
The Diverse Applications of Pancreatic Ductal Adenocarcinoma Organoids

 , , and
, , and 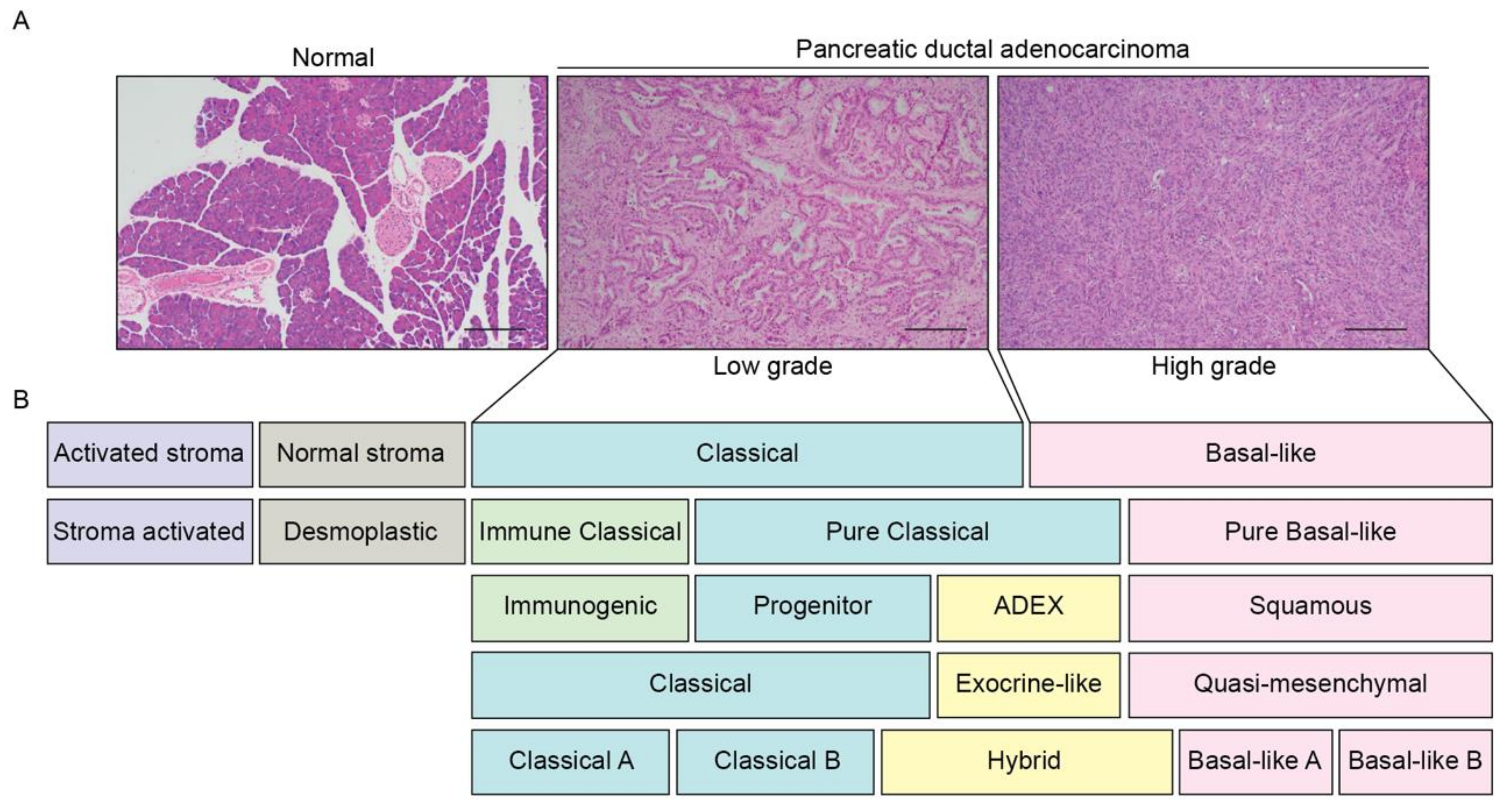{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Pancreatic Ductal Adenocarcinoma, the Most Common Pancreatic Cancer
1.2. Emerging PDAC Molecular Subtypes
1.2.1. Classical Subtype
1.2.2. Basal Subtype
1.2.3. Stromal Subtype
1.2.4. Immunogenic Subtype

1.3. Current Treatment Options
2. Preclinical Models of Pancreatic Ductal Adenocarcinoma
2.1. Genetically Engineered Mouse Models
GEMM for the Generation of Organoids
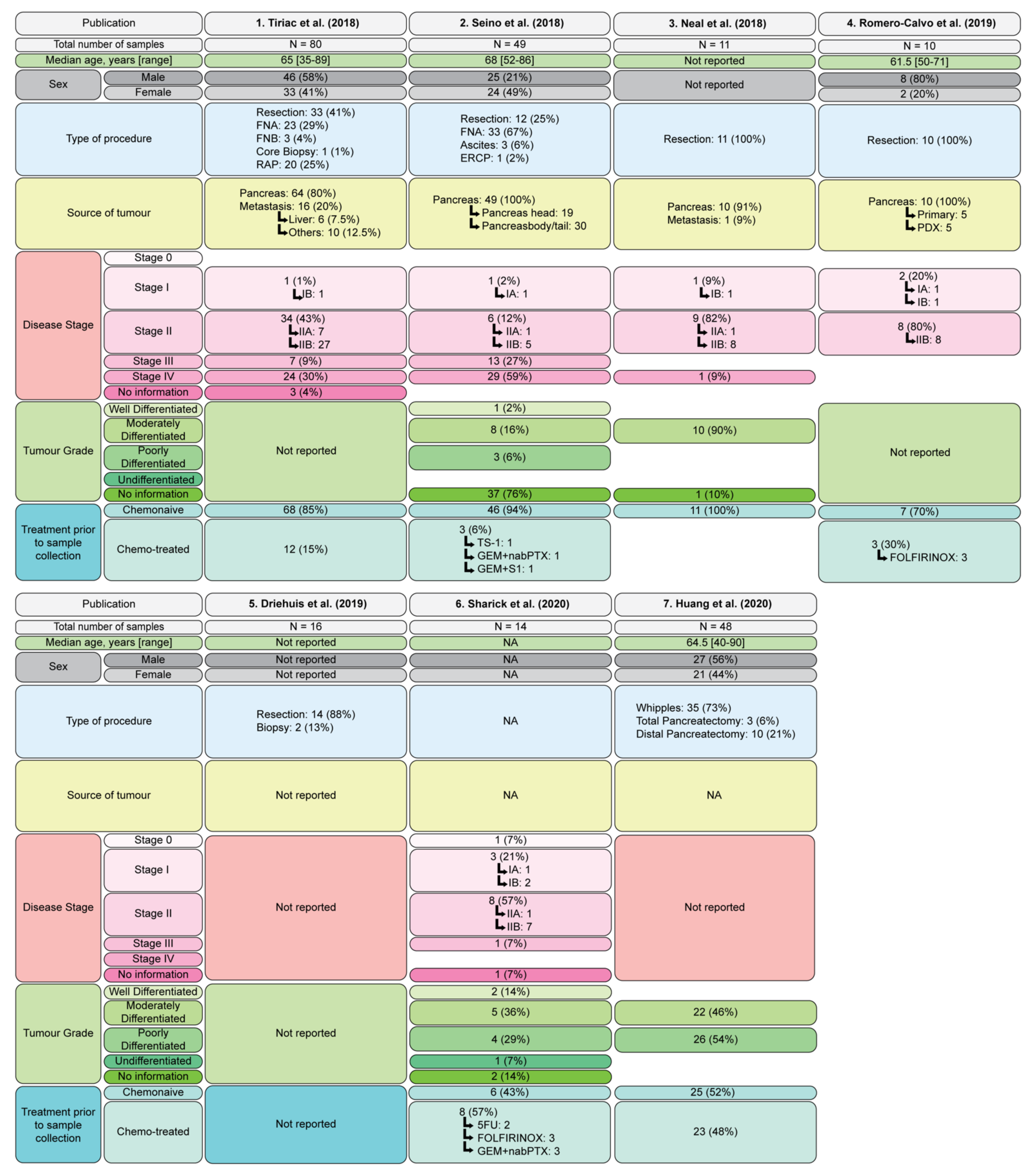
2.2. Patient Derived Models
2.2.1. Monolayer Cell Culture
2.2.2. Cell Line Derived Spheroids
2.2.3. Patient Derived Xenografts
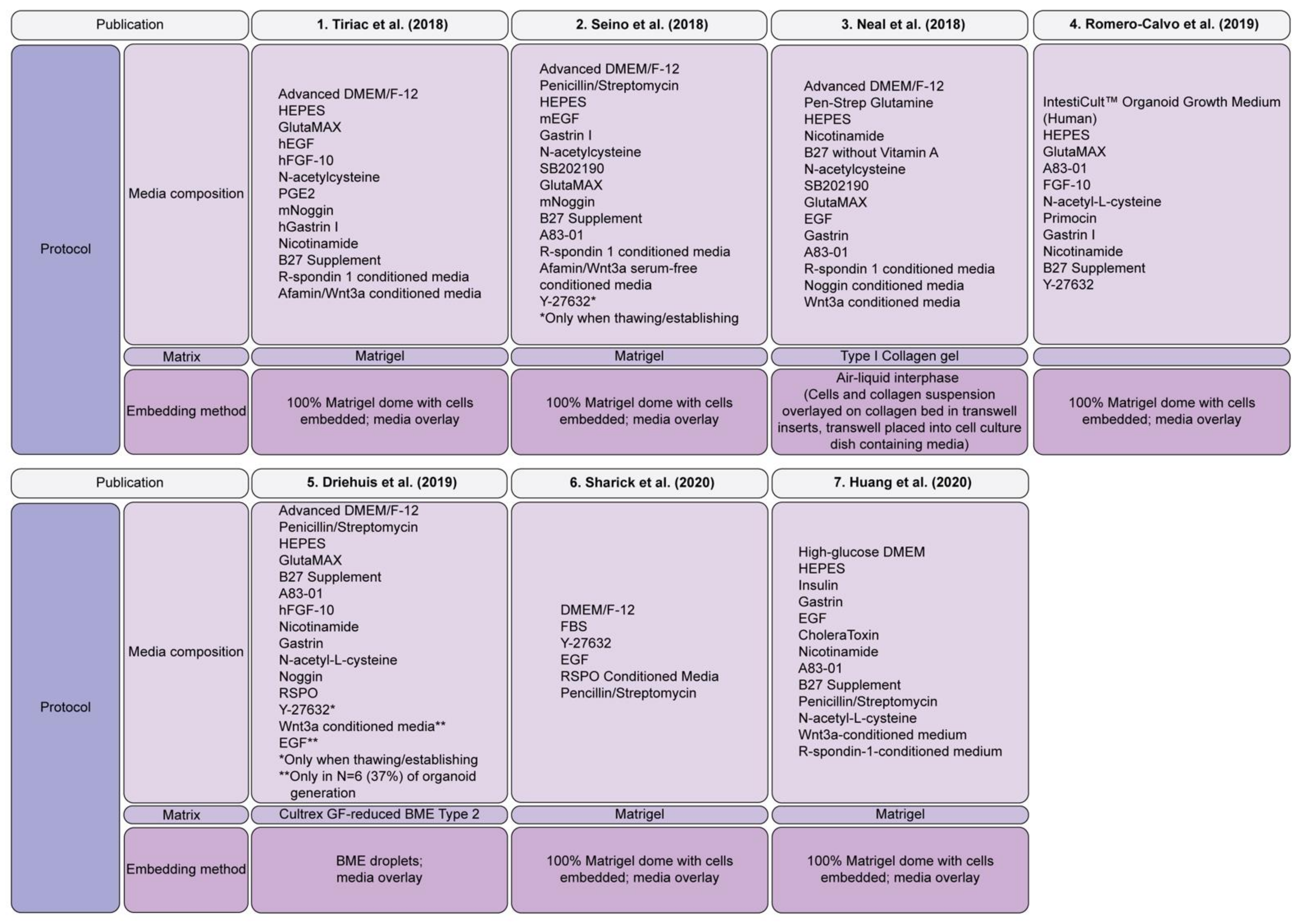
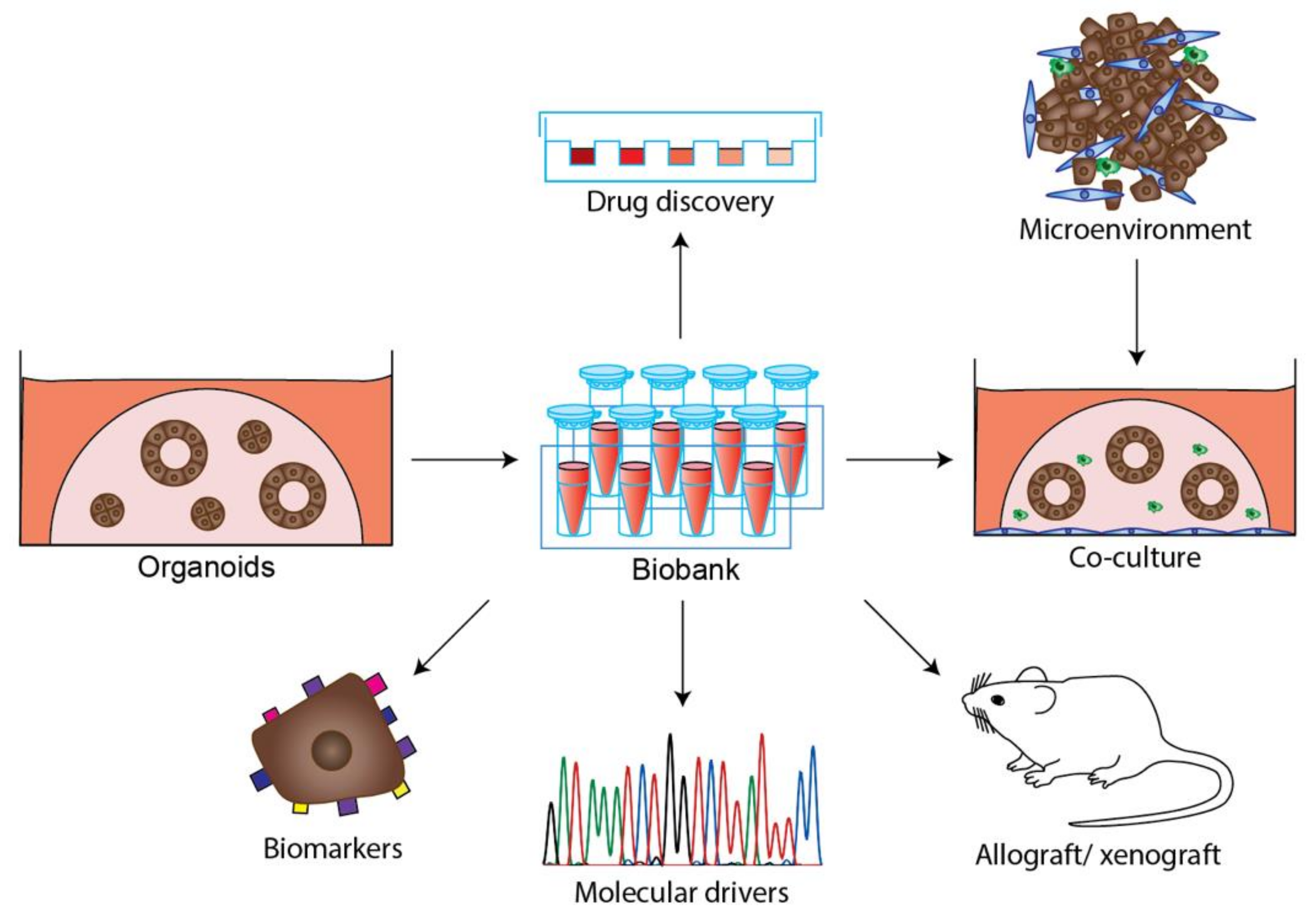
2.2.4. Patient Derived PDAC Organoids
Patient Derived Organoid Biobanks


3. Organoids, an Emerging Model System to Understand PDAC Biology
3.1. Organoids Permit a Deeper Understanding of the Molecular Features of PDAC
3.2. Organoids Provide a Unique Insight into Metastatic Processes
3.3. Modelling the Stromal Tumor Microenvironment Using Organoid Cultures
3.4. Modelling the Immune Microenvironment in Organoid Cultures

4. Patient Derived Organoids as a Model to Identify New Treatments for PDAC Patients
4.1. PDAC Organoid Treatment Sensitivity May Predict the Patient Response to Chemotherapy
4.2. Organoids Can Be Used to Identify Potential Targeted Therapies
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA A Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, D.P.; Hong, T.S.; Bardeesy, N. Pancreatic adenocarcinoma. N. Engl. J. Med. 2014, 371, 2140–2141. [Google Scholar] [CrossRef]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.-C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.-M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, G.; Beaty, R.; Hruban, R.H.; Maitra, A. Molecular genetics of pancreatic intraepithelial neoplasia. J. Hepato-Biliary-Pancreat. Surg. 2007, 14, 224–232. [Google Scholar] [CrossRef] [Green Version]
- Makohon-Moore, A.; Iacobuzio-Donahue, C.A. Pancreatic cancer biology and genetics from an evolutionary perspective, Nature reviews. Cancer 2016, 16, 553–565. [Google Scholar]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Schlitter, A.M.; Segler, A.; Steiger, K.; Michalski, C.W.; Jager, C.; Konukiewitz, B.; Pfarr, N.; Endris, V.; Bettstetter, M.; Kong, B.; et al. Molecular, morphological and survival analysis of 177 resected pancreatic ductal adenocarcinomas (PDACs): Identification of prognostic subtypes. Sci. Rep. 2017, 7, 41064. [Google Scholar] [CrossRef] [Green Version]
- Nagtegaal, I.D.; Odze, R.D.; Klimstra, D.; Paradis, V.; Rugge, M.; Schirmacher, P.; Washington, K.M.; Carneiro, F.; Cree, I.A. The 2019 WHO classification of tumours of the digestive system. Histopathology 2020, 76, 182–188. [Google Scholar] [CrossRef] [Green Version]
- Kalimuthu, S.N.; Wilson, G.W.; Grant, R.C.; Seto, M.; O’Kane, G.; Vajpeyi, R.; Notta, F.; Gallinger, S.; Chetty, R. Morphological classification of pancreatic ductal adenocarcinoma that predicts molecular subtypes and correlates with clinical outcome. Gut 2020, 69, 317–328. [Google Scholar] [CrossRef]
- Ren, B.; Liu, X.; Suriawinata, A.A. Pancreatic Ductal Adenocarcinoma and Its Precursor Lesions: Histopathology, Cytopathology, and Molecular Pathology. Am. J. Pathol. 2019, 189, 9–21. [Google Scholar] [CrossRef] [Green Version]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500. [Google Scholar] [CrossRef] [PubMed]
- Chan-Seng-Yue, M.; Kim, J.C.; Wilson, G.W.; Ng, K.; Figueroa, E.F.; O’Kane, G.M.; Connor, A.A.; Denroche, R.E.; Grant, R.C.; McLeod, J.; et al. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat. Genet. 2020, 52, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Nicolle, R.; Blum, Y.; Duconseil, P.; Vanbrugghe, C.; Brandone, N.; Poizat, F.; Roques, J.; Bigonnet, M.; Gayet, O.; Rubis, M.; et al. Establishment of a pancreatic adenocarcinoma molecular gradient (PAMG) that predicts the clinical outcome of pancreatic cancer. EBioMedicine 2020, 57, 102858. [Google Scholar] [CrossRef] [PubMed]
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev. Cell 2018, 45, 681–695.e4. [Google Scholar] [CrossRef] [Green Version]
- Puleo, F.; Nicolle, R.; Blum, Y.; Cros, J.; Marisa, L.; Demetter, P.; Quertinmont, E.; Svrcek, M.; Elarouci, N.; Iovanna, J.; et al. Stratification of Pancreatic Ductal Adenocarcinomas Based on Tumor and Microenvironment Features. Gastroenterology 2018, 155, 1999–2013.e3. [Google Scholar] [CrossRef] [Green Version]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.H.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168. [Google Scholar] [CrossRef]
- Raphael, B.J.; Hruban, R.H.; Aguirre, A.J.; Moffitt, R.A. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203.e13. [Google Scholar] [CrossRef] [Green Version]
- Noll, E.M.; Eisen, C.; Stenzinger, A.; Espinet, E.; Muckenhuber, A.; Klein, C.; Vogel, V.; Klaus, B.; Nadler, W.; Rösli, C.; et al. CYP3A5 mediates basal and acquired therapy resistance in different subtypes of pancreatic ductal adenocarcinoma. Nat. Med. 2016, 22, 278–287. [Google Scholar] [CrossRef] [Green Version]
- Knudsen, E.S.; Balaji, U.; Mannakee, B.; Vail, P.; Eslinger, C.; Moxom, C.; Mansour, J.; Witkiewicz, A.K. Pancreatic cancer cell lines as patient-derived avatars: Genetic characterisation and functional utility. Gut 2018, 67, 508–520. [Google Scholar] [CrossRef] [Green Version]
- Filippini, D.; Agosto, S.; Delfino, P.; Simbolo, M.; Piro, G.; Rusev, B.; Veghini, L.; Cantù, C.; Lupo, F.; Ugel, S.; et al. Immunoevolution of mouse pancreatic organoid isografts from preinvasive to metastatic disease. Sci. Rep. 2019, 9, 12286. [Google Scholar] [CrossRef] [Green Version]
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [Green Version]
