
Chemotherapy of HER2- and MDM2-Enriched Breast Cancer Subtypes Induces Homologous Recombination DNA Repair and Chemoresistance

 , and
, and 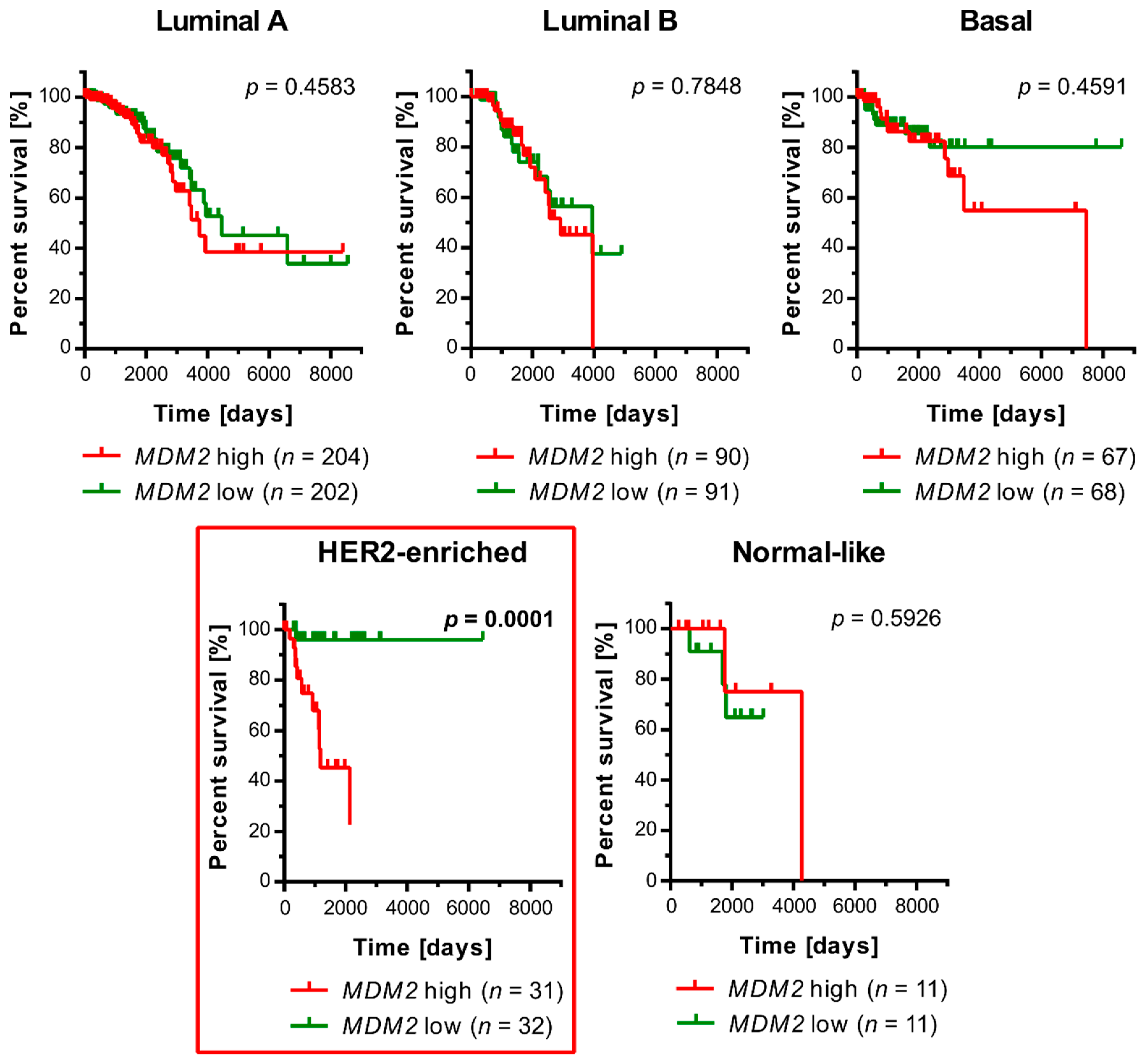{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Breast Cancer Patients Survival Analysis
2.2. Cell Culture, Transfection, and Treatment
2.3. Viability Assay
2.4. Real-Time Cell Proliferation Monitoring with the xCelligence SYSTEM
2.5. In-Cell Western
2.6. Proximity Ligation Assay (PLA)
2.7. Co-Immunoprecipitation (Co-IP)
2.8. In-Vivo Protein Ubiquitination Assay
2.9. Antibodies and Primers
2.10. Homologous Recombination Assay Kit
2.11. Homologous Recombination GFP Reporter Assay
2.12. Statistical Analysis
3. Results
3.1. An Elevated Level of MDM2 Transcript Correlates with a Worse Prognosis for Survival in Patients with an HER2-Enriched Molecular Subtype of Breast Cancer
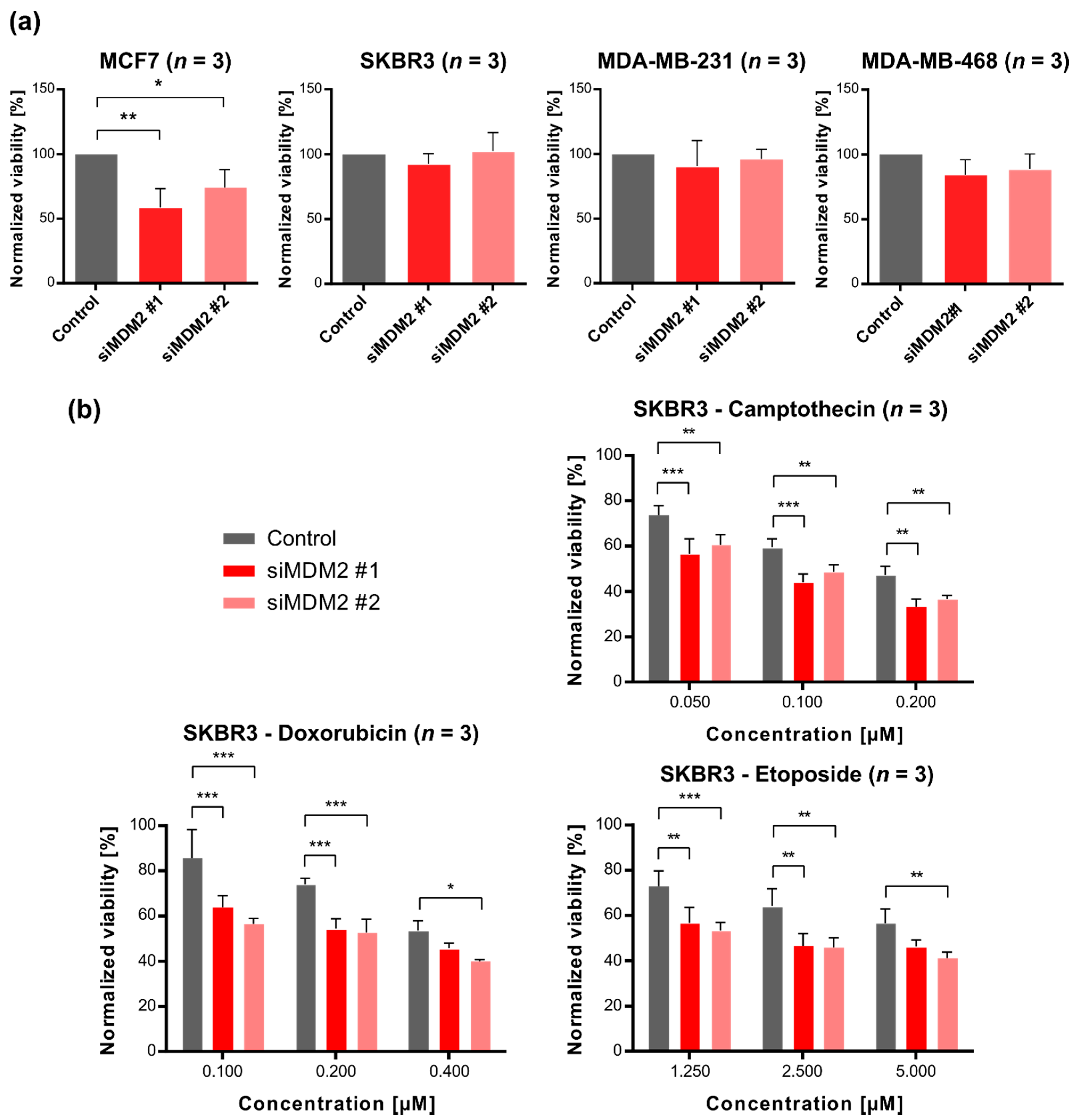
3.2. Downregulation of MDM2 Gene Expression Leads to Decreased Survival of SKBR3 Cells (HER2-Enriched Subtype) after Treatment with DNA-Damaging Chemotherapeutic Agents
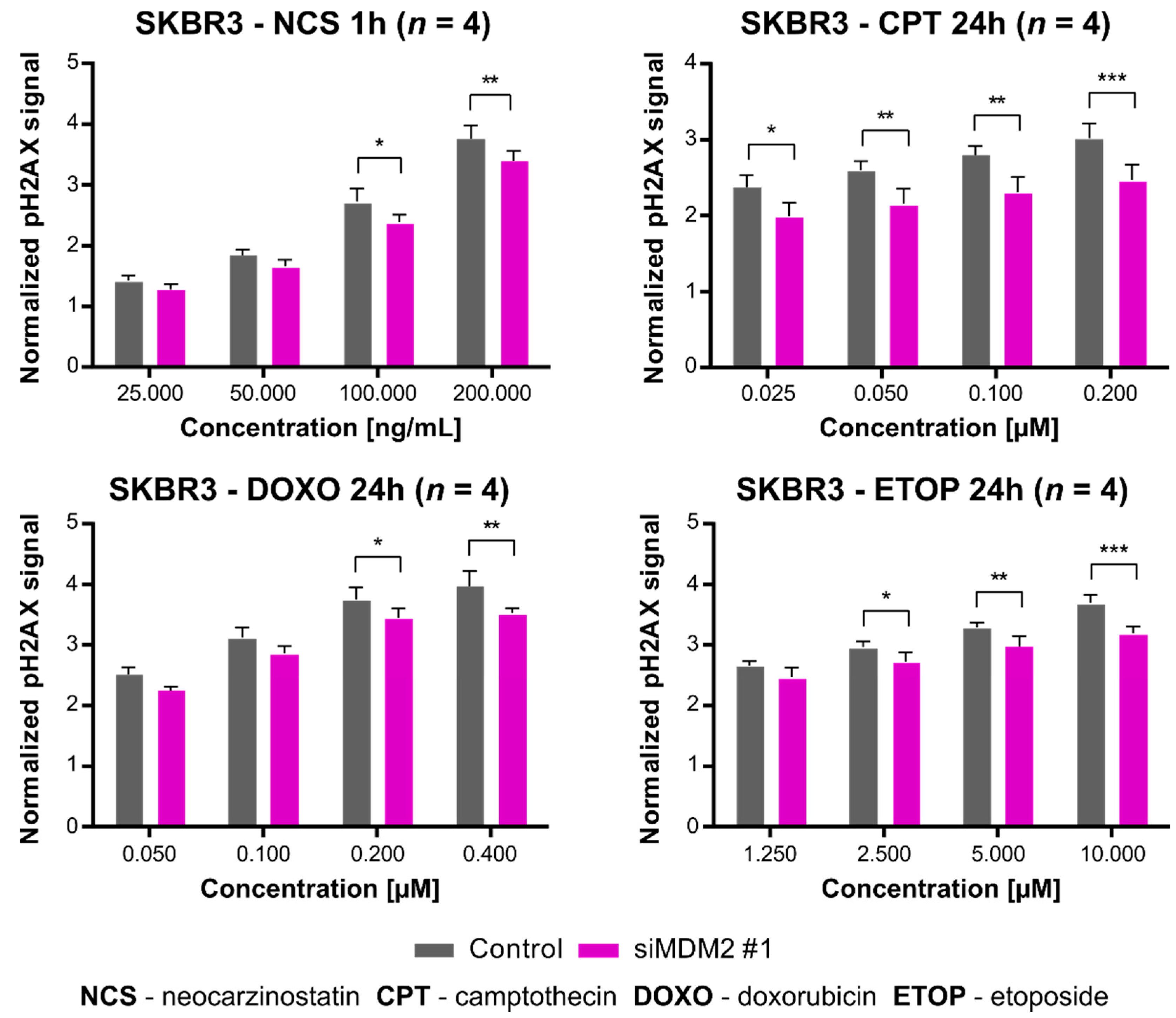
3.3. Downregulation of MDM2 Gene Expression Inhibits H2AX Histone Phosphorylation after Treatment of SKBR3 Cells with DNA-Damaging Agents
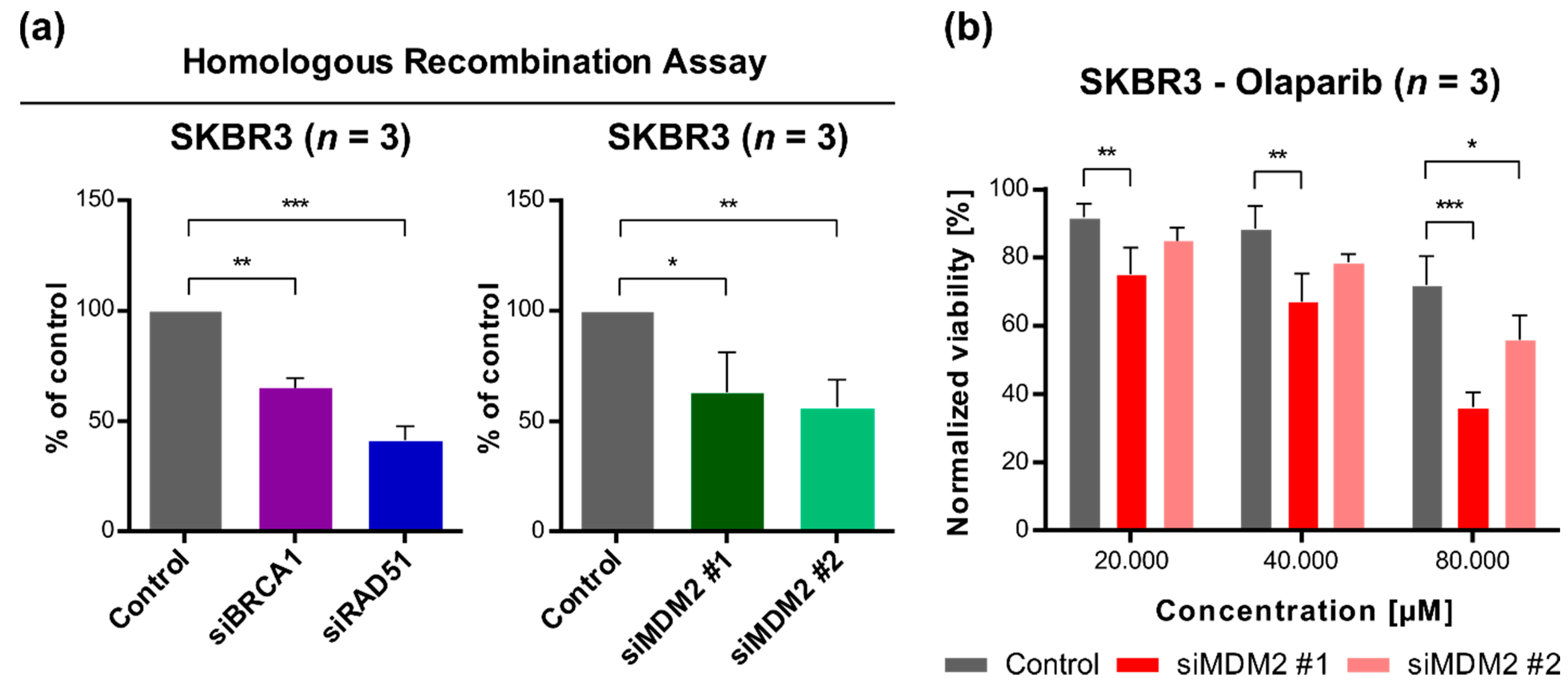
3.4. Downregulation of MDM2 Gene Expression Reduces DNA Repair Efficiency by a Homologous Recombination Mechanism and Sensitizes SKBR3 Cells to Olaparib (Poly(ADP-Ribose) Polymerase, PARP, Inhibitor)
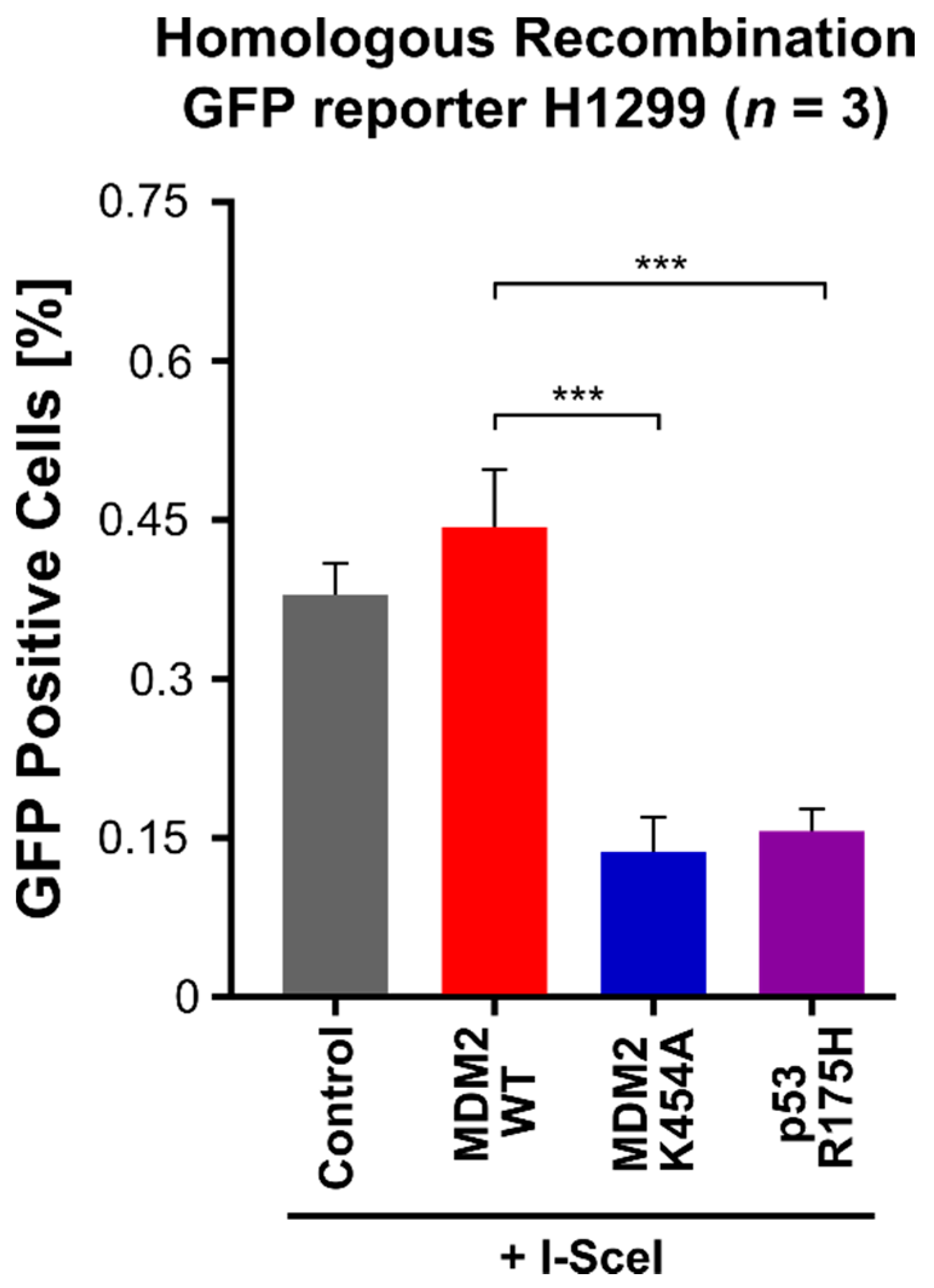
3.5. MDM2 K454A Variant, Which Lacks Chaperone-like Activity and Inhibits Homologous Recombination DNA Repair in H1299 Non-Small-Cell Lung Cancer Cells
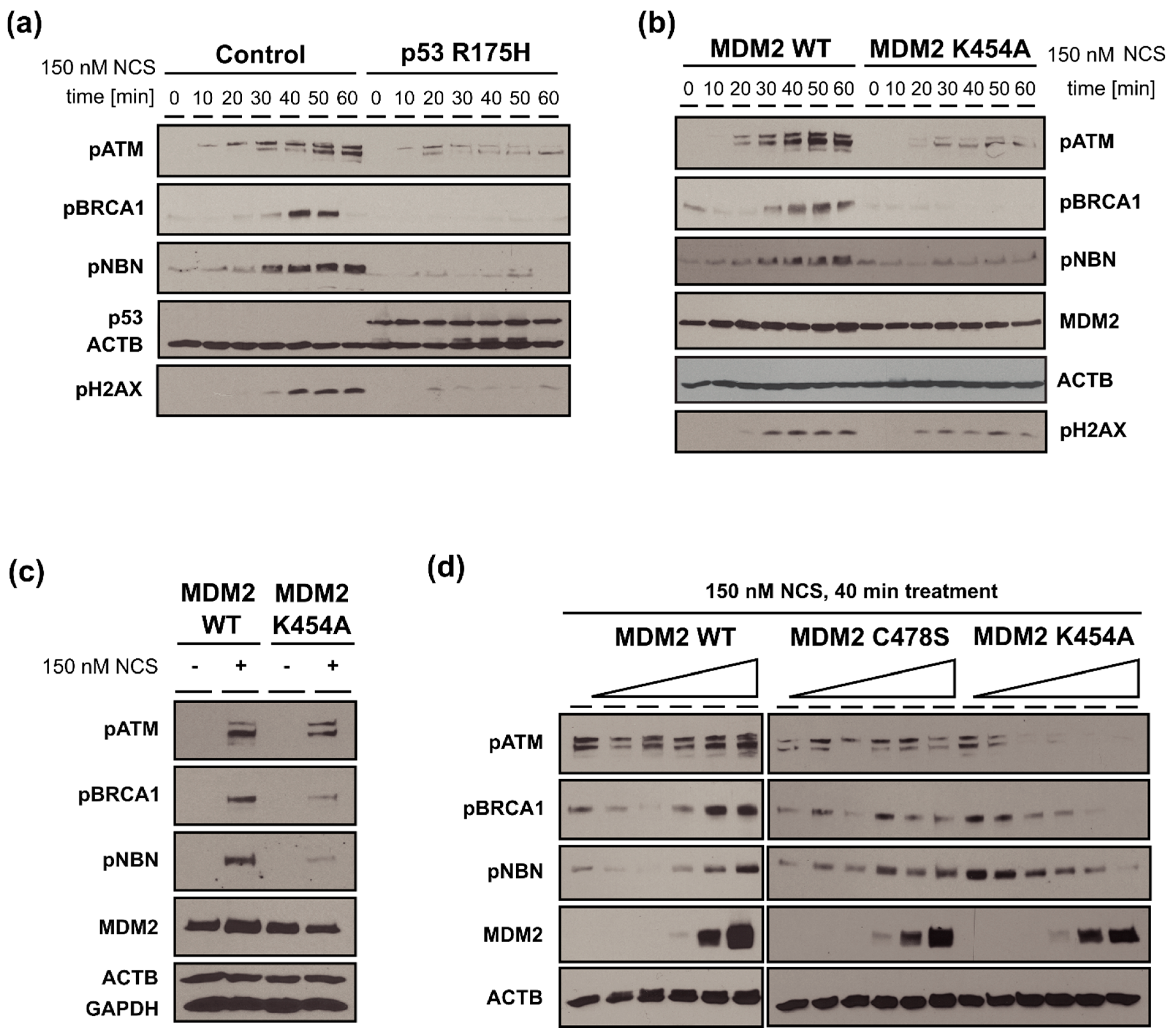
3.6. MDM2 WT Stimulates While MDM2 K454A Inhibits Phosphorylation of Multiple Proteins Involved in DDR (ATM, BRCA1, NBN) in H1299 Cells with DSBs
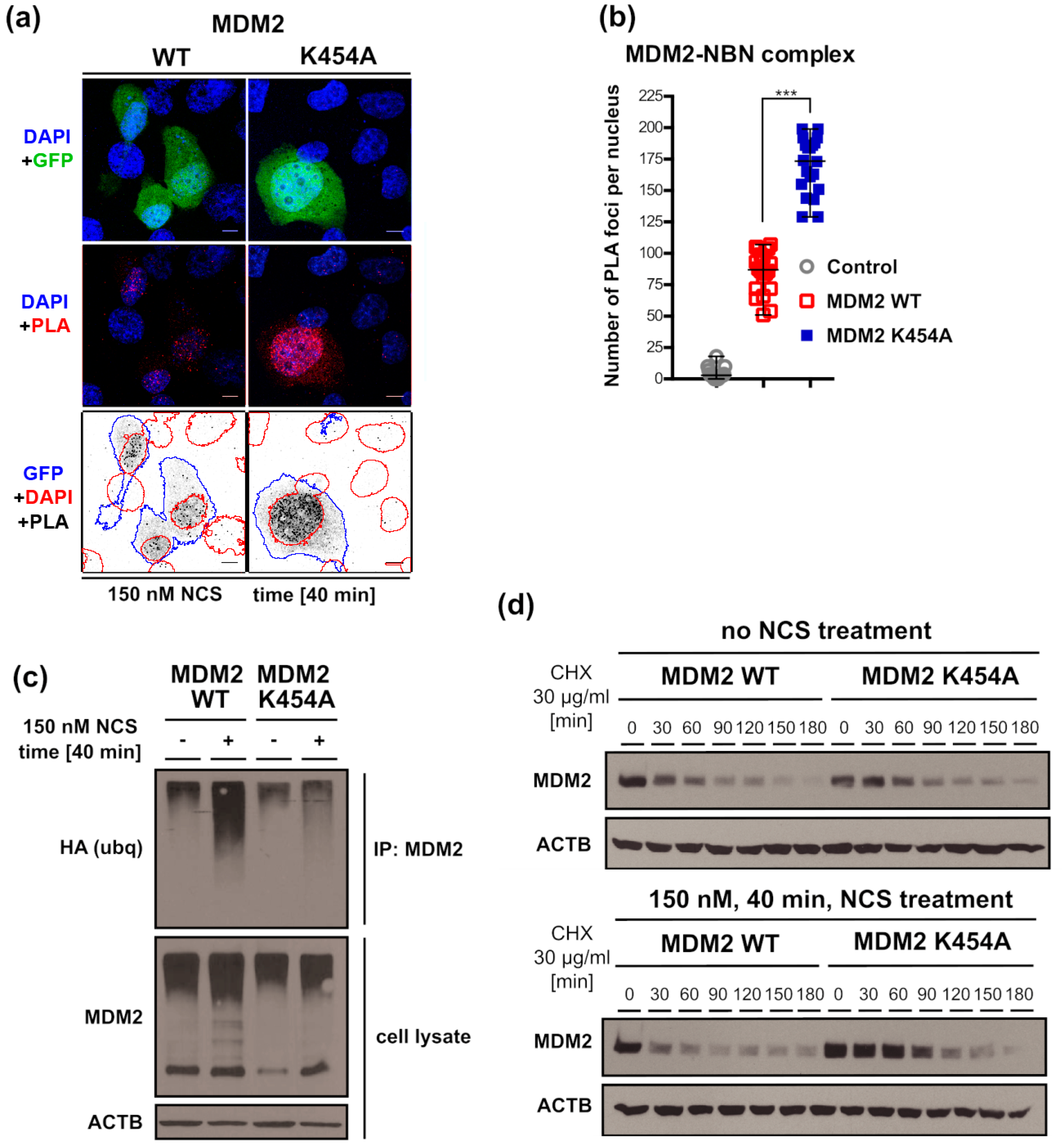
3.7. Chaperone-Dead MDM2 K454A Variant Interacts with NBN More Efficiently Than MDM2 WT
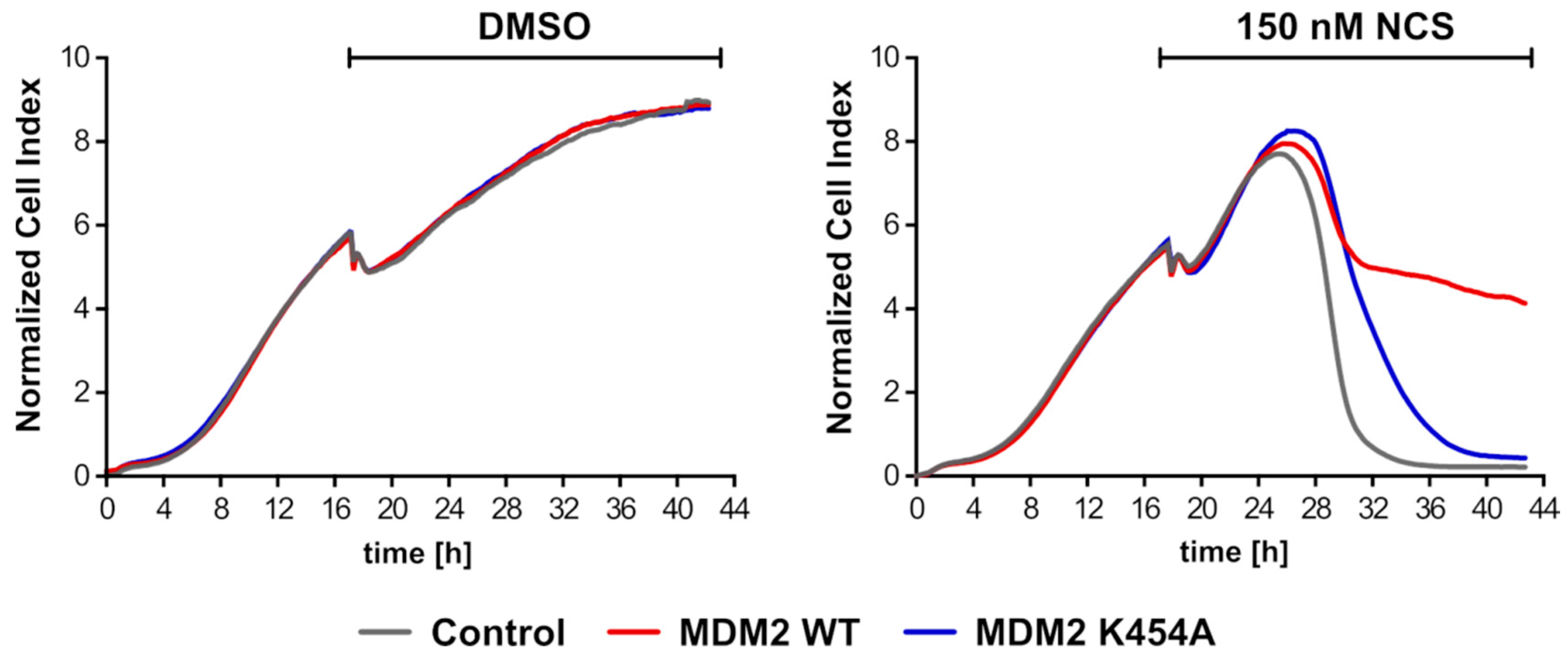
3.8. Overexpression of MDM2 WT Gene Led to Longer Proliferation of H1299 Cells (NSCLC) after Introduction of DNA Damage
