
Biology and Treatment Advances in Cutaneous Squamous Cell Carcinoma


1
Department of Medical Oncology, Peter MacCallum Cancer Centre, 305 Grattan St., Parkville, Melbourne, VIC 3000, Australia
2
The Sir Peter MacCallum Department of Oncology, University of Melbourne, Melbourne, VIC 3000, Australia
*
Author to whom correspondence should be addressed.
Cancers 2021, 13(22), 5645; https://doi.org/10.3390/cancers13225645
Submission received: 29 September 2021
/
Revised: 8 November 2021
/
Accepted: 8 November 2021
/
Published: 11 November 2021
(This article belongs to the Special Issue Treatments for Squamous Cell Cancer)
{kind=link}
Abstract
:Simple Summary
Skin cancers are the most diagnosed type of cancer worldwide. Cutaneous squamous cell carcinoma—a type of skin cancer—usually affects older people who have chronic sun exposure, as well as people with weakened immune systems. There has been significant recent progress in the treatment of this type of cancer with immune checkpoint inhibitors that utilize the immune system to target cancer. In concert with advances in treatment, our understanding of the biology of skin cancer has also deepened. The authors have reviewed the risk factors, biology, and advances in treatment in this publication.
Abstract
Cutaneous squamous cell carcinoma (CSCC) is the second most common skin cancer diagnosed worldwide. CSCC is generally localized and managed with local therapies such as excision and/or radiotherapy. For patients with unresectable or metastatic disease, recent improvements in our understanding of the underlying biology have led to significant advancements in treatment approaches—including the use of immune checkpoint inhibition (ICI)—which have resulted in substantial gains in response and survival compared to traditional cytotoxic approaches. However, there is a lack of understanding of the biology underpinning CSCC in immunocompromised patients, in whom the risk of developing CSCC is hundreds of times higher compared to immunocompetent patients. Furthermore, current ICI approaches are associated with significant risk of graft rejection in organ transplant recipients who make up a significant proportion of immunocompromised patients. Ongoing scientific and clinical research efforts are needed in order to maintain momentum to increase our understanding and refine our therapeutic approaches for patients with CSCC.
1. Introduction
CSCC is the second most common skin cancer diagnosed worldwide [1]. Important risk factors for CSCC include ultraviolet (UV) radiation and immunosuppression. Most patients have curable, localized disease, but a small proportion (2–5%) develop unresectable locally advanced or metastatic disease [2,3]. Historically, systemic therapy options for these patients were limited; however, there have been advances in our understanding of the biology of CSCC—particularly, an appreciation of the high tumor mutational burden (TMB) observed in most cases of CSCC, and the role of the immune system in tumor prevention and control. This led to a pivotal study of an immune checkpoint inhibitor (ICI) in patients with metastatic or locally advanced, unresectable CSCC, and changed the treatment paradigm for these patients. Efforts are now underway to assess the benefit of ICI in patients with high-risk localized disease, where the chance for cure with improved neoadjuvant or adjuvant approaches is greater. This review discusses the epidemiology, risk factors, and genomic alterations underlying CSCC, and summarizes treatment advances for CSCC.
2. Epidemiology
Non-melanoma skin cancers (NMSCs) comprise of basal-cell carcinomas (BCCs; ~80%), CSCC (~20%), and rarer skin cancers. The incidence of CSCC is likely underestimated, as accurate figures are difficult to ascertain, with significant variation in cancer registry practices between countries as a result of the high incidence, relatively low mortality, and multiplicity of CSCC [4,5]. However, it is clear that NMSCs are the most common types of cancer diagnosed in many regions—including Australia, North America, and Europe—and their associated public health burden is significantly underestimated [4,5,6,7,8]. The incidence of CSCC in patients aged 75 years or older is 5–10 times higher than in their younger counterparts. Men are at higher risk of CSCC than women, which was traditionally hypothesized to be a reflection of higher occupational exposure, but there is some suggestion that differences in sexual biology may be a factor in the observed disparity [9,10,11,12,13]. As UV exposure is the strongest risk factor for CSCC, most cases arise in the head and neck region, where UV exposure is highest.
3. Risk Factors
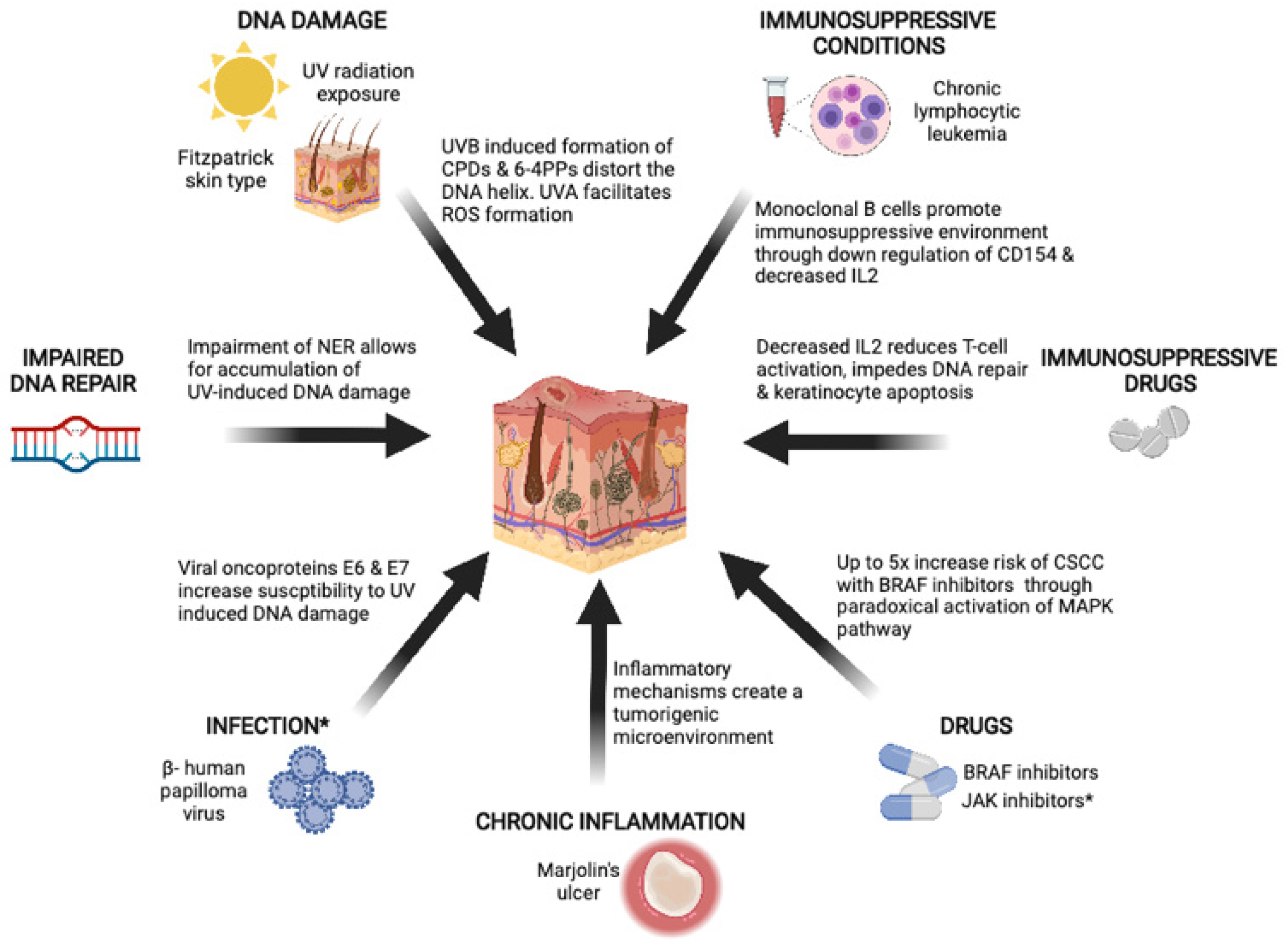
The pathogenesis of CSCC is multifactorial. Chronic UV exposure plays an important role, but other risk factors include immunosuppression, environmental exposures, chronic inflammation, and drugs (Figure 1).
3.1. Ultraviolet Radiation
Chronic UV exposure is the most important risk factor for CSCC [14]. Sunlight produces three main types of UV radiation: UVA, UVB, and UVC. UVA radiation exposure increases the risk of CSCC, but is less mutagenic than UVB. UVA radiation causes indirect DNA damage by facilitating the formation of reactive oxygen species, which can interact with DNA, lipids, and proteins to form pre-mutagenic adducts [15]. UVB radiation directly damages DNA and RNA by causing the formation of cyclobutane-pyrimidine dimers (CPDs) and 6-4 photoproducts (6-4PPs), which distort the DNA helix, impeding transcription and replication [15]. Particular genomic positions, as a result of their structure, are more vulnerable to UVB-induced DNA damage—for instance, the TP53 gene, which is the most frequently mutated gene in CSCC [16]. In vivo studies show that mice exposed to chronic UV radiation develop inactivating TP53 mutations as early as 1 week post-exposure [17]. UVC has the shortest wavelength, and is completely absorbed by the Earth’s ozone layer.