- Bernard, V.; Semaan, A.; Huang, J.; Lucas, F.A.S.; Mulu, F.C.; Stephens, B.M.; Guerrero, P.A.; Huang, Y.; Zhao, J.; Kamyabi, N.; et al. Single-Cell Transcriptomics of Pancreatic Cancer Precursors Demonstrates Epithelial and Microenvironmental Heterogeneity as an Early Event in Neoplastic Progression. Clin. Cancer Res. 2019, 25, 2194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khorana, A.A.; Mangu, P.B.; Berlin, J.; Engebretson, A.; Hong, T.S.; Maitra, A.; Mohile, S.G.; Mumber, M.; Schulick, R.; Shapiro, M.; et al. Potentially Curable Pancreatic Cancer: American Society of Clinical Oncology Clinical Practice Guideline Update. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 2324–2328. [Google Scholar] [CrossRef] [PubMed]
- Katz, M.H.; Wang, H.; Fleming, J.B.; Sun, C.C.; Hwang, R.F.; Wolff, R.A.; Varadhachary, G.; Abbruzzese, J.L.; Crane, C.H.; Krishnan, S.; et al. Long-term survival after multidisciplinary management of resected pancreatic adenocarcinoma. Ann. Surg. Oncol. 2009, 16, 836–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, B.; Lipton, L.; Cohen, J.; Tie, J.; Javed, A.A.; Li, L.; Goldstein, D.; Burge, M.; Cooray, P.; Nagrial, A.; et al. Circulating tumor DNA as a potential marker of adjuvant chemotherapy benefit following surgery for localized pancreatic cancer. Ann. Oncol. 2019, 30, 1472–1478. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Hammel, P.; Hebbar, M.; Abdelghani, M.B.; Wei, A.C.; Raoul, J.-L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- Neoptolemos, J.P.; Palmer, D.H.; Ghaneh, P.; Psarelli, E.E.; Valle, J.W.; Halloran, C.M.; Faluyi, O.; O’Reilly, D.A.; Cunningham, D.; Wadsley, J.; et al. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): A multicentre, open-label, randomised, phase 3 trial. Lancet 2017, 389, 1011–1024. [Google Scholar] [CrossRef]
- Kokkali, S.; Tripodaki, E.S.; Drizou, M.; Stefanou, D.; Magou, E.; Zylis, D.; Kapiris, M.; Nasi, D.; Georganta, C.; Ardavanis, A. Biweekly Gemcitabine/Nab-Paclitaxel as First-line Treatment for Advanced Pancreatic Cancer. In Vivo 2018, 32, 653–657. [Google Scholar] [CrossRef]
- Kang, J.; Hwang, I.; Yoo, C.; Kim, K.P.; Jeong, J.H.; Chang, H.M.; Lee, S.S.; Park, D.H.; Song, T.J.; Seo, D.W.; et al. Nab-paclitaxel plus gemcitabine versus FOLFIRINOX as the first-line chemotherapy for patients with metastatic pancreatic cancer: Retrospective analysis. Investig. New Drugs 2018, 36, 732–741. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- N.C.C. Network. Pancreatic Adenocarcinoma (Version 1.2020). Available online: https://www.nccn.org/professionals/physician_gls/pdf/pancreatic_blocks.pdf. (accessed on 30 October 2020).
- Body, A.; Wong, R.; Shapiro, J.; Jalali, A.; McLachlan, S.-A.; Ananda, S.; Lipton, L.; Cooray, P.; Gibbs, P.; Lee, B.; et al. Use and outcomes of chemotherapy for metastatic pancreatic cancer in Australia. Intern. Med. J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Goel, N.; Reddy, S.S. Randomized Clinical Trials in Pancreatic Cancer. Surg. Oncol. Clin. N. Am. 2017, 26, 767–790. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Goldstein, D.; Hamm, J.; Figer, A.; Hecht, J.R.; Gallinger, S.; Au, H.J.; Murawa, P.; Walde, D.; Wolff, R.A.; et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: A phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2007, 25, 1960–1966. [Google Scholar] [CrossRef]
- Kim, S.T.; Lim, D.H.; Jang, K.-T.; Lim, T.; Lee, J.; Choi, Y.-L.; Jang, H.-L.; Yi, J.H.; Baek, K.K.; Park, S.H.; et al. Impact of KRAS Mutations on Clinical Outcomes in Pancreatic Cancer Patients Treated with First-line Gemcitabine-Based Chemotherapy. Mol. Cancer Ther. 2011, 10, 1993. [Google Scholar]
- Sohal, D.P.S.; Kennedy, E.B.; Cinar, P.; Conroy, T.; Copur, M.S.; Crane, C.H.; Garrido-Laguna, I.; Lau, M.W.; Johnson, T.; Krishnamurthi, S.; et al. Metastatic Pancreatic Cancer: ASCO Guideline Update. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 3217–3230. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Kindler, H.L.; Niedzwiecki, D.; Hollis, D.; Sutherland, S.; Schrag, D.; Hurwitz, H.; Innocenti, F.; Mulcahy, M.F.; O’Reilly, E.; Wozniak, T.F.; et al. Gemcitabine plus bevacizumab compared with gemcitabine plus placebo in patients with advanced pancreatic cancer: Phase III trial of the Cancer and Leukemia Group B (CALGB 80303). J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 3617–3622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philip, P.A.; Benedetti, J.; Corless, C.L.; Wong, R.; O’Reilly, E.M.; Flynn, P.J.; Rowland, K.M.; Atkins, J.N.; Mirtsching, B.C.; Rivkin, S.E.; et al. Phase III study comparing gemcitabine plus cetuximab versus gemcitabine in patients with advanced pancreatic adenocarcinoma: Southwest Oncology Group-directed intergroup trial S0205. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 3605–3610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goncalves, A.; Gilabert, M.; Francois, E.; Dahan, L.; Perrier, H.; Lamy, R.; Re, D.; Largillier, R.; Gasmi, M.; Tchiknavorian, X.; et al. BAYPAN study: A double-blind phase III randomized trial comparing gemcitabine plus sorafenib and gemcitabine plus placebo in patients with advanced pancreatic cancer. Ann. Oncol. 2012, 23, 2799–2805. [Google Scholar] [CrossRef]
- Tiriac, H.; Belleau, P.; Engle, D.D.; Plenker, D.; Deschenes, A.; Somerville, T.; Froeling, F.E.M.; Burkhart, R.A.; Denroche, R.E.; Jang, G.H.; et al. Organoid profiling identifies common responders to chemotherapy in pancreatic cancer. Cancer Discov. 2018, 8, 1112–1129. [Google Scholar] [CrossRef] [Green Version]
- Seino, T.; Kawasaki, S.; Shimokawa, M.; Tamagawa, H.; Toshimitsu, K.; Fujii, M.; Ohta, Y.; Matano, M.; Nanki, K.; Kawasaki, K.; et al. Human Pancreatic Tumor Organoids Reveal Loss of Stem Cell Niche Factor Dependence during Disease Progression. Cell Stem. Cell 2018, 22, 454–467.e6. [Google Scholar] [CrossRef] [Green Version]
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018, 175, 1972–1988 e16. [Google Scholar] [CrossRef] [Green Version]
- Romero-Calvo, I.; Weber, C.; Ray, M.; Brown, M.; Kirby, K.; Nandi, R.K.; Long, T.M.; Sparrow, S.M.; Ugolkov, A.; Qiang, W.; et al. Human Organoids Share Structural and Genetic Features with Primary Pancreatic Adenocarcinoma Tumors. Mol. Cancer Res. 2018, 17, 70–83. [Google Scholar] [CrossRef] [Green Version]