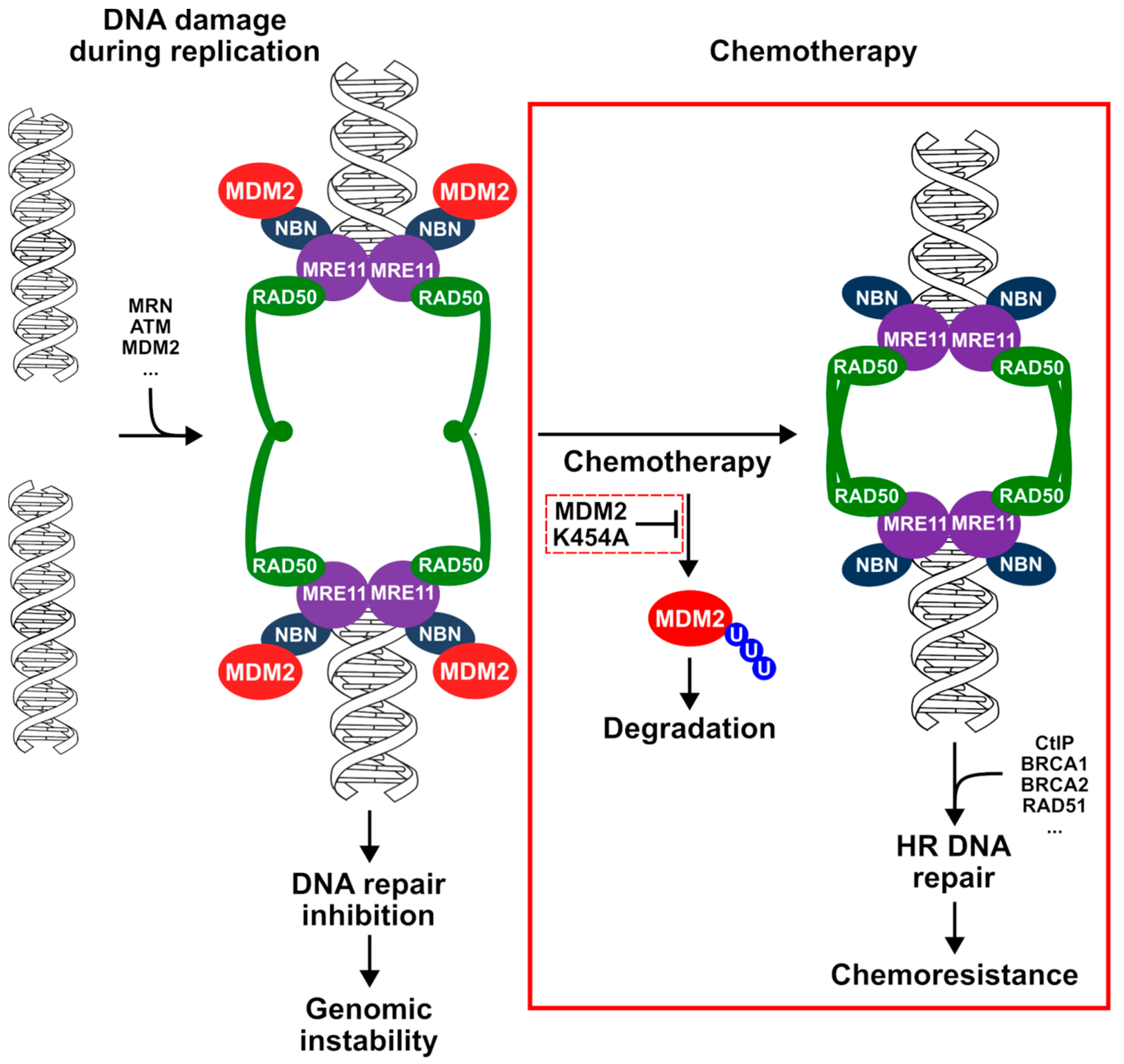
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Desai, A.; Yan, Y.; Gerson, S.L. Advances in therapeutic targeting of the DNA damage response in cancer. DNA Repair 2018, 66–67, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Lavin, M.F.; Kozlov, S.; Gatei, M.; Kijas, A.W. ATM-Dependent Phosphorylation of All Three Members of the MRN Complex: From Sensor to Adaptor. Biomolecules 2015, 5, 2877–2902. [Google Scholar] [CrossRef] [Green Version]
- Chernikova, S.; Game, J.C.; Brown, J.M. Inhibiting homologous recombination for cancer therapy. Cancer Biol. Ther. 2012, 13, 61–68. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, Y.; Kokuta, T.; Teshigahara, A.; Iijima, K.; Kitao, H.; Takata, M.; Tauchi, H. Mitotic cells can repair DNA double-strand breakes via a homology-directed pathway. J. Rad. Res. 2021, 62, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Casari, E.; Rinaldi, C.; Marsella, A.; Gnugnoli, M.; Colombo, C.V.; Bonetti, D.; Longhese, M.P. Processing of DNA double-strand breaks by the mrx complex in a chromatin context. Front. Mol. Biosci. 2019, 6, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, A.; Jeggo, P.; Lobrich, M. The pendulum of the ku-ku clock. DNA Repair 2018, 71, 164–171. [Google Scholar] [CrossRef]
- Ströbel, T.; Madlener, S.; Tuna, S.; Vose, S.; Lagerweij, T.; Wurdinger, T.; Vierlinger, K.; Wöhrer, A.; Price, B.D.; Demple, B.; et al. Ape1 guides DNA repair pathway choice that is associated with drug tolerance in glioblastoma. Sci. Rep. 2017, 7, 9674. [Google Scholar] [CrossRef] [Green Version]
- Wen, J.; Cerosaletti, K.; Schultz, K.J.; Wright, J.A.; Concannon, P. NBN Phosphorylation regulates the accumulation of MRN and ATM at sites of DNA double-strand breaks. Oncogene 2013, 32, 4448–4456. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, M.R.; Bernstein, K.A. RAD-ical New Insights into RAD51 Regulation. Genes 2018, 9, 629. [Google Scholar] [CrossRef] [Green Version]
- Maki, C.G.; Huibregtse, J.M.; Howley, P. In vivo ubiquitination and proteasome-mediated degradation of p53(1). Cancer Res. 1996, 56, 2649–2654. [Google Scholar]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef]