There is marked global variation in CSCC incidence, reflecting not only varying levels of UV exposure, but also genetic propensity to UV damage. The amount of melanin pigment in the skin can be categorized using the Fitzpatrick skin type scale, and is correlated with UV susceptibility and skin cancer risk [18]. Pale or white skin that burns easily and does not tan, classified as Fitzpatrick type 1, has a higher risk of developing skin cancers compared to people with Fitzpatrick type 6 skin, who have very pigmented skin that rarely or never burns [18]. As such, Australia, as a consequence of its location, relative lack of ozone, and high proportion of Anglo-Saxon population, has one of the highest incidences of NMSCs, including CSCC [4,19].
3.2. Immunosuppression
The role of the immune system in the development of CSCC has long been recognized from the significantly increased risk of CSCC observed in immunosuppressed patients [20]. Furthermore, the success of ICI (discussed in detail below) highlights the anticancer potency of an intact immune system.
Immunosuppression can be a result of host factors—such as chronic lymphocytic leukemia (CLL) or HIV—or extrinsic factors, such as drugs. This review will address the most common causes of immunosuppression, such as CLL and immunosuppressive drugs.
3.2.1. Chronic Lymphocytic Leukemia
CLL is a low-grade lymphoproliferative malignancy characterized by clonal proliferation of functionally incompetent B cells; it is the most common leukemia in developed countries, accounting for up to 35% of all leukemias [21]. CLL was the most common cause of immunosuppression (34%, n = 20/59) in a multicenter retrospective study of patients with CSCC receiving immune checkpoint inhibition [22]. Patients with CLL are 5–8 times more likely to develop CSCC compared to patients without CLL [23,24,25]. Furthermore, the risk of recurrence and CSCC-specific mortality is increased in patients with CLL [26,27]. The risk of metastasis at 5 years has been reported to be 18%, with a standardized mortality ratio of 17.0 (95% CI 14.4–19.8) [27].
CLL is typically diagnosed in older people, with a median age of 70 years. Older age, a higher incidence in men, and the associated immunosuppressive effects of CLL all contribute to the higher risk of CSCC. The strongest risk factor for developing CSCC in the setting of CLL is prior history of any skin cancer, but other CLL risk factors associated with a higher risk of developing CSCC include CLL international prognostic index, Rai stage, and lymphocyte doubling time [28].
The biology underlying the increased risk of CSCC is not fully understood. B cells are traditionally known for antigen presentation, antibody production, and the release of effector cytokines that modulate T-cell responses. There is growing evidence, however, that a newly identified, heterogeneous group of B cells—called regulatory B cells—can modulate the immune response to tumors [29]. In the setting of CLL, monoclonal B cells have been shown to promote an immunosuppressive environment via the downregulation of CD154 in activated T cells in preclinical models. CD154 plays a critical role in stimulating B cells, monocytes, and dendritic cells to differentiate and proliferate [30]. Additionally, in clinical tumor samples, higher levels of interleukin-2 (IL-2) receptors have been detected in patients with CLL compared to patients without CLL. IL-2 receptors are thought to be secreted from T-regulatory cells, and bind free IL-2, thus decreasing its availability [31]. Suppression of IL-2 has been shown to induce CD8+ T-cell anergy [32]. Other immune deficits have been identified in patients with CLL, such as impaired phagocytosis and functional defects in helper B cells [33]. It has also been hypothesized that in addition to the immunosuppressive effects of CLL, shared genetic risk factors between CLL and NMSC can contribute to the association between the two diseases [28,34].
3.2.2. Drugs
A number of drugs are associated with an increased risk of CSCC via different mechanisms, ranging from immunosuppression, to the paradoxical activation of pathways that lead to keratinocyte proliferation and loss of apoptosis.
Immunosuppressive Drugs
Long-term immunosuppressive drug regimens are most commonly utilized in organ transplant recipients (OTRs), and involve multiple classes of drugs to minimize graft rejection. As a result, their immunosuppressive effects can be profound, and increase the risk of CSCC by hundreds-fold. In one long-term observational study, approximately 30% of OTRs developed NMSCs, the majority of which were CSCC. The mean time from transplant to first CSCC was 9.9 years, and overall cumulative incidence increased over time to 10.6%, 24.8%, 53.9%, and 73.9% at 5, 10, 20, and 30 years post-transplant, respectively [20]. Patients who have undergone heart or lung transplantations are more susceptible to CSCC formation than renal transplant recipients, likely reflecting the more potent immunosuppressive regimens required for those organs [35,36]. CSCCs developing in OTRs have a higher risk of recurrence, metastases, and cancer-specific death compared to non-transplant patients [37].
Calcineurin inhibitors such as cyclosporine and tacrolimus are a commonly used class of immunosuppressive drugs. They reduce IL-2 production and IL-2 receptor expression, leading to reduced T-cell activation. Cyclosporine, however, also impedes the UV-induced DNA repair mechanisms and keratinocyte apoptosis, by counteracting p53 through ATF3 [38,39]. Furthermore, there is in vitro evidence that cyclosporine can induce epithelial–mesenchymal transition via the upregulation of TGF-β, thus altering the phenotype to a more invasive and aggressive tumor type [40].
Tacrolimus is a more modern calcineurin inhibitor, which has been increasingly used since the 1990s [41]. Interestingly, studies have shown that tacrolimus is not associated with an increased risk of CSCC [42,43], and does not confer resistance to UV-induced apoptosis in keratinocytes—unlike cyclosporine [44].
Since the early 2000s, there has been increasing use of mechanistic target of rapamycin (mTOR) inhibitors—such as rapamycin and sirolimus—as immunosuppressants. mTOR is a protein kinase that plays a role in cell proliferation and survival, as well as modulation of the innate and adaptive immune system [45,46]. Interestingly, however, in vivo studies of mice show that sirolimus—an mTOR inhibitor—significantly delays CSCC development and reduces its multiplicity, even if co-administered with cyclosporine, through inhibition of the transcription factor ATF3 [47,48,49]. ATF3 downregulates expression of TP53, which is one of the most commonly mutated genes in CSCC [38,50]. Furthermore, randomized controlled trials (RCTs) also show a significantly reduced incidence of CSCC in patients receiving mTOR inhibitors compared to cyclosporine [51], as well as in patients who were prescribed sirolimus after three months of treatment with cyclosporine (1.2% vs. 4.3%, p < 0.001) [43,52].
Oral glucocorticoids are a frequently used immunosuppressant, but data regarding the risk associated with the development of CSCC are inconsistent. Two studies—a case–control study, and a planned sub-study of an RCT—found no association between oral steroid use and risk of CSCC [53,54]. In contrast, a cohort study found that patients on prolonged courses of oral glucocorticoids were at higher risk of developing CSCC (standardized incidence ratio 2.45; 95% CI 1.37–4.04) [55]. Another study of OTRs found that higher cumulative immunosuppression from a combination of cyclosporine, azathioprine, and oral prednisolone increased the risk of CSCC by fourfold compared to lower cumulative doses [56]. However, there was no association between the cumulative doses of each drug alone and risk of CSCC. This highlights the possibility that the overall level and duration of immunosuppression, regardless of agent, is a factor impacting the risk of developing CSCC. Ultimately, it will be difficult to ascertain the true risk of CSCC arising in patients taking a commonly used drug such as oral glucocorticoids. There are many indications for oral glucocorticoids, thus increasing potential confounders and biases. Furthermore, accurate information regarding duration of therapy—which can vary widely, from a few days, to many months or years—is difficult to gather at a population-based level.
BRAF Inhibitors
A number of targeted therapies are associated with cutaneous side effects. Squamoproliferative lesions such as actinic keratoses and CSCC are most commonly seen with BRAF inhibitors such as vemurafenib, dabrafenib, and encorafenib, which can be used as monotherapies for patients with metastatic melanoma harboring BRAF V600E mutations. A meta-analysis of seven randomized trials found that 18% (95% CI 0.12–0.26) of patients on vemurafenib develop CSCC [57]. In patients taking dabrafenib, CSCC develops in 6–26% of patients [58,59]. BRAF-inhibitor-associated abnormal squamous proliferation is thought to be induced by the paradoxical activation of the mitogen-activated protein kinase (MAPK) pathway, and subsequent ERK-mediated transcription in wild-type BRAF keratinocytes—particularly in the presence of oncogenic RAS mutations [60,61,62,63]. CSCC arising from BRAF inhibition typically occurs within the first 3 months of treatment, and age has been identified as an independent risk factor [64]. Following the establishment of efficacy of BRAF inhibition in metastatic melanoma, overall survival and response with the combination of BRAF with MEK inhibition was found to be superior compared to BRAF monotherapy. Dual blockade of BRAF and MEK is now the standard of care for patients with metastatic BRAF V600E mutations. Fortunately, the risk of squamoproliferative lesions—including CSCC—significantly decreased with the addition of MEK inhibition, with a reported incidence of 0–2% [65,66,67].