- Driehuis, E.; van Hoeck, A.; Moore, K.; Kolders, S.; Francies, H.E.; Gulersonmez, M.C.; Stigter, E.C.A.; Burgering, B.; Geurts, V.; Gracanin, A.; et al. Pancreatic cancer organoids recapitulate disease and allow personalized drug screening. Proc. Natl. Acad. Sci. USA 2019, 116, 26580–26590. [Google Scholar] [CrossRef]
- Sharick, J.T.; Walsh, C.M.; Sprackling, C.M.; Pasch, C.A.; Pham, D.L.; Esbona, K.; Choudhary, A.; Garcia-Valera, R.; Burkard, M.E.; McGregor, S.M.; et al. Metabolic Heterogeneity in Patient Tumor-Derived Organoids by Primary Site and Drug Treatment. Front. Oncol. 2020, 10, 553. [Google Scholar] [CrossRef]
- Huang, W.; Navarro-Serer, B.; Jeong, Y.J.; Chianchiano, P.; Xia, L.; Luchini, C.; Veronese, N.; Dowiak, C.; Ng, T.; Trujillo, M.A.; et al. Pattern of Invasion in Human Pancreatic Cancer Organoids Is Associated with Loss of SMAD4 and Clinical Outcome. Cancer Res. 2020, 80, 2804. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Offield, M.F.; Jetton, T.L.; Labosky, P.A.; Ray, M.; Stein, R.W.; Magnuson, M.A.; Hogan, B.L.; Wright, C.V. PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development 1996, 122, 983–995. [Google Scholar] [CrossRef] [PubMed]
- Yachida, S.; Iacobuzio-Donahue, C.A. The pathology and genetics of metastatic pancreatic cancer. Arch. Pathol. Lab. Med. 2009, 133, 413–422. [Google Scholar] [CrossRef]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef] [Green Version]
- Aiello, N.M.; Bajor, D.L.; Norgard, R.J.; Sahmoud, A.; Bhagwat, N.; Pham, M.N.; Cornish, T.C.; Iacobuzio-Donahue, C.A.; Vonderheide, R.H.; Stanger, B.Z. Metastatic progression is associated with dynamic changes in the local microenvironment. Nat. Commun. 2016, 7, 12819. [Google Scholar] [CrossRef]
- Aiello, N.M.; Rhim, A.D.; Stanger, B.Z. Isolating Epithelial and Epithelial-to-Mesenchymal Transition Populations from Primary Tumors by Fluorescence-Activated Cell Sorting. Cold Spring Harb. Protoc. 2016. [Google Scholar] [CrossRef]
- Maddipati, R.; Stanger, B.Z. Pancreatic Cancer Metastases Harbor Evidence of Polyclonality. Cancer Discov. 2015, 5, 1086–1097. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.; Li, J.; Witkiewicz, A.K.; Brennan, D.; Neill, T.; Talarico, J.; Radice, G.L. N-cadherin haploinsufficiency increases survival in a mouse model of pancreatic cancer. Oncogene 2012, 31, 4484–4489. [Google Scholar] [CrossRef] [Green Version]
- Kaneta, Y.; Sato, T.; Hikiba, Y.; Sugimori, M.; Sue, S.; Kaneko, H.; Irie, K.; Sasaki, T.; Kondo, M.; Chuma, M.; et al. Loss of Pancreatic E-Cadherin Causes Pancreatitis-Like Changes and Contributes to Carcinogenesis. Cell Mol. Gastroenterol. Hepatol. 2020, 9, 105–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krebs, A.M.; Mitschke, J.; Losada, M.L.; Schmalhofer, O.; Boerries, M.; Busch, H.; Boettcher, M.; Mougiakakos, D.; Reichardt, W.; Bronsert, P.; et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 2017, 19, 518–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.-C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguirre, A.J.; Bardeesy, N.; Sinha, M.; Lopez, L.; Tuveson, D.A.; Horner, J.; Redston, M.S.; DePinho, R.A. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003, 17, 3112–3126. [Google Scholar] [CrossRef] [Green Version]
- Bardeesy, N.; Aguirre, A.J.; Chu, G.C.; Cheng, K.H.; Lopez, L.V.; Hezel, A.F.; Feng, B.; Brennan, C.; Weissleder, R.; Mahmood, U.; et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc. Natl. Acad. Sci. USA 2006, 103, 5947–5952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izeradjene, K.; Combs, C.; Best, M.; Gopinathan, A.; Wagner, A.; Grady, W.M.; Deng, C.X.; Hruban, R.H.; Adsay, N.V.; Tuveson, D.A.; et al. Kras(G12D) and Smad4/Dpc4 haploinsufficiency cooperate to induce mucinous cystic neoplasms and invasive adenocarcinoma of the pancreas. Cancer Cell 2007, 11, 229–243. [Google Scholar] [CrossRef] [Green Version]
- Mazur, P.K.; Einwachter, H.; Lee, M.; Sipos, B.; Nakhai, H.; Rad, R.; Zimber-Strobl, U.; Strobl, L.J.; Radtke, F.; Kloppel, G.; et al. Notch2 is required for progression of pancreatic intraepithelial neoplasia and development of pancreatic ductal adenocarcinoma. Proc. Natl. Acad. Sci. USA 2010, 107, 13438–13443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morton, J.P.; Jamieson, N.B.; Karim, S.A.; Athineos, D.; Ridgway, R.A.; Nixon, C.; McKay, C.J.; Carter, R.; Brunton, V.G.; Frame, M.C.; et al. LKB1 haploinsufficiency cooperates with Kras to promote pancreatic cancer through suppression of p21-dependent growth arrest. Gastroenterology 2010, 139, 586–597.e6. [Google Scholar] [CrossRef] [Green Version]
- Rowley, M.; Ohashi, A.; Mondal, G.; Mills, L.; Yang, L.; Zhang, L.; Sundsbak, R.; Shapiro, V.; Muders, M.H.; Smyrk, T.; et al. Inactivation of Brca2 promotes Trp53-associated but inhibits KrasG12D-dependent pancreatic cancer development in mice. Gastroenterology 2011, 140, 1303–1313.e3. [Google Scholar] [CrossRef] [Green Version]
- Beatty, G.L.; Winograd, R.; Evans, R.A.; Long, K.B.; Luque, S.L.; Lee, J.W.; Clendenin, C.; Gladney, W.L.; Knoblock, D.M.; Guirnalda, P.D.; et al. Exclusion of T Cells From Pancreatic Carcinomas in Mice Is Regulated by Ly6C(low) F4/80(+) Extratumoral Macrophages. Gastroenterology 2015, 149, 201–210. [Google Scholar] [CrossRef] [Green Version]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.B.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti–PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212. [Google Scholar] [CrossRef] [Green Version]
- Liou, G.Y.; Döppler, H.; Necela, B.; Edenfield, B.; Zhang, L.; Dawson, D.W.; Storz, P. Mutant KRAS-induced expression of ICAM-1 in pancreatic acinar cells causes attraction of macrophages to expedite the formation of precancerous lesions. Cancer Discov. 2015, 5, 52–63. [Google Scholar] [CrossRef] [Green Version]
- Keenan, B.P.; Saenger, Y.; Kafrouni, M.I.; Leubner, A.; Lauer, P.; Maitra, A.; Rucki, A.A.; Gunderson, A.J.; Coussens, L.M.; Brockstedt, D.G.; et al. A Listeria vaccine and depletion of T-regulatory cells activate immunity against early stage pancreatic intraepithelial neoplasms and prolong survival of mice. Gastroenterology 2014, 146, 1784–1794.e6. [Google Scholar] [CrossRef] [Green Version]
