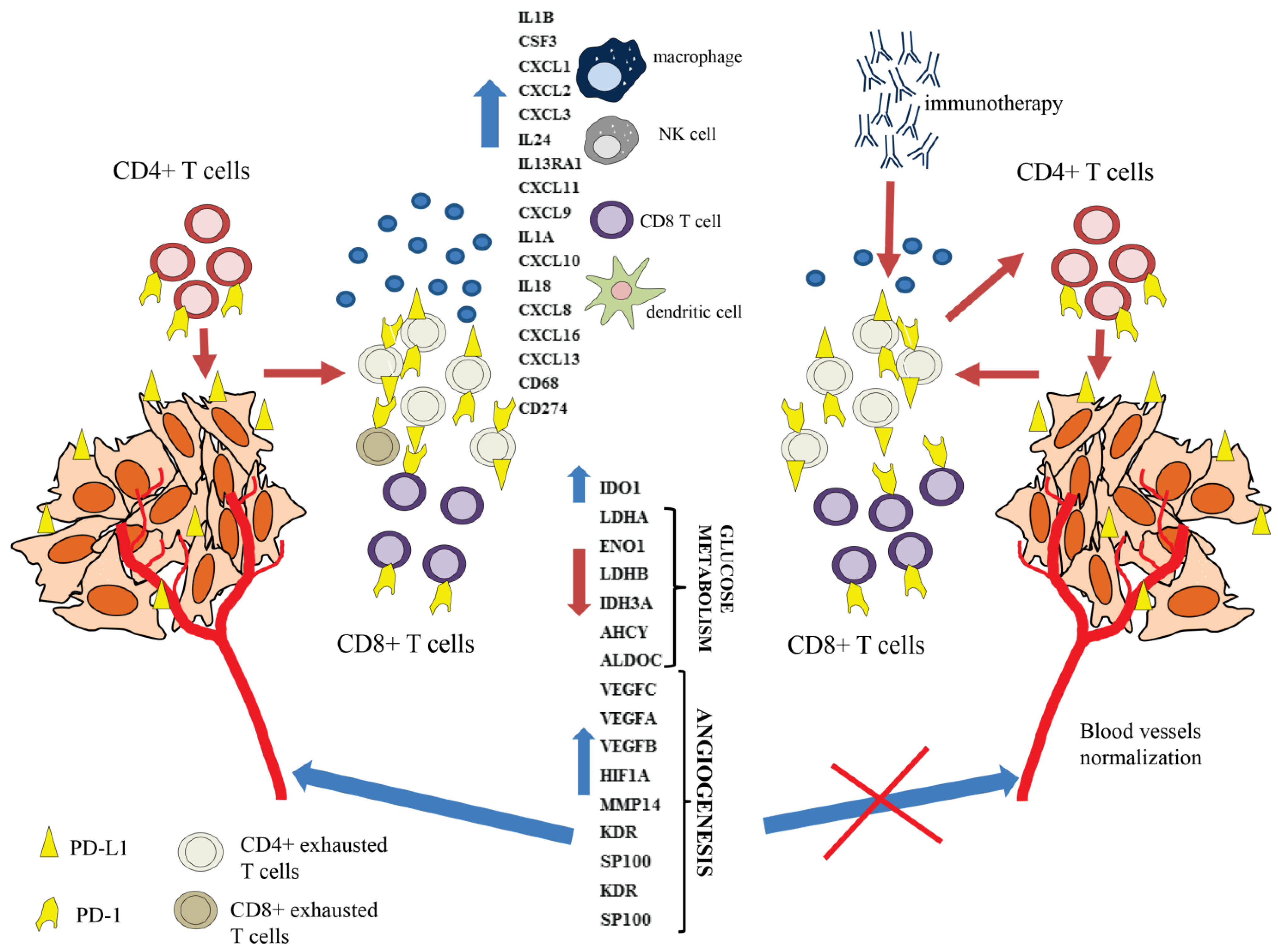
3.4. Exhaustion Process Induced by Cancer Cells Affects Expression of the SWI/SNF Chromatin Remodeling Complex, Glucose Metabolism, and Angiogenesis Genes
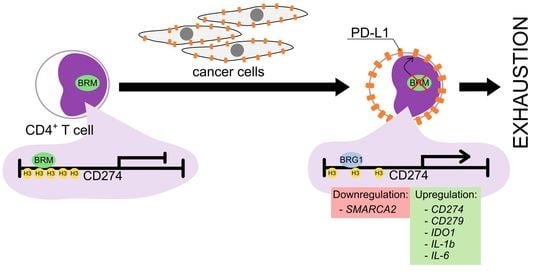
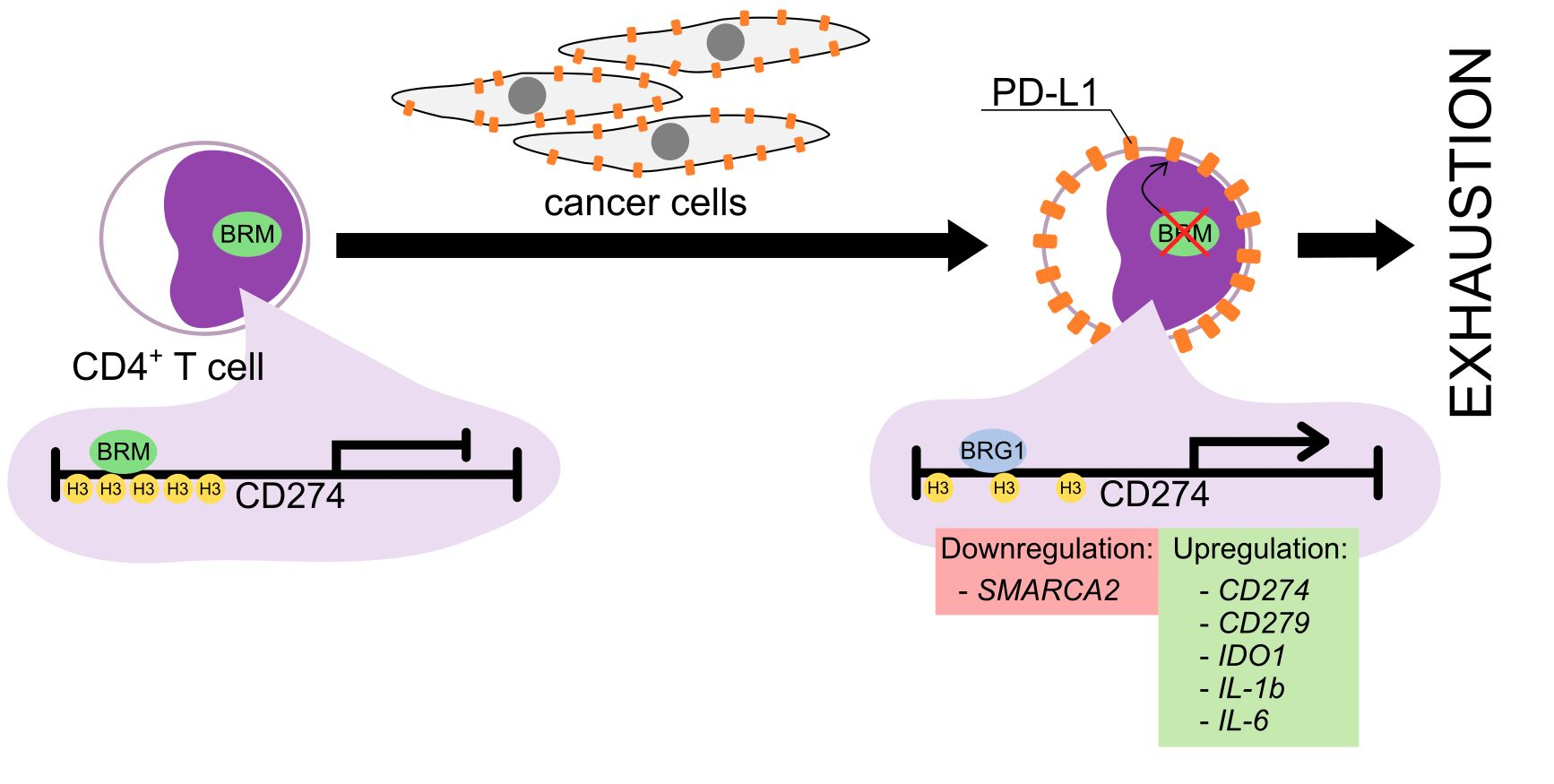
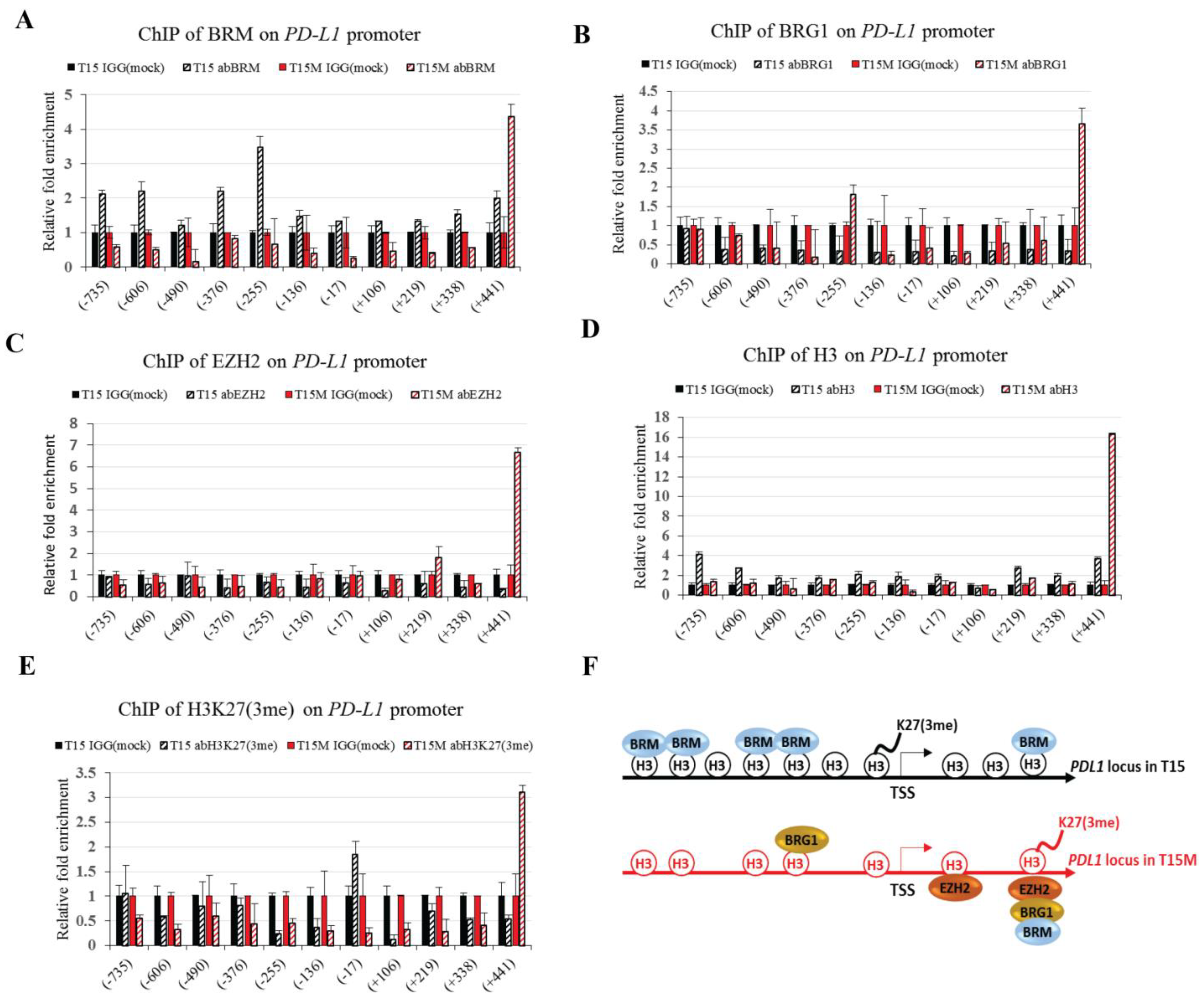
3.5. The BRM Containing the SWI/SNF Chromatin Remodeling Complex Directly Controls the PD-L1 Locus in CD4+ T Cells
3.6. The CD4+ T Cell Exhaustion Process Is Reversible
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [Green Version]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining ‘T cell exhaustion’. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef]
- Tang, L.; Zhang, Y.; Hu, Y.; Mei, H. T Cell Exhaustion and CAR-T Immunotherapy in Hematological Malignancies. Biomed. Res. Int. 2021, 2021, 6616391. [Google Scholar] [CrossRef] [PubMed]
- Balkhi, M.Y. Receptor signaling, transcriptional, and metabolic regulation of T cell exhaustion. Oncoimmunology 2020, 9, 1747349. [Google Scholar] [CrossRef] [Green Version]
- Scharping, N.E.; Rivadeneira, D.B.; Menk, A.V.; Vignali, P.D.A.; Ford, B.R.; Rittenhouse, N.L.; Peralta, R.; Wang, Y.; Wang, Y.; DePeaux, K.; et al. Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat. Immunol. 2021, 22, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Verdon, D.J.; Mulazzani, M.; Jenkins, M.R. Cellular and Molecular Mechanisms of CD8+ T Cell Differentiation, Dysfunction and Exhaustion. Int. J. Mol. Sci. 2020, 21, 7357. [Google Scholar] [CrossRef] [PubMed]