JAK1/2 Inhibitors
Janus kinase (JAK)1/2 inhibitors such as ruxolitinib are used to treat myelofibrosis or polycythemia vera. A number of cases have been reported wherein the initiation of JAK1/2 inhibitors is associated with the development of multiple, rapidly progressing CSCCs [68,69,70]. The incidence of newly diagnosed non-melanoma skin cancer was 17.1% in patients receiving ruxolitinib compared to 2.1% in those receiving best available therapy for myelofibrosis in the long-term follow-up of a phase III RCT [71]. The exact mechanism of tumorigenesis is unknown, but JAK1/2 aberrant hyperactivation has been associated with tumor proliferation and survival in different cancer types [72]. Interestingly, ruxolitinib was shown to reduce tumor progression in in vitro experiments of cyclosporine-induced CSCC cell lines [73].
3.3. Marjolin’s Ulcers
Marjolin’s ulcers describe a rare form of CSCC that arises from areas of chronic inflammation such as burn scars, venous stasis ulcers, and pressure sores [74]. Marjolin’s ulcers are more aggressive than spontaneous CSCC, with the risk of recurrence or metastases reported to be approximately 30% in case series [75,76]. There is a long latency period from initial injury to the development of CSCC, with an average time of 30 years reported [74,77,78]. The relationship between inflammation and tumorigenesis has long been appreciated, with examples of cancers arising from patients’ inflammatory bowel disease and Helicobacter-induced gastritis [79]. Inflammatory mechanisms ensure appropriate responses to infections, and promote wound healing, but can also create a microenvironment that promotes tumorigenesis via the recruitment of immune cells and subsequent release of cytokines and growth factors [80].
3.4. Environmental Exposure
Other environmental risk factors include chronic arsenic exposure, which most commonly occurs from contaminated drinking water [81]. Arsenic-induced CSCC can develop even in non-sun-exposed sites. Ionizing radiation via environmental, therapeutic, or diagnostic exposure is also a known risk factor, although the risk of BCC is higher than that of CSCC, as the basal layer of the epidermis is more affected than more superficial layers [82,83,84]. Occupational exposure to aromatic hydrocarbons such as benzene and mineral oil have also been identified as risk factors for the development of CSCC, and are of particular importance in occupations such as firefighting and petroleum work [85,86].
3.5. Inherited Bone Marrow Failure Sydromes (IBMFSs)
IBMFSs comprise of rare diseases typically characterized by genetic mutations resulting in bone marrow failure. These syndromes include Fanconi anemia and dyskeratosis congenita as the most common disorders, which are associated with defects in DNA repair and telomere function, respectively. Patients with these conditions are at increased risk of hematological and solid malignancies due to multiple factors that arise from the genetic disruption, resulting in genomic instability and bone marrow failure. The risk of CSCC is more notable in patients with Fanconi anemia and dyskeratosis congenita. Skin cancers make up approximately 10–20% of cancer cases in patients with Fanconi anemia and dyskeratosis congenita, and typically occur at a median age of approximately 30 years [87,88,89].
3.6. Beta Human Papillomavirus
HPV comprises several heterogeneous subgroups; α-papillomavirus (α-HPV) subtypes are associated with mucosal SCCs, such as cervical and oropharyngeal cancer, but it is the β-papillomavirus (β-HPV) subtypes that are hypothesized to be a risk factor for CSCCs. β-HPV was first discovered in the context of patients with a rare skin disorder—epidermodysplasia verruciformis. Patients develop pre-cancerous wart-like lesions that progress to CSCC in UV-exposed areas. Multiple β-HPV types were found in these lesions, thus raising the possibility of the carcinogenic role of β-HPV. Complicating matters, however, is the relative ubiquitousness of β-HPV in the skin. β-HPV DNA is detected in the skin of 39–91% of immunocompetent patients—particularly in hair follicles, which are considered to be a natural reservoir [90,91]. There are, however, multiple factors that suggest that β-HPV may be a risk factor for the development of CSCC.
Firstly, immunocompromised patients have an increased risk of CSCC, and have significantly higher rates of β-HPV infection and higher viral loads, suggesting a potential causal relationship between β-HPV and CSCC [92]. Second, observational studies have shown an association between β-HPV DNA and/or serum antibodies and CSCC in both immunocompromised and immunocompetent patients [93]. A meta-analysis of over 3000 immunocompetent patients found an overall association of β-HPV and CSCC (OR 1.42; 95% CI 1.18–1.72) [94]. Notably, some of these studies incorporated BCC cases, and no associations were observed between β-HPV and BCC [95,96,97]. Thirdly, there are increasing preclinical data supporting the role of β-HPV in tumor initiation, but not necessarily in tumor maintenance [98]. β-HPV DNA is detected at high levels in pre-cancerous lesions such as actinic keratoses, whereas lower levels are detected in CSCC lesions [99,100]. In vitro and in vivo studies have shown that the HPV oncoproteins E6 and/or E7 from HPV types 5, 8, and 38 can increase susceptibility to UV-induced oncogenesis via alterations in p53 and Notch1 signaling [101,102,103,104,105,106,107,108]. β-HPV is also thought to infect and expand adult tissue stem cells, thus enabling cells to persist and accumulate mutations [109]. It is hypothesized that once cells have accumulated mutations such as TP53 and Notch, which allow for ongoing cell proliferation, expression of viral oncogenes becomes redundant, and they are no longer positively selected.
4. Biology and Pathogenesis
There have been significant advances in our understanding of the biological pathways in CSCC development, with multiple genes identified as playing a critical role in tumor initiation and persistence. CSCC, however, has one of the highest median TMBs of any tumor type; thus, hundreds of mutations can be found per megabase [110]. One of the challenges in understanding the biological pathways involved in CSCC is separating true oncogenic mutations from passenger mutations. Here, we discuss the oncogenic roles of selected commonly mutated genes such as TP53, Notch, and CDKN2A. No specific oncogenic drivers of CSCC have been identified.
p53 functions predominantly as a transcription factor, and can activate or repress a large number of target genes. In particular, p53 plays an important role in modulating nucleotide excision repair (NER) and other DNA repair pathways that are essential in the repair of UV-induced DNA damage [111]. Mutations in TP53 allow for ongoing, unrepaired UV-induced DNA damage. As an example, the risk of CSCCs is significantly higher if there are genetically impaired DNA repair mechanisms, such as in patients with xeroderma pigmentosum who develop NMSCs during childhood [112].
Mutations in TP53 occur early in CSCC development, and are often found in normal keratinocytes [113,114,115] and pre-malignant lesions [116]. Whole-exome sequencing of CSCC has identified bi-allelic TP53 mutations in nearly all tumors, again suggesting that the loss of wild-type TP53 is an early step in carcinogenesis [117,118,119]. This is in contrast to other solid malignancies, where TP53 gene mutations occur later in tumor evolution [120,121,122]. Further evidence of the oncogenic role of TP53 in CSCC has been shown in in vivo studies, where homozygous p53-knockout mice rapidly developed CSCC after UV exposure [123,124]. p53-mutant cells are more resistant to UV-induced apoptosis, and have a proliferative advantage over wild-type keratinocytes [125].
The Notch signaling pathway is commonly affected, and Notch mutations are found in 60–80% of CSCCs [118,119,126]. Notch is a highly conserved intercellular signaling mechanism that plays a critical role in the development and maintenance of tissue homeostasis [127]. Genes of the Notch family encode four transmembrane receptors (Notch1–4). In the epidermis, Notch signaling is involved in the terminal differentiation of keratinocytes [128]. Interestingly, Notch can have oncogenic or tumor-suppressive functions depending on the cell context [129]. Constitutive Notch1 signaling as a result of activating mutations is the initiating step in almost all T-cell acute lymphoblastic leukemia (T-ALL) cases [130,131]. In contrast, loss of Notch signaling—particularly Notch1 and Notch2—is associated with carcinogenesis in keratinocytes [118,132,133]. Preclinical studies have shown that Notch1 is a downstream positive target of p53 in keratinocytes; thus, inactivating TP53 mutations can further lead to reduced Notch1 expression [134]. Several in vivo experiments have shown Notch1 deficiency or Notch1 inhibition in mice can result in the spontaneous development of CSCC [135]. A possible mechanism is via upregulation of the Wnt/β-catenin pathway [132]. Intriguingly, Notch deficiency or loss does not purely exert its effect autonomously on cells, but can also create a pro-tumorigenic microenvironment. Loss of Notch signaling disrupts skin barrier function, creating a chronic wound-like environment [136]. As a result, mesenchymal components are recruited for repair, which also stimulates a vascularized and growth-factor-rich stroma, providing an ideal environment for tumor formation [136]. There is also clinical evidence of Notch inactivation resulting in increased CSCC risk. Semagacestat, a γ-secretase inhibitor, was developed as a drug for Alzheimer’s disease. A phase III RCT of semagacestat was halted early due to lack of efficacy as well as an increased risk of CSCC. γ-Secretase, in addition to converting amyloid precursor protein to amyloid-β is also responsible for cleaving and activating Notch1; thus, its inhibition indirectly inactivates Notch1 [137].