- Haupt, S.; Buckley, D.; Pang, J.-M.; Panimaya, J.; Paul, P.J.; Gamell, C.; Takano, E.; Lee, Y.Y.; Hiddingh, S.; Rogers, T.-M.; et al. Targeting Mdmx to treat breast cancers with wild-type p53. Cell Death Dis. 2015, 6, e1821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haupt, S.; Vijayakumaran, R.; Miranda, P.J.; Burgess, A.; Lim, E.; Haupt, Y. The role of mdm2 and mdm4 in breast can-cer development and prevention. J. Mol. Cell Biol. 2017, 9, 53–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manfredi, J.J. The Mdm2-p53 relationship evolves: Mdm2 swings both ways as an oncogene and a tumor suppressor. Genes Dev. 2010, 24, 1580–1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacob, A.G.; Singh, R.K.; Comiskey, D.F., Jr.; Rouhier, M.F.; Mohammad, F.; Bebee, T.W.; Chandler, D.S. Stress-Induced Alternative Splice Forms of MDM2 and MDMX Modulate the p53-Pathway in Distinct Ways. PLoS ONE 2014, 9, e104444. [Google Scholar] [CrossRef] [Green Version]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef] [Green Version]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. p53 mutations in human cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oren, M.; Rotter, V. Mutant p53 Gain-of-Function in Cancer. Cold Spring Harb. Perspect. Biol. 2010, 2, a001107. [Google Scholar] [CrossRef]
- Honda, R.; Tanaka, H.; Yasuda, H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997, 420, 25–27. [Google Scholar] [CrossRef] [Green Version]
- Wawrzynow, B.; Zylicz, A.; Wallace, M.; Hupp, T.; Zylicz, M. MDM2 Chaperones the p53 Tumor Suppressor. J. Biol. Chem. 2007, 282, 32603–32612. [Google Scholar] [CrossRef] [Green Version]
- Oliner, J.D.; Pietenpol, J.A.; Thiagalingam, S.; Gyuris, J.; Kinzler, K.W.; Vogelstein, B. Oncoprotein mdm2 conceals the ac-tivation domain of tumour suppressor p53. Nature 1993, 362, 857–860. [Google Scholar] [CrossRef] [PubMed]
- Momand, J.; Zambetti, G.P.; Olson, D.C.; George, D.; Levine, A.J. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992, 69, 1237–1245. [Google Scholar] [CrossRef]