- Leone, R.D.; Emens, L.A. Targeting adenosine for cancer immunotherapy. J. Immunother. Cancer 2018, 6, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eil, R.; Vodnala, S.K.; Clever, D.; Klebanoff, C.A.; Sukumar, M.; Pan, J.H.; Palmer, D.C.; Gros, A.; Yamamoto, T.N.; Patel, S.J.; et al. Ionic immune suppression within the tumour microenvironment limits T cell effector function. Nature 2016, 537, 539–543. [Google Scholar] [CrossRef] [Green Version]
- Perez-Diez, A.; Joncker, N.T.; Choi, K.; Chan, W.F.N.; Anderson, C.C.; Lantz, O.; Matzinger, P. CD4 cells can be more efficient at tumor rejection than CD8 cells. Blood 2007, 109, 5346–5354. [Google Scholar] [CrossRef]
- Schietinger, A.; Philip, M.; Liu, R.B.; Schreiber, K.; Schreiber, H. Bystander killing of cancer requires the cooperation of CD4(+) and CD8(+) T cells during the effector phase. J. Exp. Med. 2010, 207, 2469–2477. [Google Scholar] [CrossRef]
- Zuazo, M.; Arasanz, H.; Bocanegra, A.; Fernandez, G.; Chocarro, L.; Vera, R.; Kochan, G.; Escors, D. Systemic CD4 Immunity as a Key Contributor to PD-L1/PD-1 Blockade Immunotherapy Efficacy. Front. Immunol. 2020, 11, 1–10. [Google Scholar] [CrossRef]
- Lichawska-Cieslar, A.; Pietrzycka, R.; Ligeza, J.; Kulecka, M.; Paziewska, A.; Kalita, A.; Dolicka, D.D.; Wilamowski, M.; Miekus, K.; Ostrowski, J.; et al. RNA sequencing reveals widespread transcriptome changes in a renal carcinoma cell line. Oncotarget 2018, 9, 8597–8613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. Genome analysis HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 2014, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, X.; Huang, X.; Zhou, Z.; Lin, X. An improvement of the 2ˆ(-delta delta CT) method for quantitative real-time polymerase chain reaction data analysis. Biostat. Bioinform. Biomath. 2013, 3, 71–85. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Gonzalez-Aparicio, M.; Alfaro, C. Significance of the IL-8 pathway for immunotherapy. Hum. Vaccin. Immunother. 