The cyclin-dependent kinase inhibitor 2A (CDKN2A) gene encodes two tumor-suppressor genes: p16INK4a and p14ARF. Both genes regulate cell cycling: p16INK4A binds to CDK4 and CDK6, thus preventing Rb protein phosphorylation and G1-S phase progression, while p14ARF binds to MDM2, preventing p53 degradation and Rb inactivation, causing cell arrest. Methylation of the promoter region is the most common mechanism of p16 and p14 inactivation in CSCC, followed by point mutations and loss of heterozygosity [138]. Alterations in CDKN2A are found in up to 80% of CSCCs [119]. Inactivating mutations of CDKN2A result in uncontrolled cell cycling and proliferation. A recent analysis, however, consistently found upregulation of CDKN2A in gene expression profiles and cell lines, in contrast to the pre-existing literature; the authors hypothesized that ERK signaling in CSCC may upregulate CDKN2A as a stress response to induce senescence rather than stimulating cell cycling [139].
RAS gene mutations are among the most common activating mutations found in human cancers, and also present a significant therapeutic challenge due to their molecular characteristics. The RAS gene encodes four RAS proteins: HRAS, NRAS, and two splice variants of KRAS. RAS proteins belong to a family of small GTPases that cycle between “off” and “on” states [140]. Activating RAS mutations can result in oncogenic constitutive activation of the RAF-MEK-ERK and PI3K-AKT pathways, leading to cell proliferation [141]. In CSCC, HRAS mutations are most common, and are found in 3–20% of CSCCs [118,133]. In keratinocytes, upregulated expression of RAS alone is not sufficient to induce tumorigenesis [142]. Concomitant Notch1 deletion, IκBα co-expression, or CDK4-mediated bypass of Rb cell cycle restraints increase CSCC formation in the presence of activated RAS [132,143,144].
5. Tumor Mutation Burden
As a result of the chronic nature of UV exposure and the mechanism of DNA damage, there are cumulative DNA aberrations in CSCCs. In a study examining TMB in over 100,000 tumor samples, CSCC had the highest median TMB (45.2 mutations/Mb) compared to other tumor types [110]. High TMB is predictive of response to ICI, although prospective validation is lacking [145]. The impressive and durable responses observed with ICI in CSCC are thought to be due to high TMB representing a large number of immunostimulatory neoantigens.
6. Tumor Mutational Signatures
Somatic mutations in cancer cells can create a characteristic mutational signature, which reflects the mutational process involved in carcinogenesis. The UV mutation signature—the first characterized signature—is found in the great majority of CSCCs, and even in immunocompromised hosts. UV radiation damage most commonly results in cytosine to thymine or cytosine–cytosine to thymine–thymine changes—i.e., C > T or CC > TT—at dipyrimidine sites [146,147]. Recent studies have also identified signatures in CSCC associated with azathioprine exposure [139] and hyperactivity of endogenous cytidine deaminases (APOBEC)—specifically in patients with epidermolysis bullosa [148].
7. Treatment Advances
7.1. Localized Resectable High-Risk Disease
Most CSCCs are small, indolent, and surgically resectable, and adjuvant therapy is often not required. Post-operative radiotherapy, however, is considered for patients with resected high-risk localized disease—usually defined as tumors showing involved resection margins, depth of invasion of more than 2–6 mm, extensive perineural invasion, or large nerve involvement [2,149]. Other indications include lymph node involvement and large primary tumors [150,151].
7.1.1. Post-Operative Chemoradiotherapy
Platinum chemotherapy agents such as cisplatin and carboplatin are often used concurrently with postoperative radiotherapy in patients with mucosal head and neck squamous cell carcinoma (HNSCC), as several studies have shown a survival benefit [152,153,154,155]. The results of these trials have been extrapolated and applied to patients with cutaneous SCC. Until recently, there was no definitive prospective study supporting its use in this population.
A phase III trial randomized patients with high-risk resected CSCC to postoperative radiotherapy alone, or with concurrent weekly carboplatin chemotherapy [156]. Concurrent cisplatin is considered the gold standard in HNSCC, but its significant toxicity profile often precludes its use in patients with CSCC who are generally older, and with significant comorbidities. Thus, carboplatin is more frequently used. High-risk disease was defined as patients with primary tumors > 5 cm or T4 disease, resected intra-parotid nodal disease, two or more cervical nodal diseases, or with a node ≥ 3 cm or extranodal extension. Contrary to the results of mucosal HNSCC, no benefit was observed in freedom from locoregional relapse, nor in disease-free or overall survival, in patients with CSCC receiving concurrent chemotherapy. Based on the results of this trial, and the lack of evidence with other regimens, concurrent chemotherapy is generally not recommended in the adjuvant treatment of CSCC outside of clinical trials [151].
7.1.2. Neo/Adjuvant Immunotherapy
The success of ICI in patients with advanced disease has driven efforts to incorporate treatment into earlier stages of disease in order to reduce (a) the morbidity associated with resections of large tumors, and (b) the risk of locoregional relapse or metastasis.
Neoadjuvant immunotherapy is particularly appealing for clinical and translational purposes. Immune activation may be potentiated by the presence of neoantigens and intra-tumoral immune cells within the unresected cancer, and changes in the tumor and stroma can be compared between pretreatment biopsies and the resection specimen [157,158,159]. Furthermore, neoadjuvant studies allow for earlier assessment, using pathologic response, compared to adjuvant studies, where survival data can take many years to mature. A pilot phase II study of two doses of neoadjuvant cemiplimab for patients with locally advanced, curable CSCC resulted in 14/20 patients (70%; 95% CI 45.7–88.1) with a pathological complete response (n = 11) or major pathological response (n = 3) [160]; this was despite only 30% (95% CI 11.9–54.3) showing a partial response by RECIST, highlighting the challenges of assessing ICI response using current radiological criteria. Neoadjuvant studies of other ICIs, as well as combination neoadjuvant treatment with dual anti-PD(L)1 with anti-cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) blockade, are ongoing [NCT04154943] [161,162]. Furthermore, there are two large phase III adjuvant studies of pembrolizumab or cemiplimab [163,164].
7.2. Unresectable Locally Advanced or Metastatic Disease
Historically, no standard of care for systemic therapies existed for patients with unresectable or metastatic CSCC. Cytotoxic chemotherapies such as platinums, fluoropyrimidines, and taxanes have shown activity in retrospective analyses. Response rates are generally low, and the toxicity profiles of therapies often preclude their use in elderly patients with CSCC [165]. ICI with monoclonal antibodies against PD1 and PD-L1 has transformed the treatment landscape for many solid tumors, including CSCC. Other treatment approaches include targeting the epidermal growth factor receptor (EGFR) pathway.
7.2.1. Immunotherapy
A practice-changing phase II study demonstrated the efficacy of cemiplimab—an anti-PD-1 monoclonal antibody—in patients with unresectable or metastatic CSCC [166,167]. Responses were observed in 54.4% (95% CI 47.1–61.6) of patients (both previously treated and untreated) [167]. In patients with initial response, 76% (95% CI 64.1–84.4%) had ongoing response at 24 months, demonstrating the excellent durability of disease response; estimated overall survival at 24 months was 73.3% (95% CI 66.1–79.2) [167,168,169]. As a result of this study, cemiplimab was approved by the FDA, and became the standard treatment for patients with locally advanced or metastatic CSCC who are not candidates for curative surgery or radiation.
Other ICIs, such as pembrolizumab, have shown comparable activity. Two studies—CARSKIN, and KEYNOTE-629—assessed the efficacy of pembrolizumab in advanced CSCC. The objective response rate was 34.3% (95% CI 25.3–44.2%) in KEYNOTE-629 in a heavily pretreated population, and median overall survival has not been ascertained [170]. Based on KEYNOTE-629, pembrolizumab has also been approved by the FDA for advanced CSCC.
The CARKSIN study enrolled treatment-naïve patients with unresectable or metastatic CSCC to receive pembrolizumab [171]. Response rate (RR) at 15 weeks was the primary objective of the study, and was 41% (95% CI 26–58%), including 13 partial and 3 complete responses. Similarly, nivolumab has shown robust results in a phase II first-line study of patients with advanced CSCC [172]. Recently, real-world data regarding the use of ICIs in 245 patients—including immunocompromised patients—were reported to be comparable to trial data [22]. The estimated 12-month OS was 63% (95% CI 51–70); 50% of patients achieved a complete response or partial response (95% CI 44–57), and there were no unexpected toxicities. In univariate and multivariate analysis, ECOG score > 2 was the only clinical factor that was significantly associated with poor OS and PS in the first 6 months.