- Malbert-Colas, L.; Ponnuswamy, A.; Olivares-Illana, V.; Tournillon, A.-S.; Naski, N.; Fåhraeus, R. HDMX Folds the Nascent p53 mRNA following Activation by the ATM Kinase. Mol. Cell 2014, 54, 500–511. [Google Scholar] [CrossRef] [Green Version]
- Bohlman, S.; Manfredi, J.J. Mdm2-RNA Interactions as a Target for Cancer Therapy: It’s Not All About p53. Cancer Cell 2016, 30, 513–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eischen, C.M. Role of Mdm2 and Mdmx in DNA repair. J. Mol. Cell Biol. 2017, 9, 69–73. [Google Scholar] [CrossRef]
- Bohlman, S.; Manfredi, J.J. p53-Independent Effects of Mdm2. Subcell Biochem. 2014, 85, 235–246. [Google Scholar] [CrossRef] [Green Version]
- Cordon-Cardo, C.; Latres, E.; Drobnjak, M.; Oliva, M.R.; Pollack, D.; Woodruff, J.M.; Marechal, V.; Chen, J.; Brennan, M.F.; Levine, A.J. Molecular abnormalities of mdm2 and p53 genes in adult soft tissue sarcomas. Cancer Res. 1994, 54, 794–799. [Google Scholar] [PubMed]
- Dworakowska, D.; Jassem, E.; Jassem, J.; Peters, B.; Dziadziuszko, R.; Zylicz, M.; Jakobkiewicz-Banecka, J.; Kobierska-Gulida, G.; Szymanowska, A.; Skokowski, J.; et al. Mdm2 gene amplification: A new independent factor of ad-verse prognosis in non-small cell lung cancer (nsclc). Lung Cancer 2004, 43, 285–295. [Google Scholar] [CrossRef]
- Sheikh, M.S.; Shao, Z.M.; Hussain, A.; Fontana, J. The p53-binding protein MDM2 gene is differentially expressed in human breast carcinoma. Cancer Res. 1993, 53, 3226–3228. [Google Scholar]
- Watanabe, T.; Ichikawa, A.; Saito, H.; Hotta, T. Overexpression of the MDM2 Oncogene in Leukemia and Lymphoma. Leuk. Lymphoma 1996, 21, 391–397. [Google Scholar] [CrossRef]
- Momand, J.; Jung, D.; Wilczynski, S.; Niland, J. The MDM2 gene amplification database. Nucleic Acids Res. 1998, 26, 3453–3459. [Google Scholar] [CrossRef]
- Onel, K.; Cordon-Cardo, C. MDM2 and prognosis. Mol. Cancer Res. 2004, 2, 1–8. [Google Scholar] [PubMed]