- McAllister, F.; Bailey, J.M.; Alsina, J.; Nirschl, C.J.; Sharma, R.; Fan, H.; Rattigan, Y.; Roeser, J.C.; Lankapalli, R.H.; Zhang, H.; et al. Oncogenic Kras activates a hematopoietic-to-epithelial IL-17 signaling axis in preinvasive pancreatic neoplasia. Cancer Cell 2014, 25, 621–637. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.W.; Komar, C.A.; Bengsch, F.; Graham, K.; Beatty, G.L. Genetically Engineered Mouse Models of Pancreatic Cancer: The KPC Model (LSL-Kras(G12D/+); LSL-Trp53(R172H/+); Pdx-1-Cre), Its Variants, and Their Application in Immuno-oncology Drug Discovery. Curr. Protoc. Pharm. 2016, 73, 14.39.1–14.39.20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werb, Z.; Lu, P. The Role of Stroma in Tumor Development. Cancer J. 2015, 21, 250–253. [Google Scholar] [CrossRef] [Green Version]
- Neesse, A.; Bauer, C.A.; Ohlund, D.; Lauth, M.; Buchholz, M.; Michl, P.; Tuveson, D.A.; Gress, T.M. Stromal biology and therapy in pancreatic cancer: Ready for clinical translation? Gut 2019, 68, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Briest, F.; Berndt, A.; Clement, J.; Junker, K.; Eggeling, F.; Grimm, S.; Friedrich, K. Tumor-stroma interactions in tumorigenesis: Lessons from stem cell biology. Front. Biosci. (Elite Ed.) 2012, 4, 1871–1887. [Google Scholar] [CrossRef]
- Greggio, C.; de Franceschi, F.; Figueiredo-Larsen, M.; Gobaa, S.; Ranga, A.; Semb, H.; Lutolf, M.; Grapin-Botton, A. Artificial three-dimensional niches deconstruct pancreas development in vitro. Development 2013, 140, 4452–4462. [Google Scholar] [CrossRef] [Green Version]
- Huch, M.; Koo, B.-K. Modeling mouse and human development using organoid cultures. Development 2015, 142, 3113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, L.A.; Tiriac, H.; Clevers, H.; Tuveson, D.A. Modeling pancreatic cancer with organoids. Trends Cancer 2016, 2, 176–190. [Google Scholar] [CrossRef] [Green Version]
- Boj, S.F.; Hwang, C.I.; Baker, L.A.; Chio, I.I.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Nadauld, L.; Ootani, A.; Corney, D.C.; Pai, R.K.; Gevaert, O.; Cantrell, M.A.; Rack, P.G.; Neal, J.T.; Chan, C.W.; et al. Oncogenic transformation of diverse gastrointestinal tissues in primary organoid culture. Nat. Med. 2014, 20, 769–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roe, J.S.; Hwang, C.I.; Somerville, T.D.D.; Milazzo, J.P.; Lee, E.J.; da Silva, B.; Maiorino, L.; Tiriac, H.; Young, C.M.; Miyabayashi, K.; et al. Enhancer Reprogramming Promotes Pancreatic Cancer Metastasis. Cell 2017, 170, 875–888 e20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehlen, L.; Arndt, J.; Treue, D.; Bischoff, P.; Loch, F.N.; Hahn, E.M.; Kotsch, K.; Klauschen, F.; Beyer, K.; Margonis, G.A.; et al. Novel methods for in vitro modeling of pancreatic cancer reveal important aspects for successful primary cell culture. BMC Cancer 2020, 20, 417. [Google Scholar] [CrossRef] [PubMed]
- Engle, D.D.; Tiriac, H.; Rivera, K.D.; Pommier, A.; Whalen, S.; Oni, T.E.; Alagesan, B.; Lee, E.J.; Yao, M.A.; Lucito, M.S.; et al. The glycan CA19-9 promotes pancreatitis and pancreatic cancer in mice. Science 2019, 364, 1156–1162. [Google Scholar] [CrossRef] [PubMed]
- Lupo, F.; Piro, G.; Torroni, L.; Delfino, P.; Trovato, R.; Rusev, B.; Fiore, A.; Filippini, D.; de Sanctis, F.; Manfredi, M.; et al. Organoid-Transplant Model Systems to Study the Effects of Obesity on the Pancreatic Carcinogenesis in vivo. Front. Cell Dev. Biol. 2020, 8, 308. [Google Scholar] [CrossRef]
- Miyabayashi, K.; Baker, L.A.; Deschenes, A.; Traub, B.; Caligiuri, G.; Plenker, D.; Alagesan, B.; Belleau, P.; Li, S.; Kendall, J.; et al. Intraductal transplantation models of human pancreatic ductal adenocarcinoma reveal progressive transition of molecular subtypes. Cancer Discov. 2020, 10, 1566–1589. [Google Scholar] [CrossRef]
- Deer, E.L.; González-Hernández, J.; Coursen, J.D.; Shea, J.E.; Ngatia, J.; Scaife, C.L.; Firpo, M.A.; Mulvihill, S.J. Phenotype and genotype of pancreatic cancer cell lines. Pancreas 2010, 39, 425–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, M.H.; Nowak, N.J.; Loor, R.; Ochi, H.; Sandberg, A.A.; Lopez, C.; Pickren, J.W.; Berjian, R.; Douglass, H.O.; Chu, T.M., Jr. Characterization of a new primary human pancreatic tumor line. Cancer Investig. 1986, 4, 15–23. [Google Scholar] [CrossRef]
- Kyriazis, A.A.; Kyriazis, A.P.; Sternberg, C.N.; Sloane, N.H.; Loveless, J.D. Morphological, biological, biochemical, and karyotypic characteristics of human pancreatic ductal adenocarcinoma Capan-2 in tissue culture and the nude mouse. Cancer Res. 1986, 46, 5810–5815. [Google Scholar]
- Gower, W.R., Jr.; Risch, R.M.; Godellas, C.V.; Fabri, P.J. HPAC, a new human glucocorticoid-sensitive pancreatic ductal adenocarcinoma cell line. In Vitro Cell Dev. Biol. Anim. 1994, 30A, 151–161. [Google Scholar] [CrossRef]
- Yunis, A.A.; Arimura, G.K.; Russin, D.J. Human pancreatic carcinoma (MIA PaCa-2) in continuous culture: Sensitivity to asparaginase. Int. J. Cancer 1977, 19, 128–135. [Google Scholar] [CrossRef]
- Lieber, M.; Mazzetta, J.; Nelson-Rees, W.; Kaplan, M.; Todaro, G. Establishment of a continuous tumor-cell line (panc-1) from a human carcinoma of the exocrine pancreas. Int. J. Cancer 1975, 15, 741–747. [Google Scholar] [CrossRef]
- Chen, W.H.; Horoszewicz, J.S.; Leong, S.S.; Shimano, T.; Penetrante, R.; Sanders, W.H.; Berjian, R.; Douglass, H.O.; Martin, E.W.; Chu, T.M. Human pancreatic adenocarcinoma: In vitro and in vivo morphology of a new tumor line established from ascites. In Vitro Cell. Dev. Biol. Plant 1982, 18, 24–34. [Google Scholar] [CrossRef]
- Kyriazis, A.P.; Kyriazis, A.A.; Scarpelli, D.G.; Fogh, J.; Rao, M.S.; Lepera, R. Human pancreatic adenocarcinoma line Capan-1 in tissue culture and the nude mouse: Morphologic, biologic, and biochemical characteristics. Am. J. Pathol 1982, 106, 250–260. [Google Scholar] [PubMed]