More aggressive approaches are also being considered for select patients with unresectable localized disease where cure may be possible. A phase II study of neoadjuvant avelumab, followed by curative-dose radiotherapy with concurrent avelumab, is ongoing [173].
Combination strategies of ICIs with cetuximab and oncolytic viruses are being investigated in order to address the challenges of resistance and improve durability of response. CSCC and melanoma share similar features, such as chronic UV exposure and high TMB. Studies of immunotherapy have been established longer in melanoma than in CSCC; thus, approaches that are efficacious or promising in melanoma are being tested in patients with CSCC. Talimogene laherparepvec (T-VEC) is a modified attenuated oncolytic herpes simplex virus containing the granulocyte macrophage colony-stimulating factor (GM-CSF) gene. Production of intratumoral GM-CSF can induce cellular immunity, and the direct oncolytic effect from viral infection of tumor cells can cause an antitumor response. Early-phase studies in metastatic melanoma show that intralesional injections of T-VEC combined with immune checkpoint blockade resulted in an objective response rate of 39% and 50% with concurrent ipilimumab and pembrolizumab, respectively [174,175,176]. Currently, there are studies in CSCC combining oncolytic viruses such as T-VEC and RP1 with ICI or EGFR antibodies [177,178]. EGFR antibodies, which are discussed in more detail below, are also being investigated in combination with anti-PD(L)1 antibodies. A phase II trial will randomize immunocompetent patients with unresectable/metastatic CSCC to avelumab alone, or in combination with cetuximab [179].
7.2.2. EGFR Pathway Inhibition
EGFR is a transmembrane glycoprotein with an extracellular binding domain, along with an intracellular tyrosine kinase domain that regulates cell proliferation via pathways such as MAPK and PI3K. The EGFR protein is highly expressed in CSCC [180,181,182]. EGFR monoclonal antibodies such as cetuximab and panitumumab have shown activity in CSCC in small phase II trials. In a phase II trial of cetuximab in patients with locally advanced unresectable or metastatic CSCC, 28% achieved a response, while 41% had stable disease [183]. An ORR of 31% was observed in a study of panitumumab [184].
Cetuximab has also been used in the neoadjuvant setting. Five out of nine patients receiving neoadjuvant cetuximab alone had a response that allowed for surgical resection and, of these, three had a complete pathological response [185].
Oral tyrosine inhibitors such as gefitinib and dacomitinib, which target the intracellular tyrosine kinase domain, are typically used in patients with EGFR-driven non-small-cell lung cancer, where responses are seen in approximately 75% of patients. These agents have activity in CSCC, with overall response rates of 16% and 28% observed in early phase trials of gefitinib and dacomitinib, respectively [186,187,188].
Combination therapies with drugs known to target common EGFR resistance mechanisms such as fibroblast growth factor receptor (FGFR) signaling are also being investigated. A phase I study of cetuximab with lenvatinib—a multitarget tyrosine kinase inhibitor that has activity against FGFR—in patients with metastatic CSCC or HNSCC is underway [189].
7.2.3. Other Approaches
The risk of BRAF-induced CSCC is abrogated with the addition of MEK inhibition, forming the rationale for investigating the potential role of MEK inhibition in the treatment of CSCC. In vivo studies have shown that MEK induces CSCC cell senescence, but not apoptosis. Interestingly, MEK inhibition also significantly delayed or prevented CSCC development in murine models [190]. Currently, there is a phase II study investigating the efficacy of cobimetinib—an MEK inhibitor—with atezolizumab [191].
Future therapeutic approaches may include novel small molecule inhibitors of both PI3K and mTOR. The oral dual PI3K/mTOR inhibitors—GDC-0084 and LY3023414—have been shown to inhibit proliferation and promote apoptosis in CSCC cell lines [192,193]. GDC-0084 and LY3023414 have been shown to be safe and tolerable in early-phase studies in patients with solid tumors, and there were promising signals of activity [194,195].
8. Therapeutic Options for Immunocompromised Patients
Immunocompromised patients have historically been excluded from clinical trials, but with the success of ICI for immunocompetent patients with CSCC, it became apparent that high-level data to guide treatment for immunocompromised patients were lacking. There are several ongoing studies investigating approaches in different groups of immunocompromised patients. A major concern with the use of ICIs in solid organ transplant patents is graft rejection. Case reports and case series have reported up to a 40% risk of graft rejection with the use of anti-PD(L)1 antibodies [196,197]. To potentially ameliorate that risk, two studies are investigating the combination of tacrolimus—an immunosuppressant—with ipilimumab plus nivolumab, and sirolimus with cemiplimab [198]. Tacrolimus and sirolimus—both mTOR inhibitors—may reduce the risk of CSCC development, as discussed earlier in the review. Cemiplimab is also being investigated in patients with CLL, HIV, or allogenic hematopoietic stem cell transplants [199,200].
Recently, real-world data of patients with CSCC receiving cemiplimab included a cohort of immunocompromised patients. Somewhat surprisingly, given the poor prognosis of immunocompromised patients with CSCC compared to immunocompetent patients, ORR and OS did not differ between immunocompetent and immunocompromised patients. Several patients experienced graft rejection as expected. The causes of immunosuppression in this cohort were heterogeneous, ranging from CLL to OTRs and patients with HIV.
Given the increased risk of developing CSCC and its increased lethality in immunocompromised patients, there is an urgent need to better understand the underlying biology driving this disparity, and to identify potential novel treatment approaches for this cohort.
9. Conclusions
Our understanding of the underlying biology of CSCC—such as the mechanisms and sequelae of UV-induced DNA damage—has resulted in significant advances in the management of patients with CSCC. ICI is established as the first-line management of advanced CSCC, but focus has now shifted to more challenging questions. Can we reduce the risk of patients with localized disease developing recurrent or metastatic disease? How can we improve current treatment paradigms, particularly in immunocompromised patients, where the risk of treatment-related adverse events—particularly graft rejection in OTRs—is high? Finally, what do we do for patients who do not respond to—or have progressed despite—immunotherapy? Current and future scientific research efforts towards identifying predictive biomarkers and understanding the biology behind clinically disparate groups will hopefully address these clinical challenges.
Author Contributions
All authors were involved in the conceptualization, writing, and review of the published manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
A.A.T. has no conflict of interest to declare. A.M.L. has served on uncompensated advisory boards for Merck Sharp & Dohme and Bristol-Myers Squibb, with travel and accommodation expenses; and uncompensated consultancy for Eisai. B.S. has served on advisory boards for AstraZeneca, Roche-Genentech, Sanofi-Regeneron, Pfizer, Novartis, Merck, Bristol Myers Squibb, Lilly, and Amgen, and received institutional research funding from Sanofi/Regeneron. D.R. has received institutional research grants and funding from Regeneron Pharmaceuticals, Inc., Roche, Merck Sharp & Dohme, Bristol-Myers Squibb, GlaxoSmithKline, Sanofi, Replimune, and Kura Oncology, and has served on uncompensated scientific committees and advisory boards for Merck Sharp & Dohme, Regeneron Pharmaceuticals, Inc., Sanofi, GlaxoSmithKline, and Bristol-Myers Squibb.