- Bond, G.L.; Hu, W.; Bond, E.E.; Robins, H.; Lutzker, S.G.; Arva, N.C.; Bargonetti, J.; Bartel, F.; Taubert, H.; Wuerl, P.; et al. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and acceler-ates tumor formation in humans. Cell 2004, 119, 591–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rayburn, E.; Zhang, R.; He, J.; Wang, H. MDM2 and Human Malignancies: Expression, Clinical Pathology, Prognostic Markers, and Implications for Chemotherapy. Curr. Cancer Drug Targets 2005, 5, 27–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bond, G.L.; Hirshfield, K.M.; Kirchhoff, T.; Alexe, G.; Bond, E.E.; Robins, H.; Bartel, F.; Taubert, H.; Wuerl, P.; Hait, W.; et al. MDM2 SNP309 Accelerates Tumor Formation in a Gender-Specific and Hormone-Dependent Manner. Cancer Res. 2006, 66, 5104–5110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brekman, A.; Singh, K.; Polotskaia, A.; Kundu, N.; Bargonetti, J. A p53-independent role of MDM2 in estrogen-mediated activation of breast cancer cell proliferation. Breast Cancer Res. 2011, 13, R3. [Google Scholar] [CrossRef] [Green Version]
- Deb, S.P.; Singh, S.; Deb, S. MDM2 overexpression, activation of signaling networks, and cell proliferation. Subcell Biochem. 2014, 85, 215–234. [Google Scholar] [PubMed]
- Yu, Q.; Li, Y.; Mu, K.; Li, Z.; Meng, Q.; Wu, X.; Wang, Y.; Li, L. Amplification of Mdmx and overexpression of MDM2 contribute to mammary carcinogenesis by substituting for p53 mutations. Diagn. Pathol. 2014, 9, 71. [Google Scholar] [CrossRef] [Green Version]
- Tracz-Gaszewska, Z.; Klimczak, M.; Biecek, P.; Herok, M.; Kosinski, M.; Olszewski, M.; Czerwińska, P.; Wiech, M.; Wiznerowicz, M.; Zylicz, A.; et al. Molecular chaperones in the acquisition of cancer cell chemoresistance with mutated TP53 and MDM2 up-regulation. Oncotarget 2017, 8, 82123–82143. [Google Scholar] [CrossRef] [Green Version]
- Park, H.S.; Park, J.M.; Park, S.; Cho, J.; Kim, S.I.; Park, B.-W. Subcellular localization of Mdm2 expression and prognosis of breast cancer. Int. J. Clin. Oncol. 2014, 19, 842–851. [Google Scholar] [CrossRef]
- Hernández-Monge, J.; Rousset-Roman, A.B.; Medina-Medina, I.; Olivares-Illana, V. Dual function of MDM2 and MDMX toward the tumor suppressors p53 and RB. Genes Cancer 2016, 7, 278–287. [Google Scholar] [CrossRef]