- Schoumacher, R.A.; Ram, J.; Iannuzzi, M.C.; Bradbury, N.A.; Wallace, R.W.; Hon, C.T.; Kelly, D.R.; Schmid, S.M.; Gelder, F.B.; Rado, T.A.; et al. A cystic fibrosis pancreatic adenocarcinoma cell line. Proc. Natl. Acad. Sci. USA 1990, 87, 4012–4016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metzgar, R.S.; Gaillard, M.T.; Levine, S.J.; Tuck, F.L.; Bossen, E.H.; Borowitz, M.J. Antigens of human pancreatic adenocarcinoma cells defined by murine monoclonal antibodies. Cancer Res. 1982, 42, 601–608. [Google Scholar] [PubMed]
- Smith, H.S. In vitro properties of epithelial cell lines established from human carcinomas and nonmalignant tissue. J. Natl. Cancer Inst. 1979, 62, 225–230. [Google Scholar] [PubMed]
- Drucker, B.J.; Marincola, F.M.; Siao, D.Y.; Donlon, T.A.; Bangs, C.D.; Holder, W.D., Jr. A new human pancreatic carcinoma cell line developed for adoptive immunotherapy studies with lymphokine-activated killer cells in nude mice. In Vitro Cell Dev. Biol. 1988, 24, 1179–1187. [Google Scholar] [CrossRef]
- Yu, K.; Chen, B.; Aran, D.; Charalel, J.; Yau, C.; Wolf, D.M.; Veer, L.J.v.; Butte, A.J.; Goldstein, T.; Sirota, M. Comprehensive transcriptomic analysis of cell lines as models of primary tumors across 22 tumor types. Nat. Commun. 2019, 10, 3574. [Google Scholar] [CrossRef] [Green Version]
- Gadaleta, E.; Cutts, R.J.; Kelly, G.P.; Crnogorac-Jurcevic, T.; Kocher, H.M.; Lemoine, N.R.; Chelala, C. A global insight into a cancer transcriptional space using pancreatic data: Importance, findings and flaws. Nucleic. Acids. Res. 2011, 39, 7900–7907. [Google Scholar] [CrossRef]
- Froeling, F.E.; Marshall, J.F.; Kocher, H.M. Pancreatic cancer organotypic cultures. J. Biotechnol. 2010, 148, 16–23. [Google Scholar] [CrossRef]
- Stock, K.; Estrada, M.F.; Vidic, S.; Gjerde, K.; Rudisch, A.; Santo, V.E.; Barbier, M.; Blom, S.; Arundkar, S.C.; Selvam, I.; et al. Capturing tumor complexity in vitro: Comparative analysis of 2D and 3D tumor models for drug discovery. Sci. Rep. 2016, 6, 28951. [Google Scholar] [CrossRef] [Green Version]
- Fogh, J.; Fogh, J.M.; Orfeo, T. One hundred and twenty-seven cultured human tumor cell lines producing tumors in nude mice. J. Natl. Cancer Inst. 1977, 59, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Voskoglou-Nomikos, T.; Pater, J.L.; Seymour, L. Clinical predictive value of the in vitro cell line, human xenograft, and mouse allograft preclinical cancer models. Clin. Cancer Res. 2003, 9, 4227–4239. [Google Scholar] [PubMed]
- Hwang, C.I.; Boj, S.F.; Clevers, H.; Tuveson, D.A. Preclinical models of pancreatic ductal adenocarcinoma. J. Pathol. 2016, 238, 197–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrel, A.; Lindbergh, C. The Culture of Organs; Paul B. Hoeber, Inc.: New York, NY, USA, 1938. [Google Scholar]
- Jones, R.T.; Trump, B.F. Cellular and subcellular effects of ischemia on the pancreatic acinar cell. Virchows Arch. B 1975, 19, 325–336. [Google Scholar]
- Jones, R.T.; Hudson, E.A.; Resau, J.H. A review of in vitro and in vivo culture techniques for the study of pancreatic carcinogenesis. Cancer 1981, 47 (Suppl. 6), 1490–1496. [Google Scholar] [CrossRef]
- Huch, M.; Bonfanti, P.; Boj, S.F.; Sato, T.; Loomans, C.J.; van de Wetering, M.; Sojoodi, M.; Li, V.S.; Schuijers, J.; Gracanin, A.; et al. Unlimited in vitro expansion of adult bi-potent pancreas progenitors through the Lgr5/R-spondin axis. EMBO J. 2013, 32, 2708–2721. [Google Scholar] [CrossRef] [Green Version]
- Fanjul, M.; Hollande, E. Morphogenesis of “duct-like” structures in three-dimensional cultures of human cancerous pancreatic duct cells (Capan-1). In Vitro Cell Dev. Biol. Anim. 1993, 29A, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, Y.; Ishiwata, T.; Kawamoto, Y.; Kawahara, K.; Peng, W.X.; Yamamoto, T.; Naito, Z. Morphological and cytoskeletal changes of pancreatic cancer cells in three-dimensional spheroidal culture. Med. Mol. Morphol. 2010, 43, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Broekgaarden, M.; Rizvi, I.; Bulin, A.L.; Petrovic, L.; Goldschmidt, R.; Massodi, I.; Celli, J.P.; Hasan, T. Neoadjuvant photodynamic therapy augments immediate and prolonged oxaliplatin efficacy in metastatic pancreatic cancer organoids. Oncotarget 2018, 9, 13009–13022. [Google Scholar] [CrossRef] [PubMed]
- Yeon, S.-E.; No, D.Y.; Lee, S.-H.; Nam, S.W.; Oh, I.-H.; Lee, J.; Kuh, H.-J. Application of Concave Microwells to Pancreatic Tumor Spheroids Enabling Anticancer Drug Evaluation in a Clinically Relevant Drug Resistance Model. PLoS ONE 2013, 8, e73345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ware, M.J.; Colbert, K.; Keshishian, V.; Ho, J.; Corr, S.J.; Curley, S.A.; Godin, B. Generation of Homogenous Three-Dimensional Pancreatic Cancer Cell Spheroids Using an Improved Hanging Drop Technique. Tissue Eng. Part. CMethods 2016, 22, 312–321. [Google Scholar] [CrossRef] [Green Version]
- Kapalczynska, M.; Kolenda, T.; Przybyla, W.; Zajaczkowska, M.; Teresiak, A.; Filas, V.; Ibbs, M.; Blizniak, R.; Luczewski, L.; Lamperska, K. 2D and 3D cell cultures—A comparison of different types of cancer cell cultures. Arch. Med. Sci. 2018, 14, 910–919. [Google Scholar] [PubMed]
- Longati, P.; Jia, X.; Eimer, J.; Wagman, A.; Witt, M.R.; Rehnmark, S.; Verbeke, C.; Toftgard, R.; Lohr, M.; Heuchel, R.L. 3D pancreatic carcinoma spheroids induce a matrix-rich, chemoresistant phenotype offering a better model for drug testing. BMC Cancer 2013, 13, 95. [Google Scholar] [CrossRef] [Green Version]
- Gagliano, N.; Sforza, C.; Sommariva, M.; Menon, A.; Conte, V.; Sartori, P.; Procacci, P. 3D-spheroids: What can they tell us about pancreatic ductal adenocarcinoma cell phenotype? Exp. Cell Res. 2017, 357, 299–309. [Google Scholar] [CrossRef]
- Shoval, H.; Karsch-Bluman, A.; Brill-Karniely, Y.; Stern, T.; Zamir, G.; Hubert, A.; Benny, O. Tumor cells and their crosstalk with endothelial cells in 3D spheroids. Sci. Rep. 2017, 7, 10428. [Google Scholar] [CrossRef] [Green Version]
- Wen, Z.; Liao, Q.; Hu, Y.; You, L.; Zhou, L.; Zhao, Y. A spheroid-based 3-D culture model for pancreatic cancer drug testing, using the acid phosphatase assay. Braz. J. Med. Biol. Res. Rev. Bras. Pesqui. Med. Biol. 2013, 46, 634–642. [Google Scholar] [CrossRef] [Green Version]