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Brantsch, K.D.; Meisner, C.; Schönfisch, B.; Trilling, B.; Wehner-Caroli, J.; Röcken, M.; Breuninger, H. Analysis of risk factors determining prognosis of cutaneous squamous cell carcinoma: A prospective study. Lancet Oncol. 2008, 9, 713–720. [Google Scholar] [CrossRef]
- Brougham, N.D.; Dennett, E.R.; Cameron, R.; Tan, S.T. The incidence of metastasis from cutaneous squamous cell carcinoma and the impact of its risk factors. J. Surg. Oncol. 2012, 106, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Lomas, A.; Leonardi-Bee, J.; Bath-Hextall, F. A systematic review of worldwide incidence of nonmelanoma skin cancer. Br. J. Dermatol. 2012, 166, 1069–1080. [Google Scholar] [CrossRef]
- Pandeya, N.; Olsen, C.M.; Whiteman, D.C. The incidence and multiplicity rates of keratinocyte cancers in Australia. Med. J. Aust. 2017, 207, 339–343. [Google Scholar] [CrossRef]
- Goon, P.K.C.; Greenberg, D.C.; Igali, L.; Levell, N.J. Predicted cases of U.K. skin squamous cell carcinoma and basal cell carcinoma in 2020 and 2025: Horizon planning for National Health Service dermatology and dermatopathology. Br. J. Dermatol. 2017, 176, 1351–1353. [Google Scholar] [CrossRef] [PubMed]
- Karia, P.S.; Han, J.; Schmults, C.D. Cutaneous squamous cell carcinoma: Estimated incidence of disease, nodal metastasis, and deaths from disease in the United States, 2012. J. Am. Acad. Dermatol. 2013, 68, 957–966. [Google Scholar] [CrossRef] [PubMed]
- Ronconi, G.; Piccinni, C.; Dondi, L.; Calabria, S.; Pedrini, A.; Esposito, I.; Ascierto, P.A.; Naldi, L.; Martini, N. Identification of cases and estimate of direct costs of unresectable and advanced cutaneous squamous cell carcinoma: Real-world data from a large Italian database. Br. J. Dermatol. 2020, 183, 172–174. [Google Scholar] [CrossRef]
- Foote, J.A.; Harris, R.B.; Giuliano, A.R.; Roe, D.J.; Moon, T.E.; Cartmel, B.; Alberts, D.S. Predictors for cutaneous basal-and squamous cell carcinoma among actinically damaged adults. Int. J. Cancer 2001, 95, 7–11. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, B.K.; Kricker, A. The epidemiology of UV induced skin cancer. J. Photochem. Photobiol. B Biol. 2001, 63, 8–18. [Google Scholar] [CrossRef]
- Hall, H.I.; May, D.S.; Lew, R.A.; Koh, H.K.; Nadel, M. Sun protection behaviors of the US white population. Prev. Med. 1997, 26, 401–407. [Google Scholar] [CrossRef]
- McCarthy, E.M.; Ethridge, K.P.; Wagner Jr, R. Beach holiday sunburn: The sunscreen paradox and gender differences. Cutis 1999, 64, 37–42. [Google Scholar]
- Thomas-Ahner, J.M.; Wulff, B.C.; Tober, K.L.; Kusewitt, D.F.; Riggenbach, J.A.; Oberyszyn, T.M. Gender Differences in UVB-Induced Skin Carcinogenesis, Inflammation, and DNA Damage. Cancer Res. 2007, 67, 3468–3474. [Google Scholar] [CrossRef] [Green Version]
- Rosso, S.; Zanetti, R.; Martinez, C.; Tormo, M.J.; Schraub, S.; Sancho-Garnier, H.; Franceschi, S.; Gafà, L.; Perea, E.; Navarro, C.; et al. The multicentre south European study ‘Helios’. II: Different sun exposure patterns in the aetiology of basal cell and squamous cell carcinomas of the skin. Br. J. Cancer 1996, 73, 1447–1454. [Google Scholar] [CrossRef] [Green Version]
- Sinha, R.P.; Häder, D.P. UV-induced DNA damage and repair: A review. Photochem. Photobiol. Sci. 2002, 1, 225–236. [Google Scholar] [CrossRef]
- Benjamin, C.L.; Ananthaswamy, H.N. p53 and the pathogenesis of skin cancer. Toxicol. Appl. Pharm. 2007, 224, 241–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benavides, F.; Oberyszyn, T.M.; VanBuskirk, A.M.; Reeve, V.E.; Kusewitt, D.F. The hairless mouse in skin research. J. Dermatol. Sci. 2009, 53, 10–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzpatrick, T.B. The validity and practicality of sun-reactive skin types I through VI. Arch. Dermatol. 1988, 124, 869–871. [Google Scholar] [CrossRef] [PubMed]
- Staples, M.P.; Elwood, M.; Burton, R.C.; Williams, J.L.; Marks, R.; Giles, G.G. Non-melanoma skin cancer in Australia: The 2002 national survey and trends since 1985. Med. J. Aust. 2006, 184, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Harwood, C.A.; Mesher, D.; McGregor, J.M.; Mitchell, L.; Leedham-Green, M.; Raftery, M.; Cerio, R.; Leigh, I.M.; Sasieni, P.; Proby, C.M. A Surveillance Model for Skin Cancer in Organ Transplant Recipients: A 22-Year Prospective Study in an Ethnically Diverse Population. Am. J. Transplant. 2013, 13, 119–129. [Google Scholar] [CrossRef]
- Chiorazzi, N.; Rai, K.R.; Ferrarini, M. Chronic lymphocytic leukemia. N. Engl. J. Med. 2005, 352, 804–815. [Google Scholar] [CrossRef] [Green Version]
- Hober, C.; Fredeau, L.; Pham-Ledard, A.; Boubaya, M.; Herms, F.; Celerier, P.; Aubin, F.; Beneton, N.; Dinulescu, M.; Jannic, A.; et al. Cemiplimab for Locally Advanced and Metastatic Cutaneous Squamous Cell Carcinomas: Real-Life Experience from the French CAREPI Study Group. Cancers 2021, 13, 3547. [Google Scholar] [CrossRef]
- Brewer, J.D.; Shanafelt, T.D.; Khezri, F.; Seda, I.M.S.; Zubair, A.S.; Baum, C.L.; Arpey, C.J.; Cerhan, J.R.; Call, T.G.; Roenigk, R.K. Increased incidence and recurrence rates of nonmelanoma skin cancer in patients with non-Hodgkin lymphoma: A Rochester Epidemiology Project population-based study in Minnesota. J. Am. Acad. Dermatol. 2015, 72, 302–309. [Google Scholar] [CrossRef] [Green Version]
- Levi, F.; Randimbison, L.; Te, V.C.; La Vecchia, C. Non-Hodgkin’s lymphomas, chronic lymphocytic leukaemias and skin cancers. Br. J. Cancer 1996, 74, 1847–1850. [Google Scholar] [CrossRef] [Green Version]
- Que, S.K.T.; Zwald, F.O.; Schmults, C.D. Cutaneous squamous cell carcinoma: Incidence, risk factors, diagnosis, and staging. J. Am. Acad. Dermatol. 2018, 78, 237–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehrany, K.; Weenig, R.H.; Lee, K.K.; Pittelkow, M.R.; Otley, C.C. Increased metastasis and mortality from cutaneous squamous cell carcinoma in patients with chronic lymphocytic leukemia. J. Am. Acad. Dermatol. 2005, 53, 1067–1071. [Google Scholar] [CrossRef] [PubMed]
- Royle, J.A.; Baade, P.D.; Joske, D.; Girschik, J.; Fritschi, L. Second cancer incidence and cancer mortality among chronic lymphocytic leukaemia patients: A population-based study. Br. J. Cancer 2011, 105, 1076–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinstern, G.; Rishi, A.; Achenbach, S.J.; Rabe, K.G.; Kay, N.E.; Shanafelt, T.D.; Ding, W.; Leis, J.F.; Norman, A.D.; Call, T.G.; et al. Delineation of clinical and biological factors associated with cutaneous squamous cell carcinoma among patients with chronic lymphocytic leukemia. J. Am. Acad. Dermatol. 2020, 83, 1581–1589. [Google Scholar] [CrossRef]
- Sarvaria, A.; Madrigal, J.A.; Saudemont, A. B cell regulation in cancer and anti-tumor immunity. Cell. Mol. Immunol. 2017, 14, 662–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantwell, M.; Hua, T.; Pappas, J.; Kipps, T.J. Acquired CD40-ligand deficiency in chronic lymphocytic leukemia. Nat. Med. 1997, 3, 984–989. [Google Scholar] [CrossRef]
- Lindqvist, C.A.; Christiansson, L.H.; Simonsson, B.; Enblad, G.; Olsson-Strömberg, U.; Loskog, A.S.I. T regulatory cells control T-cell proliferation partly by the release of soluble CD25 in patients with B-cell malignancies. Immunology 2010, 131, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Chikuma, S.; Terawaki, S.; Hayashi, T.; Nabeshima, R.; Yoshida, T.; Shibayama, S.; Okazaki, T.; Honjo, T. PD-1-mediated suppression of IL-2 production induces CD8+ T cell anergy in vivo. J. Immunol. 2009, 182, 6682–6689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riches, J.C.; Ramsay, A.G.; Gribben, J.G. Immune reconstitution in chronic lymphocytic leukemia. Curr. Hematol. Malig. Rep. 2012, 7, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Besson, C.; Moore, A.; Wu, W.; Camp, N.J.; Vajdic, C.; Morton, L.M.; Smedby, K.E.; de Sanjose, S.; Shanafelt, T.D.; Brewer, J.; et al. Common Genetic Polymorphisms Contribute to the Association between Non-Melanoma Skin Cancer and Chronic Lymphocytic Leukemia. Blood 2017, 130, 1454. [Google Scholar] [CrossRef]