- Stevens, C.; Pettersson, S.; Wawrzynow, B.; Wallace, M.; Ball, K.; Zylicz, A.; Hupp, T.R. ATP stimulates MDM2-mediated inhibition of the DNA-binding function of E2F1. FEBS J. 2008, 275, 4875–4886. [Google Scholar] [CrossRef]
- Alt, J.R.; Bouska, A.; Fernandez, M.; Cerny, R.L.; Xiao, H.; Eischen, C.M. Mdm2 Binds to Nbs1 at Sites of DNA Damage and Regulates Double Strand Break Repair. J. Biol. Chem. 2005, 280, 18771–18781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouska, A.; Lushnikova, T.; Plaza, S.; Eischen, C.M. Mdm2 Promotes Genetic Instability and Transformation Independent of p53. Mol. Cell. Biol. 2008, 28, 4862–4874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senturk, J.C.; Bohlman, S.; Manfredi, J.J. Mdm2 selectively suppresses DNA damage arising from inhibition of topoiso-merase ii independent of p53. Oncogene 2017, 36, 6085–6096. [Google Scholar] [CrossRef]
- Conradt, L.; Henrich, A.; Wirth, M.; Reichert, M.; Lesina, M.; Algül, H.; Schmid, R.M.; Krämer, O.H.; Saur, D.; Schneider, G. Mdm2 inhibitors synergize with topoisomerase II inhibitors to induce p53-independent pancreatic cancer cell death. Int. J. Cancer 2013, 132, 2248–2257. [Google Scholar] [CrossRef] [PubMed]
- Feeley, K.P.; Adams, C.M.; Mitra, R.; Eischen, C.M. Mdm2 Is Required for Survival and Growth of p53-Deficient Cancer Cells. Cancer Res. 2017, 77, 3823–3833. [Google Scholar] [CrossRef] [Green Version]
- Lyons, T.G.; Robson, M.E. Resurrection of PARP Inhibitors in Breast Cancer. J. Natl. Compr. Cancer Netw. 2018, 16, 1150–1156. [Google Scholar] [CrossRef]
- Liu, D.-P.; Song, H.; Xu, Y. A common gain of function of p53 cancer mutants in inducing genetic instability. Oncogene 2010, 29, 949–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Słabicki, M.; Theis, M.; Krastev, D.B.; Samsonov, S.; Mundwiller, E.; Junqueira, M.; Paszkowski-Rogacz, M.; Teyra, J.; Heninger, A.-K.; Poser, I.; et al. A Genome-Scale DNA Repair RNAi Screen Identifies SPG48 as a Novel Gene Associated with Hereditary Spastic Paraplegia. PLoS Biol. 2010, 8, e1000408. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.S.; Truong, L.N.; Aslanian, A.; Shi, L.Z.; Li, Y.; Hwang, P.Y.; Koh, K.H.; Hunter, T.; Yates, J.R., 3rd; Berns, M.W.; et al. The ring finger protein rnf8 ubiquitinates nbs1 to promote DNA double-strand break repair by homologous recombination. J. Biol. Chem. 2012, 287, 43984–43994. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Zhang, X.; Zhang, L.; Wu, C.Y.; Rezaeian, A.H.; Chan, C.H.; Li, J.M.; Wang, J.; Gao, Y.; Han, F.; et al. Skp2 e3 lig-ase integrates atm activation and homologous recombination repair by ubiquitinating nbs1. Mol. Cell 2012, 46, 351–361. [Google Scholar] [CrossRef] [Green Version]
- Ha, G.H.; Ji, J.H.; Chae, S.; Park, J.; Kim, S.; Lee, J.K.; Kim, Y.; Min, S.; Park, J.M.; Kang, T.H.; et al. Pellino1 regulates re-versible atm activation via nbs1 ubiquitination at DNA double-strand breaks. Nat. Commun. 2019, 10, 1577. [Google Scholar] [CrossRef] [Green Version]
- Eischen, C.M.; Lozano, G. The Mdm network and its regulation of p53 activities: A rheostat of cancer risk. Hum. Mutat. 2014, 35, 728–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turbin, D.; Cheang, M.C.U.; Bajdik, C.D.; Gelmon, K.; Yorida, E.; De Luca, A.; Nielsen, T.; Huntsman, D.G.; Gilks, C.B. MDM2 protein expression is a negative prognostic marker in breast carcinoma. Mod. Pathol. 2006, 19, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Campillo-Marcos, I.; Lazo, P.A. Implication of the VRK1 chromatin kinase in the signaling responses to DNA damage: A therapeutic target? Cell. Mol. Life Sci. 2018, 75, 2375–2388. [Google Scholar] [CrossRef] [Green Version]
- Scully, R.; Xie, A. Double strand break repair functions of histone H2AX. Mutat. Res. Mol. Mech. Mutagen. 2013, 750, 5–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiloh, Y. The ATM-mediated DNA-damage response: Taking shape. Trends Biochem. Sci. 2006, 31, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Paull, T.T. Direct activation of the atm protein kinase by the mre11/rad50/nbs1 complex. Science 2004, 304, 93–96. [Google Scholar] [CrossRef]