- Sirri, E.; Castro, F.A.; Kieschke, J.; Jansen, L.; Emrich, K.; Gondos, A.; Holleczek, B.; Katalinic, A.; Urbschat, I.; Vohmann, C.; et al. Recent Trends in Survival of Patients With Pancreatic Cancer in Germany and the United States. Pancreas 2016, 45, 908–914. [Google Scholar] [CrossRef]
- Mackay, T.M.; van der Geest, L.G.M.; Koerkamp, B.G.; van Laarhoven, H.W.M.; Bonsing, B.A.; Wilmink, J.W.; van Santvoort, H.C.; de Vos-Geelen, J.; van Eijck, C.H.J.; Busch, O.R.; et al. Association of the location of pancreatic ductal adenocarcinoma (head, body, tail) with tumor stage, treatment, and survival: A population-based analysis AU—van Erning, Felice N. Acta Oncol. 2018, 57, 1655–1662. [Google Scholar]
- Pergolini, I.; Morales-Oyarvide, V.; Mino-Kenudson, M.; Honselmann, K.C.; Rosenbaum, M.W.; Nahar, S.; Kem, M.; Ferrone, C.R.; Lillemoe, K.D.; Bardeesy, N.; et al. Tumor engraftment in patient-derived xenografts of pancreatic ductal adenocarcinoma is associated with adverse clinicopathological features and poor survival. PLoS ONE 2017, 12, e0182855. [Google Scholar] [CrossRef]
- Damhofer, H.; Ebbing, E.A.; Steins, A.; Welling, L.; Tol, J.A.; Krishnadath, K.K.; van Leusden, T.; van de Vijver, M.J.; Besselink, M.G.; Busch, O.R.; et al. Establishment of patient-derived xenograft models and cell lines for malignancies of the upper gastrointestinal tract. J. Transl. Med. 2015, 13, 115. [Google Scholar] [CrossRef] [Green Version]
- Perez-Torras, S.; Vidal-Pla, A.; Miquel, R.; Almendro, V.; Fernandez-Cruz, L.; Navarro, S.; Maurel, J.; Carbo, N.; Gascon, P.; Mazo, A. Characterization of human pancreatic orthotopic tumor xenografts suitable for drug screening. Cell Oncol. (Dordr) 2011, 34, 511–521. [Google Scholar] [CrossRef] [Green Version]
- Nicolle, R.; Blum, Y.; Marisa, L.; Loncle, C.; Gayet, O.; Moutardier, V.; Turrini, O.; Giovannini, M.; Bian, B.; Bigonnet, M.; et al. Pancreatic Adenocarcinoma Therapeutic Targets Revealed by Tumor-Stroma Cross-Talk Analyses in Patient-Derived Xenografts. Cell Rep. 2017, 21, 2458–2470. [Google Scholar] [CrossRef] [Green Version]
- Chou, A.; Froio, D.; Nagrial, A.M.; Parkin, A.; Murphy, K.J.; Chin, V.T.; Wohl, D.; Steinmann, A.; Stark, R.; Drury, A.; et al. Tailored first-line and second-line CDK4-targeting treatment combinations in mouse models of pancreatic cancer. Gut 2018, 67, 2142–2155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, J.; Lee, C.H.; Seol, H.S.; Choi, Y.S.; Kim, E.; Lee, E.J.; Rhee, J.K.; Singh, S.R.; Jun, E.S.; Han, B.; et al. Generation and molecular characterization of pancreatic cancer patient-derived xenografts reveals their heterologous nature. Oncotarget 2016, 7, 62533–62546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delitto, D.; Pham, K.; Vlada, A.C.; Sarosi, G.A.; Thomas, R.M.; Behrns, K.E.; Liu, C.; Hughes, S.J.; Wallet, S.M.; Trevino, J.G. Patient-derived xenograft models for pancreatic adenocarcinoma demonstrate retention of tumor morphology through incorporation of murine stromal elements. Am. J. Pathol. 2015, 185, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, J.; Seol, H.S.; Chang, S. The Generation and Application of Patient-Derived Xenograft Model for Cancer Research. Cancer Res. Treat. 2018, 50, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinska, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; de Jong, S.; Jonkers, J.; Maelandsmo, G.M.; et al. Patient-derived xenograft models: An emerging platform for translational cancer research. Cancer Discov. 2014, 4, 998–1013. [Google Scholar] [CrossRef] [Green Version]
- Hidalgo, M.; Bruckheimer, E.; Rajeshkumar, N.V.; Garrido-Laguna, I.; de Oliveira, E.; Rubio-Viqueira, B.; Strawn, S.; Wick, M.J.; Martell, J.; Sidransky, D. A pilot clinical study of treatment guided by personalized tumorgrafts in patients with advanced cancer. Mol. Cancer 2011, 10, 1311–1316. [Google Scholar] [CrossRef] [Green Version]
- Sebastiani, V.; Ricci, F.; Rubio-Viqueira, B.; Kulesza, P.; Yeo, C.J.; Hidalgo, M.; Klein, A.; Laheru, D.; Iacobuzio-Donahue, C.A. Immunohistochemical and genetic evaluation of deoxycytidine kinase in pancreatic cancer: Relationship to molecular mechanisms of gemcitabine resistance and survival. Clin. Cancer Res. 2006, 12, 2492–2497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrido-Laguna, I.; Uson, M.; Rajeshkumar, N.V.; Tan, A.C.; de Oliveira, E.; Karikari, C.; Villaroel, M.C.; Salomon, A.; Taylor, G.; Sharma, R.; et al. Tumor engraftment in nude mice and enrichment in stroma- related gene pathways predict poor survival and resistance to gemcitabine in patients with pancreatic cancer. Clin. Cancer Res. 2011, 17, 5793–5800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutta, P.; Perez, M.R.; Lee, J.; Kang, Y.; Pratt, M.; Salzillo, T.C.; Weygand, J.; Zacharias, N.M.; Gammon, S.T.; Koay, E.J.; et al. Combining Hyperpolarized Real-Time Metabolic Imaging and NMR Spectroscopy To Identify Metabolic Biomarkers in Pancreatic Cancer. J. Proteome Res. 2019, 18, 2826–2834. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Viqueira, B.; Jimeno, A.; Cusatis, G.; Zhang, X.; Iacobuzio-Donahue, C.; Karikari, C.; Shi, C.; Danenberg, K.; Danenberg, P.V.; Kuramochi, H.; et al. An in vivo platform for translational drug development in pancreatic cancer. Clin. Cancer Res. 2006, 12, 4652–4661. [Google Scholar] [CrossRef] [Green Version]
- Willey, C.D.; Gilbert, A.N.; Anderson, J.C.; Gillespie, G.Y. Patient-Derived Xenografts as a Model System for Radiation Research. Semin. Radiat. Oncol. 2015, 25, 273–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Holtzinger, A.; Jagan, I.; BeGora, M.; Lohse, I.; Ngai, N.; Nostro, C.; Wang, R.; Muthuswamy, L.B.; Crawford, H.C.; et al. Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell- and patient-derived tumor organoids. Nat. Med. 2015, 21, 1364–1371. [Google Scholar] [CrossRef]
- Frappart, P.O.; Walter, K.; Gout, J.; Beutel, A.K.; Morawe, M.; Arnold, F.; Breunig, M.; Barth, T.F.; Marienfeld, R.; Schulte, L.; et al. Pancreatic cancer-derived organoids—A disease modeling tool to predict drug response. United Eur. Gastroenterol. J. 2020, 8, 594–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorrell, C.; Tarlow, B.; Wang, Y.; Canaday, P.S.; Haft, A.; Schug, J.; Streeter, P.R.; Finegold, M.J.; Shenje, L.T.; Kaestner, K.H.; et al. The organoid-initiating cells in mouse pancreas and liver are phenotypically and functionally similar. Stem. Cell Res. 2014, 13, 275–283. [Google Scholar] [CrossRef] [Green Version]