- Veness, M.J.; Quinn, D.I.; Ong, C.S.; Keogh, A.M.; Macdonald, P.S.; Cooper, S.G.; Morgan, G.W. Aggressive cutaneous malignancies following cardiothoracic transplantation: The Australian experience. Cancer Interdiscip. Int. J. Am. Cancer Soc. 1999, 85, 1758–1764. [Google Scholar] [CrossRef]
- Jensen, P.; Hansen, S.; Møller, B.; Leivestad, T.; Pfeffer, P.; Geiran, O.; Fauchald, P.; Simonsen, S. Skin cancer in kidney and heart transplant recipients and different long-term immunosuppressive therapy regimens. J. Am. Acad. Dermatol. 1999, 40, 177–186. [Google Scholar] [CrossRef]
- Garrett, G.L.; Lowenstein, S.E.; Singer, J.P.; He, S.Y.; Arron, S.T. Trends of skin cancer mortality after transplantation in the United States: 1987 to 2013. J. Am. Acad. Dermatol. 2016, 75, 106–112. [Google Scholar] [CrossRef]
- Wu, X.; Nguyen, B.-C.; Dziunycz, P.; Chang, S.; Brooks, Y.; Lefort, K.; Hofbauer, G.F.L.; Dotto, G.P. Opposing roles for calcineurin and ATF3 in squamous skin cancer. Nature 2010, 465, 368–372. [Google Scholar] [CrossRef] [Green Version]
- Yarosh, D.B.; Pena, A.V.; Nay, S.L.; Canning, M.T.; Brown, D.A. Calcineurin Inhibitors Decrease DNA Repair and Apoptosis in Human Keratinocytes Following Ultraviolet B Irradiation. J. Investig. Dermatol. 2005, 125, 1020–1025. [Google Scholar] [CrossRef] [Green Version]
- Walsh, S.B.; Xu, J.; Xu, H.; Kurundkar, A.R.; Maheshwari, A.; Grizzle, W.E.; Timares, L.; Huang, C.C.; Kopelovich, L.; Elmets, C.A.; et al. Cyclosporine a mediates pathogenesis of aggressive cutaneous squamous cell carcinoma by augmenting epithelial-mesenchymal transition: Role of TGFβ signaling pathway. Mol. Carcinog. 2011, 50, 516–527. [Google Scholar] [CrossRef] [Green Version]
- Hong, J.C.; Kahan, B.D. Immunosuppressive agents in organ transplantation: Past, present, and future. Semin. Nephrol. 2000, 20, 108–125. [Google Scholar] [PubMed]
- Coghill, A.E.; Johnson, L.G.; Berg, D.; Resler, A.J.; Leca, N.; Madeleine, M.M. Immunosuppressive Medications and Squamous Cell Skin Carcinoma: Nested Case-Control Study Within the Skin Cancer after Organ Transplant (SCOT) Cohort. Am. J. Transplant. 2016, 16, 565–573. [Google Scholar] [CrossRef]
- Salgo, R.; Gossmann, J.; Schöfer, H.; Kachel, H.; Kuck, J.; Geiger, H.; Kaufmann, R.; Scheuermann, E. Switch to a sirolimus-based immunosuppression in long-term renal transplant recipients: Reduced rate of (pre-) malignancies and nonmelanoma skin cancer in a prospective, randomized, assessor-blinded, controlled clinical trial. Am. J. Transplant. 2010, 10, 1385–1393. [Google Scholar] [CrossRef] [PubMed]
- Norman, K.G.; Canter, J.A.; Shi, M.; Milne, G.L.; Morrow, J.D.; Sligh, J.E. Cyclosporine A suppresses keratinocyte cell death through MPTP inhibition in a model for skin cancer in organ transplant recipients. Mitochondrion 2010, 10, 94–101. [Google Scholar] [CrossRef]
- Thomson, A.W.; Turnquist, H.R.; Raimondi, G. Immunoregulatory functions of mTOR inhibition. Nat. Rev. Immunol. 2009, 9, 324–337. [Google Scholar] [CrossRef] [Green Version]
- Hackstein, H.; Taner, T.; Zahorchak, A.F.; Morelli, A.E.; Logar, A.J.; Gessner, A.; Thomson, A.W. Rapamycin inhibits IL-4—Induced dendritic cell maturation in vitro and dendritic cell mobilization and function in vivo. Blood 2003, 101, 4457–4463. [Google Scholar] [CrossRef]
- de Gruijl, F.R.; Koehl, G.E.; Voskamp, P.; Strik, A.; Rebel, H.G.; Gaumann, A.; de Fijter, J.W.; Tensen, C.P.; Bavinck, J.N.B.; Geissler, E.K. Early and late effects of the immunosuppressants rapamycin and mycophenolate mofetil on UV carcinogenesis. Int. J. Cancer 2010, 127, 796–804. [Google Scholar] [CrossRef]
- Wulff, B.C.; Kusewitt, D.F.; VanBuskirk, A.M.; Thomas-Ahner, J.M.; Duncan, F.J.; Oberyszyn, T.M. Sirolimus reduces the incidence and progression of UVB-induced skin cancer in SKH mice even with co-administration of cyclosporine A. J. Investig. Dermatol. 2008, 128, 2467–2473. [Google Scholar] [CrossRef] [Green Version]
- Schaper-Gerhardt, K.; Walter, A.; Schmitz-Rode, C.; Satzger, I.; Gutzmer, R. The mTOR-inhibitor Sirolimus decreases the cyclosporine-induced expression of the oncogene ATF3 in human keratinocytes. J. Dermatol. Sci. 2018, 92, 172–180. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.S.; In, S.G.; Park, O.J.; Won, C.H.; Lee, M.W.; Choi, J.H.; Kim, C.W.; Kim, S.E.; Moon, K.C.; Chang, S. Increased expression of activating transcription factor 3 is related to the biologic behavior of cutaneous squamous cell carcinomas. Hum. Pathol. 2011, 42, 954–959. [Google Scholar] [CrossRef] [PubMed]
- Euvrard, S.; Kanitakis, J.; Decullier, E.; Butnaru, A.C.; Lefrançois, N.; Boissonnat, P.; Sebbag, L.; Garnier, J.-L.; Pouteil-Noble, C.; Cahen, R. Subsequent skin cancers in kidney and heart transplant recipients after the first squamous cell carcinoma. Transplantation 2006, 81, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Alberú, J.; Pascoe, M.D.; Campistol, J.M.; Schena, F.P.; del Carmen Rial, M.; Polinsky, M.; Neylan, J.F.; Korth-Bradley, J.; Goldberg-Alberts, R.; Maller, E.S. Lower malignancy rates in renal allograft recipients converted to sirolimus-based, calcineurin inhibitor-free immunotherapy: 24-month results from the CONVERT trial. Transplantation 2011, 92, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Jensen, A.; Thomsen, H.F.; Engebjerg, M.C.; Olesen, A.B.; Friis, S.; Karagas, M.R.; Sørensen, H.T. Use of oral glucocorticoids and risk of skin cancer and non-Hodgkin’s lymphoma: A population-based case–control study. Br. J. Cancer 2009, 100, 200–205. [Google Scholar] [CrossRef] [Green Version]
- Baibergenova, A.T.; Weinstock, M.A.; Group, V.T. Oral prednisone use and risk of keratinocyte carcinoma in non-transplant population. The VATTC trial. J. Eur. Acad. Dermatol. Venereol. 2012, 26, 1109–1115. [Google Scholar] [CrossRef]
- Sørensen, H.T.; Mellemkjær, L.; Nielsen, G.L.; Baron, J.A.; Olsen, J.H.; Karagas, M.R. Skin Cancers and Non-Hodgkin Lymphoma Among Users of Systemic Glucocorticoids: A Population-Based Cohort Study. JNCI J. Natl. Cancer Inst. 2004, 96, 709–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fortina, A.B.; Piaserico, S.; Caforio, A.L.P.; Abeni, D.; Alaibac, M.; Angelini, A.; Iliceto, S.; Peserico, A. Immunosuppressive Level and Other Risk Factors for Basal Cell Carcinoma and Squamous Cell Carcinoma in Heart Transplant Recipients. Arch. Dermatol. 2004, 140, 1079–1085. [Google Scholar] [CrossRef]
- Chen, P.; Chen, F.; Zhou, B. Systematic review and meta-analysis of prevalence of dermatological toxicities associated with vemurafenib treatment in patients with melanoma. Clin. Exp. Dermatol. 2019, 44, 243–251. [Google Scholar] [CrossRef] [Green Version]
- Anforth, R.; Fernandez-Peñas, P.; Long, G.V. Cutaneous toxicities of RAF inhibitors. Lancet. Oncol. 2013, 14, e11–e18. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Minor, D.; Ribas, A.; Lebbe, C.; O’Hagan, A.; Arya, N.; Guckert, M.; Schadendorf, D.; Kefford, R.F.; Grob, J.J.; et al. Phase II trial (BREAK-2) of the BRAF inhibitor dabrafenib (GSK2118436) in patients with metastatic melanoma. J. Clin. Oncol. 2013, 31, 3205–3211. [Google Scholar] [CrossRef]