2020, 16, 2312–2317. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wang, X.; Wang, L.; Ma, X.; Gong, Z.; Zhang, S.; Li, Y. Targeting the IDO1 pathway in cancer: From bench to bedside. J. Hematol. Oncol. 2018, 11, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettonville, M.; D’Aria, S.; Braun, M.Y. Metabolic programming in chronically stimulated T cells: Lessons from cancer and viral infections. Eur. J. Immunol. 2016, 46, 1574–1582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Bourgeois, T.; Strauss, L.; Aksoylar, H.I.; Daneshmandi, S.; Seth, P.; Patsoukis, N.; Boussiotis, V.A. Targeting T cell metabolism for improvement of cancer immunotherapy. Front. Oncol. 2018, 8, 237. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Long, G.V.; Scolyer, R.A.; Teng, M.W.L.; Smyth, M.J. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat. Rev. 2017, 52, 71–81. [Google Scholar] [CrossRef] [Green Version]
- Shen, C.; Zhang, Z.; Zhang, Y. Chimeric antigen receptor T cell exhaustion during treatment for hematological malignancies. Biomed Res. Int. 2020, 2020, 8765028. [Google Scholar] [CrossRef]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. ‘Off-the-shelf’ allogeneic CAR T cells: Development and challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef]
- He, Q.-F.; Xu, Y.; Li, J.; Huang, Z.-M.; Li, X.-H.; Wang, X. CD8+ T-cell exhaustion in cancer: Mechanisms and new area for cancer immunotherapy. Brief. Funct. Genom. 2019, 18, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, S.; Zhang, B.; Qiao, L.; Zhang, Y.; Zhang, Y. T Cell Dysfunction and Exhaustion in Cancer. Front. Cell Dev. Biol. 2020, 8, 17. [Google Scholar] [CrossRef] [Green Version]