- You, Z.; Chahwan, C.; Bailis, J.; Hunter, T.; Russell, P. ATM Activation and Its Recruitment to Damaged DNA Require Binding to the C Terminus of Nbs1. Mol. Cell. Biol. 2005, 25, 5363–5379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falck, J.; Coates, J.; Jackson, S.P. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 2005, 434, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, R.; Antoccia, A.; Leone, S.; Ascenzi, P.; Di Masi, A. Hsp90α regulates ATM and NBN functions in sensing and repair of DNA double-strand breaks. FEBS J. 2017, 284, 2378–2395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stracker, T.H. Chaperoning the DNA damage response. FEBS J. 2017, 284, 2375–2377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dote, H.; Burgan, W.E.; Camphausen, K.; Tofilon, P.J. Inhibition of hsp90 compromises the DNA damage response to radiation. Cancer Res. 2006, 66, 9211–9220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Toledo, S.M.; Azzam, E.I.; Dahlberg, W.K.; Gooding, T.B.; Little, J.B. Atm complexes with hdm2 and promotes its rap-id phosphorylation in a p53-independent manner in normal and tumor human cells exposed to ionizing radiation. Oncogene 2000, 19, 6185–6193. [Google Scholar] [CrossRef] [Green Version]
- Inuzuka, H.; Fukushima, H.; Shaik, S.; Wei, W. Novel insights into the molecular mechanisms governing mdm2 ubiquiti-nation and destruction. Oncotarget 2010, 1, 685–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herok, M.; Wawrzynow, B.; Maluszek, M.J.; Olszewski, M.B.; Zylicz, A.; Zylicz, M. Chemotherapy of HER2- and MDM2-Enriched Breast Cancer Subtypes Induces Homologous Recombination DNA Repair and Chemoresistance. Cancers 2021, 13, 4501. https://doi.org/10.3390/cancers13184501
Herok M, Wawrzynow B, Maluszek MJ, Olszewski MB, Zylicz A, Zylicz M. Chemotherapy of HER2- and MDM2-Enriched Breast Cancer Subtypes Induces Homologous Recombination DNA Repair and Chemoresistance. Cancers. 2021; 13(18):4501. https://doi.org/10.3390/cancers13184501
Chicago/Turabian StyleHerok, Marcin, Bartosz Wawrzynow, Marta J. Maluszek, Maciej B. Olszewski, Alicja Zylicz, and Maciej Zylicz. 2021. "Chemotherapy of HER2- and MDM2-Enriched Breast Cancer Subtypes Induces Homologous Recombination DNA Repair and Chemoresistance" Cancers 13, no. 18: 4501. https://doi.org/10.3390/cancers13184501