- Eser, S.; Schnieke, A.; Schneider, G.; Saur, D. Oncogenic KRAS signalling in pancreatic cancer. Br. J. Cancer 2014, 111, 817–822. [Google Scholar] [CrossRef]
- Jiramongkolchai, P.; Owens, P.; Hong, C.C. Emerging roles of the bone morphogenetic protein pathway in cancer: Potential therapeutic target for kinase inhibition. Biochem. Soc. Trans. 2016, 44, 1117–1134. [Google Scholar] [CrossRef] [Green Version]
- Nanki, K.; Toshimitsu, K.; Takano, A.; Fujii, M.; Shimokawa, M.; Ohta, Y.; Matano, M.; Seino, T.; Nishikori, S.; Ishikawa, K.; et al. Divergent Routes toward Wnt and R-spondin Niche Independency during Human Gastric Carcinogenesis. Cell 2018, 174, 856–869.e17. [Google Scholar] [CrossRef] [Green Version]
- Mihara, E.; Hirai, H.; Yamamoto, H.; Tamura-Kawakami, K.; Matano, M.; Kikuchi, A.; Sato, T.; Takagi, J. Active and water-soluble form of lipidated Wnt protein is maintained by a serum glycoprotein afamin/α-albumin. eLife 2016, 5, e11621. [Google Scholar] [CrossRef]
- Gendoo, D.M.A.; Denroche, R.E.; Zhang, A.; Radulovich, N.; Jang, G.H.; Lemire, M.; Fischer, S.; Chadwick, D.; Lungu, I.M.; Ibrahimov, E.; et al. Whole genomes define concordance of matched primary, xenograft, and organoid models of pancreas cancer. PLoS Comput. Biol. 2019, 15, e1006596. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Huang, S.; Sun, Y.L. Epithelial-Mesenchymal Transition in Pancreatic Cancer: A Review. BioMed Res. Int. 2017, 2017, 2646148. [Google Scholar] [CrossRef] [Green Version]
- Kuo, T.L.; Cheng, K.H.; Shan, Y.S.; Chen, L.T.; Hung, W.C. β-catenin-activated autocrine PDGF/Src signaling is a therapeutic target in pancreatic cancer. Theranostics 2019, 9, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Messal, H.A.; Alt, S.; Ferreira, R.M.M.; Gribben, C.; Wang, V.M.-Y.; Cotoi, C.G.; Salbreux, G.; Behrens, A. Tissue curvature and apicobasal mechanical tension imbalance instruct cancer morphogenesis. Nature 2019, 566, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Morgani, S.M.; David, C.J.; Wang, Q.; Er, E.E.; Huang, Y.H.; Basnet, H.; Zou, Y.; Shu, W.; Soni, R.K.; et al. TGF-β orchestrates fibrogenic and developmental EMTs via the RAS effector RREB1. Nature 2020, 577, 566–571. [Google Scholar] [CrossRef]
- David, C.J.; Huang, Y.H.; Chen, M.; Su, J.; Zou, Y.; Bardeesy, N.; Iacobuzio-Donahue, C.A.; Massagué, J. TGF-β Tumor Suppression through a Lethal EMT. Cell 2016, 164, 1015–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dougan, S.K. The Pancreatic Cancer Microenvironment. Cancer J. 2017, 23, 321–325. [Google Scholar] [CrossRef]
- Erkan, M.; Michalski, C.W.; Rieder, S.; Reiser-Erkan, C.; Abiatari, I.; Kolb, A.; Giese, N.A.; Esposito, I.; Friess, H.; Kleeff, J. The activated stroma index is a novel and independent prognostic marker in pancreatic ductal adenocarcinoma. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2008, 6, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Apte, M.V.; Park, S.; Phillips, P.A.; Santucci, N.; Goldstein, D.; Kumar, R.K.; Ramm, G.A.; Buchler, M.; Friess, H.; McCarroll, J.A.; et al. Desmoplastic reaction in pancreatic cancer: Role of pancreatic stellate cells. Pancreas 2004, 29, 179–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamoto, H.; Murakami, T.; Tsuchida, K.; Sugino, H.; Miyake, H.; Tashiro, S. Tumor-stroma interaction of human pancreatic cancer: Acquired resistance to anticancer drugs and proliferation regulation is dependent on extracellular matrix proteins. Pancreas 2004, 28, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, P.; Gunzer, F.; Gunzer, M. Analyzing the physicodynamics of immune cells in a three-dimensional collagen matrix. Methods Mol. Biol. 2007, 380, 253–269. [Google Scholar] [PubMed]
- Weniger, M.; Honselmann, K.C.; Liss, A.S. The Extracellular Matrix and Pancreatic Cancer: A Complex Relationship. Cancers 2018, 10, 316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, J.Y.; Tan, S.-J.; Wu, Y.-C.; Yang, Z.; Hoang, B.X.; Han, B. From competency to dormancy: A 3D model to study cancer cells and drug responsiveness. J. Transl. Med. 2016, 14, 38. [Google Scholar] [CrossRef] [Green Version]
- Gjorevski, N.; Sachs, N.; Manfrin, A.; Giger, S.; Bragina, M.E.; Ordóñez-Morán, P.; Clevers, H.; Lutolf, M.P. Designer matrices for intestinal stem cell and organoid culture. Nature 2016, 539, 560. [Google Scholar] [CrossRef]
- Lee, J.-H.; Kim, S.-K.; Khawar, I.A.; Jeong, S.-Y.; Chung, S.; Kuh, H.-J. Microfluidic co-culture of pancreatic tumor spheroids with stellate cells as a novel 3D model for investigation of stroma-mediated cell motility and drug resistance. J. Exp. Clin. Cancer Res. 2018, 37, 4. [Google Scholar] [CrossRef] [Green Version]
- Ware, M.J.; Keshishian, V.; Law, J.J.; Ho, J.C.; Favela, C.A.; Rees, P.; Smith, B.; Mohammad, S.; Hwang, R.F.; Rajapakshe, K.; et al. Generation of an in vitro 3D PDAC stroma rich spheroid model. Biomaterials 2016, 108, 129–142. [Google Scholar] [CrossRef] [Green Version]
- Tsai, S.; McOlash, L.; Palen, K.; Johnson, B.; Duris, C.; Yang, Q.; Dwinell, M.B.; Hunt, B.; Evans, D.B.; Gershan, J.; et al. Development of primary human pancreatic cancer organoids, matched stromal and immune cells and 3D tumor microenvironment models. BMC Cancer 2018, 18, 335. [Google Scholar] [CrossRef]
- Broekgaarden, M.; Anbil, S.; Bulin, A.-L.; Obaid, G.; Mai, Z.; Baglo, Y.; Rizvi, I.; Hasan, T. Modulation of redox metabolism negates cancer-associated fibroblasts-induced treatment resistance in a heterotypic 3D culture platform of pancreatic cancer. Biomaterials 2019, 222, 119421. [Google Scholar] [CrossRef]
- Walsh, A.J.; Castellanos, J.A.; Nagathihalli, N.S.; Merchant, N.B.; Skala, M.C. Optical Imaging of Drug-Induced Metabolism Changes in Murine and Human Pancreatic Cancer Organoids Reveals Heterogeneous Drug Response. Pancreas 2016, 45, 863–869. [Google Scholar] [CrossRef]