- Kohli, K.; Pillarisetty, V.G.; Kim, T.S. Key chemokines direct migration of immune cells in solid tumors. Cancer Gene Ther. 2021, 1–12. [Google Scholar] [CrossRef]
- Zhang, Y.; Guan, X.Y.; Jiang, P. Cytokine and Chemokine Signals of T-Cell Exclusion in Tumors. Front. Immunol. 2020, 11, 594609. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.T.; Appenheimer, M.M.; Evans, S.S. The two faces of IL-6 in the tumor microenvironment. Semin. Immunol. 2014, 26, 38–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplanov, I.; Carmi, Y.; Kornetsky, R.; Shemesh, A.; Shurin, G.V.; Shurin, M.R.; Dinarello, C.A.; Voronov, E.; Apte, R.N. Blocking IL-1β reverses the immunosuppression in mouse breast cancer and synergizes with anti–PD-1 for tumor abrogation. Proc. Natl. Acad. Sci. USA 2019, 116, 1361–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frumento, G.; Rotondo, R.; Tonetti, M.; Damonte, G.; Benatti, U.; Ferrara, G.B. Tryptophan-derived Catabolites Are Responsible for Inhibition of T and Natural Killer Cell Proliferation Induced by Indoleamine 2,3-Dioxygenase. J. Exp. Med. 2002, 196, 459–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rad Pour, S.; Morikawa, H.; Kiani, N.A.; Yang, M.; Azimi, A.; Shafi, G.; Shang, M.; Baumgartner, R.; Ketelhuth, D.F.J.; Kamleh, M.A.; et al. Exhaustion of CD4+ T-cells mediated by the Kynurenine Pathway in Melanoma. Sci. Rep. 2019, 9, 12150. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Wang, Y.; Huang, Y.; Kim, B.Y.S.; Shan, H.; Wu, D.; Jiang, W. Tumor Vasculatures: A New Target for Cancer Immunotherapy. Trends Pharmacol. Sci. 2019, 40, 613–623. [Google Scholar] [CrossRef]