- Shi, Y.; Gao, W.; Lytle, N.K.; Huang, P.; Yuan, X.; Dann, A.M.; Ridinger-Saison, M.; DelGiorno, K.E.; Antal, C.E.; Liang, G.; et al. Targeting LIF-mediated paracrine interaction for pancreatic cancer therapy and monitoring. Nature 2019, 569, 131–135. [Google Scholar] [CrossRef]
- Zhang, Y.; Yan, W.; Collins, M.A.; Bednar, F.; Rakshit, S.; Zetter, B.R.; Stanger, B.Z.; Chung, I.; Rhim, A.D.; di Magliano, M.P. Interleukin-6 Is Required for Pancreatic Cancer Progression by Promoting MAPK Signaling Activation and Oxidative Stress Resistance. Cancer Res. 2013, 73, 6359. [Google Scholar] [CrossRef] [Green Version]
- Lesina, M.; Kurkowski, M.U.; Ludes, K.; Rose-John, S.; Treiber, M.; Klöppel, G.; Yoshimura, A.; Reindl, W.; Sipos, B.; Akira, S.; et al. Stat3/Socs3 Activation by IL-6 Transsignaling Promotes Progression of Pancreatic Intraepithelial Neoplasia and Development of Pancreatic Cancer. Cancer Cell 2011, 19, 456–469. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, A.; Wang, S.C.; Morris, J.P.T.; Folias, A.E.; Liou, A.; Kim, G.E.; Akira, S.; Boucher, K.M.; Firpo, M.A.; Mulvihill, S.J.; et al. Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer Cell 2011, 19, 441–455. [Google Scholar] [CrossRef] [Green Version]
- Koikawa, K.; Ohuchida, K.; Ando, Y.; Kibe, S.; Nakayama, H.; Takesue, S.; Endo, S.; Abe, T.; Okumura, T.; Iwamoto, C.; et al. Basement membrane destruction by pancreatic stellate cells leads to local invasion in pancreatic ductal adenocarcinoma. Cancer Lett. 2018, 425, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Skelton, R.A.; Javed, A.; Zheng, L.; He, J. Overcoming the resistance of pancreatic cancer to immune checkpoint inhibitors. J. Surg. Oncol. 2017, 116, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Clark, C.E.; Hingorani, S.R.; Mick, R.; Combs, C.; Tuveson, D.A.; Vonderheide, R.H. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007, 67, 9518–9527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuen, J.; Darowski, D.; Kluge, T.; Majety, M. Pancreatic cancer cell/fibroblast co-culture induces M2 like macrophages that influence therapeutic response in a 3D model. PLoS ONE 2017, 12, e0182039. [Google Scholar] [CrossRef] [PubMed]
- Holokai, L.; Chakrabarti, J.; Lundy, J.; Croagh, D.; Adhikary, P.; Richards, S.S.; Woodson, C.; Steele, N.; Kuester, R.; Scott, A.; et al. Murine- and Human-Derived Autologous Organoid/Immune Cell Co-Cultures as Pre-Clinical Models of Pancreatic Ductal Adenocarcinoma. Cancers 2020, 12, 3816. [Google Scholar] [CrossRef] [PubMed]
- Aung, K.L.; Fischer, S.E.; Denroche, R.E.; Jang, G.-H.; Dodd, A.; Creighton, S.; Southwood, B.; Liang, S.-B.; Chadwick, D.; Zhang, A.; et al. Genomics-Driven Precision Medicine for Advanced Pancreatic Cancer: Early Results from the COMPASS Trial. Clin. Cancer Res. 2018, 24, 1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganesh, K.; Wu, C.; O’Rourke, K.P.; Szeglin, B.C.; Zheng, Y.; Sauvé, C.-E.G.; Adileh, M.; Wasserman, I.; Marco, M.R.; Kim, A.S.; et al. A rectal cancer organoid platform to study individual responses to chemoradiation. Nat. Med. 2019, 25, 1607–1614. [Google Scholar] [CrossRef]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernandez-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.H.N.; Siu, H.C.; Law, S.; Ho, S.L.; Yue, S.S.K.; Tsui, W.Y.; Chan, D.; Chan, A.S.; Ma, S.; Lam, K.O.; et al. A Comprehensive Human Gastric Cancer Organoid Biobank Captures Tumor Subtype Heterogeneity and Enables Therapeutic Screening. Cell Stem. Cell 2018, 23, 882–897. [Google Scholar] [CrossRef] [Green Version]
- Gilles, M.E.; Hao, L.; Huang, L.; Rupaimoole, R.; Lopez-Casas, P.P.; Pulver, E.; Jeong, J.C.; Muthuswamy, S.K.; Hidalgo, M.; Bhatia, S.N.; et al. Personalized RNA Medicine for Pancreatic Cancer. Clin. Cancer Res. 2018, 24, 1734–1747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, K.; Chow, C.R.; Ebine, K.; Arslan, A.D.; Kwok, B.; Bentrem, D.J.; Eckerdt, F.D.; Platanias, L.C.; Munshi, H.G. Differential Regulation of ZEB1 and EMT by MAPK-Interacting Protein Kinases (MNK) and eIF4E in Pancreatic Cancer. Mol. Cancer Res. 2016, 14, 216–227. [Google Scholar] [CrossRef] [Green Version]
- Chio, I.I.C.; Jafarnejad, S.M.; Ponz-Sarvise, M.; Park, Y.; Rivera, K.; Palm, W.; Wilson, J.; Sangar, V.; Hao, Y.; Ohlund, D.; et al. NRF2 Promotes Tumor Maintenance by Modulating mRNA Translation in Pancreatic Cancer. Cell 2016, 166, 963–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponz-Sarvise, M.; Corbo, V.; Tiriac, H.; Engle, D.D.; Frese, K.K.; Oni, T.E.; Hwang, C.-I.; Öhlund, D.; Chio, I.I.C.; Baker, L.A.; et al. Identification of Resistance Pathways Specific to Malignancy Using Organoid Models of Pancreatic Cancer. Clin. Cancer Res. 2019, 25, 6742. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Sanagapalli, S.; Stoita, A. Challenges in diagnosis of pancreatic cancer. World J. Gastroenterol. 2018, 24, 2047–2060. [Google Scholar] [CrossRef] [PubMed]
- Tiriac, H.; Bucobo, J.C.; Tzimas, D.; Grewel, S.; Lacomb, J.F.; Rowehl, L.M.; Nagula, S.; Wu, M.; Kim, J.; Sasson, A.; et al. Successful creation of pancreatic cancer organoids by means of EUS-guided fine-needle biopsy sampling for personalized cancer treatment. Gastrointest. Endosc. 2018, 87, 1474–1480. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Low, R.R.J.; Lim, W.W.; Nguyen, P.M.; Lee, B.; Christie, M.; Burgess, A.W.; Gibbs, P.; Grimmond, S.M.; Hollande, F.; Putoczki, T.L. The Diverse Applications of Pancreatic Ductal Adenocarcinoma Organoids. Cancers 2021, 13, 4979. https://doi.org/10.3390/cancers13194979
Low RRJ, Lim WW, Nguyen PM, Lee B, Christie M, Burgess AW, Gibbs P, Grimmond SM, Hollande F, Putoczki TL. The Diverse Applications of Pancreatic Ductal Adenocarcinoma Organoids. Cancers. 2021; 13(19):4979. https://doi.org/10.3390/cancers13194979
Chicago/Turabian StyleLow, Ronnie Ren Jie, Wei Wen Lim, Paul M. Nguyen, Belinda Lee, Michael Christie, Antony W. Burgess, Peter Gibbs, Sean M. Grimmond, Frédéric Hollande, and Tracy L. Putoczki. 2021. "The Diverse Applications of Pancreatic Ductal Adenocarcinoma Organoids" Cancers 13, no. 19: 4979. https://doi.org/10.3390/cancers13194979