- Tian, L.; Goldstein, A.; Wang, H.; Ching Lo, H.; Sun Kim, I.; Welte, T.; Sheng, K.; Dobrolecki, L.E.; Zhang, X.; Putluri, N.; et al. Mutual regulation of tumour vessel normalization and immunostimulatory reprogramming. Nature 2017, 544, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Fang, Z.; Liu, X.; Deng, S.; Zhou, P.; Wang, X.; Zhang, C.; Yin, R.; Hu, H.; Chen, X.; et al. Increased vessel perfusion predicts the efficacy of immune checkpoint blockade. J. Clin. Investig. 2018, 128, 2104–2115. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Shi, H. Unlocking the epigenetic code of T cell exhaustion. Transl. Cancer Res. 2017, 6, S384–S387. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jancewicz, I.; Szarkowska, J.; Konopinski, R.; Stachowiak, M.; Swiatek, M.; Blachnio, K.; Kubala, S.; Oksinska, P.; Cwiek, P.; Rusetska, N.; et al. PD-L1 Overexpression, SWI/SNF Complex Deregulation, and Profound Transcriptomic Changes Characterize Cancer-Dependent Exhaustion of Persistently Activated CD4+ T Cells. Cancers 2021, 13, 4148. https://doi.org/10.3390/cancers13164148
Jancewicz I, Szarkowska J, Konopinski R, Stachowiak M, Swiatek M, Blachnio K, Kubala S, Oksinska P, Cwiek P, Rusetska N, et al. PD-L1 Overexpression, SWI/SNF Complex Deregulation, and Profound Transcriptomic Changes Characterize Cancer-Dependent Exhaustion of Persistently Activated CD4+ T Cells. Cancers. 2021; 13(16):4148. https://doi.org/10.3390/cancers13164148
Chicago/Turabian StyleJancewicz, Iga, Joanna Szarkowska, Ryszard Konopinski, Malgorzata Stachowiak, Monika Swiatek, Katarzyna Blachnio, Szymon Kubala, Paulina Oksinska, Pawel Cwiek, Natalia Rusetska, and et al. 2021. "PD-L1 Overexpression, SWI/SNF Complex Deregulation, and Profound Transcriptomic Changes Characterize Cancer-Dependent Exhaustion of Persistently Activated CD4+ T Cells" Cancers 13, no. 16: 4148. https://doi.org/10.3390/cancers13164148