- Gibney, G.T.; Messina, J.L.; Fedorenko, I.V.; Sondak, V.K.; Smalley, K.S.M. Paradoxical oncogenesis—The long-term effects of BRAF inhibition in melanoma. Nat. Rev. Clin. Oncol. 2013, 10, 390–399. [Google Scholar] [CrossRef] [Green Version]
- Heidorn, S.J.; Milagre, C.; Whittaker, S.; Nourry, A.; Niculescu-Duvas, I.; Dhomen, N.; Hussain, J.; Reis-Filho, J.S.; Springer, C.J.; Pritchard, C. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 2010, 140, 209–221. [Google Scholar] [CrossRef]
- Poulikakos, P.I.; Zhang, C.; Bollag, G.; Shokat, K.M.; Rosen, N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010, 464, 427–430. [Google Scholar] [CrossRef] [Green Version]
- Su, F.; Viros, A.; Milagre, C.; Trunzer, K.; Bollag, G.; Spleiss, O.; Reis-Filho, J.S.; Kong, X.; Koya, R.C.; Flaherty, K.T. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N. Engl. J. Med. 2012, 366, 207–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anforth, R.; Menzies, A.; Byth, K.; Carlos, G.; Chou, S.; Sharma, R.; Scolyer, R.A.; Kefford, R.; Long, G.V.; Fernandez-Peñas, P. Factors influencing the development of cutaneous squamous cell carcinoma in patients on BRAF inhibitor therapy. J. Am. Acad. Dermatol. 2015, 72, 809–815.e801. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Flaherty, K.T.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: Long-term survival and safety analysis of a phase 3 study. Ann. Oncol. 2017, 28, 1631–1639. [Google Scholar] [CrossRef] [PubMed]
- Carlos, G.; Anforth, R.; Clements, A.; Menzies, A.M.; Carlino, M.S.; Chou, S.; Fernandez-Peñas, P. Cutaneous Toxic Effects of BRAF Inhibitors Alone and in Combination with MEK Inhibitors for Metastatic Melanoma. JAMA Derm. 2015, 151, 1103–1109. [Google Scholar] [CrossRef] [PubMed]
- March-Rodriguez, Á.; Bellosillo, B.; Álvarez-Larrán, A.; Besses, C.; Pujol, R.M.; Toll, A. Rapidly Growing and Aggressive Cutaneous Squamous Cell Carcinomas in a Patient Treated with Ruxolitinib. Ann. Derm. 2019, 31, 204–208. [Google Scholar] [CrossRef]
- Aboul-Fettouh, N.; Nijhawan, R.I. Aggressive squamous cell carcinoma in a patient on the Janus kinase inhibitor ruxolitinib. JAAD Case Rep. 2018, 4, 455–457. [Google Scholar] [CrossRef]
- Aleisa, A.I.; Plante, J.G.; Hsia, L.-L.B. A case of aggressive squamous cell carcinoma with lymphovascular invasion during treatment with the Janus kinase inhibitor tofacitinib. JAAD Case Rep. 2020, 6, 727–730. [Google Scholar] [CrossRef]
- Harrison, C.N.; Vannucchi, A.M.; Kiladjian, J.J.; Al-Ali, H.K.; Gisslinger, H.; Knoops, L.; Cervantes, F.; Jones, M.M.; Sun, K.; McQuitty, M.; et al. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia 2016, 30, 1701–1707. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Abikhair Burgo, M.; Roudiani, N.; Chen, J.; Santana, A.L.; Doudican, N.; Proby, C.; Felsen, D.; Carucci, J.A. Ruxolitinib inhibits cyclosporine-induced proliferation of cutaneous squamous cell carcinoma. JCI Insight 2020, 3, e120750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerr-Valentic, M.A.; Samimi, K.; Rohlen, B.H.; Agarwal, J.P.; Rockwell, W.B. Marjolin’s Ulcer: Modern Analysis of an Ancient Problem. Plast. Reconstr. Surg. 2009, 123, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Xiang, F.; Song, H.-P.; Huang, Y.-S. Clinical features and treatment of 140 cases of Marjolin’s ulcer at a major burn center in southwest China. Exp. Med. 2019, 17, 3403–3410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadir, A.R. Burn scar neoplasm. Ann. Burn. Fire Disasters 2007, 20, 185–188. [Google Scholar]
- Das, K.K.; Chakaraborty, A.; Rahman, A.; Khandkar, S. Incidences of malignancy in chronic burn scar ulcers: Experience from Bangladesh. Burns 2015, 41, 1315–1321. [Google Scholar] [CrossRef]
- Yu, N.; Long, X.; Lujan-Hernandez, J.R.; Hassan, K.Z.; Bai, M.; Wang, Y.; Wang, X.; Zhao, R. Marjolin’s ulcer: A preventable malignancy arising from scars. World J. Surg. Oncol. 2013, 11, 313. [Google Scholar] [CrossRef] [Green Version]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [Green Version]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Yu, H.S.; Liao, W.T.; Chai, C.Y. Arsenic carcinogenesis in the skin. J. Biomed. Sci. 2006, 13, 657–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishikawa, M.; Koyama, K.; Iseki, M.; Kobuke, T.; Yonehara, S.; Soda, M.; Ron, E.; Tokunaga, M.; Preston, D.L.; Mabuchi, K.; et al. Histologic characteristics of skin cancer in Hiroshima and Nagasaki: Background incidence and radiation effects. Int. J. Cancer 2005, 117, 363–369. [Google Scholar] [CrossRef]
- Yoshinaga, S.; Hauptmann, M.; Sigurdson, A.J.; Doody, M.M.; Freedman, D.M.; Alexander, B.H.; Linet, M.S.; Ron, E.; Mabuchi, K. Nonmelanoma skin cancer in relation to ionizing radiation exposure among U.S. radiologic technologists. Int. J. Cancer 2005, 115, 828–834. [Google Scholar] [CrossRef]
- Lichter, M.D.; Karagas, M.R.; Mott, L.A.; Spencer, S.K.; Stukel, T.A.; Greenberg, E.R. Therapeutic ionizing radiation and the incidence of basal cell carcinoma and squamous cell carcinoma. The New Hampshire Skin Cancer Study Group. Arch. Dermatol. 2000, 136, 1007–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenehjem, J.S.; Robsahm, T.E.; Bråtveit, M.; Samuelsen, S.O.; Kirkeleit, J.; Grimsrud, T.K. Aromatic hydrocarbons and risk of skin cancer by anatomical site in 25 000 male offshore petroleum workers. Am. J. Ind. Med. 2017, 60, 679–688. [Google Scholar] [CrossRef] [Green Version]
- Pukkala, E.; Martinsen, J.I.; Weiderpass, E.; Kjaerheim, K.; Lynge, E.; Tryggvadottir, L.; Sparén, P.; Demers, P.A. Cancer incidence among firefighters: 45 years of follow-up in five Nordic countries. Occup. Environ. Med. 2014, 71, 398–404. [Google Scholar] [CrossRef] [Green Version]
- Alter, B.P.; Giri, N.; Savage, S.A.; Rosenberg, P.S. Cancer in dyskeratosis congenita. Blood 2009, 113, 6549–6557. [Google Scholar] [CrossRef] [Green Version]
- Alter, B.P.; Giri, N.; Savage, S.A.; Peters, J.A.; Loud, J.T.; Leathwood, L.; Carr, A.G.; Greene, M.H.; Rosenberg, P.S. Malignancies and survival patterns in the National Cancer Institute inherited bone marrow failure syndromes cohort study. Br. J. Haematol. 2010, 150, 179–188. [Google Scholar] [CrossRef] [Green Version]
- Risitano, A.M.; Marotta, S.; Calzone, R.; Grimaldi, F.; Zatterale, A. Twenty years of the Italian Fanconi Anemia Registry: Where we stand and what remains to be learned. Haematologica 2016, 101, 319–327. [Google Scholar] [CrossRef] [Green Version]
- de Koning, M.N.C.; Weissenborn, S.J.; Abeni, D.; Bouwes Bavinck, J.N.; Euvrard, S.; Green, A.C.; Harwood, C.A.; Naldi, L.; Neale, R.; Nindl, I.; et al. Prevalence and associated factors of betapapillomavirus infections in individuals without cutaneous squamous cell carcinoma. J. Gen. Virol. 2009, 90, 1611–1621. [Google Scholar] [CrossRef] [PubMed]
- Antonsson, A.; Forslund, O.; Ekberg, H.; Sterner, G.; Hansson, B.G. The ubiquity and impressive genomic diversity of human skin papillomaviruses suggest a commensalic nature of these viruses. J. Virol. 2000, 74, 11636–11641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neale, R.E.; Weissenborn, S.; Abeni, D.; Bavinck, J.N.B.; Euvrard, S.; Feltkamp, M.C.W.; Green, A.C.; Harwood, C.; de Koning, M.; Naldi, L.; et al. Human Papillomavirus Load in Eyebrow Hair Follicles and Risk of Cutaneous Squamous Cell Carcinoma. Cancer Epidemiol. Biomark. Prev. 2013, 22, 719–727. [Google Scholar] [CrossRef] [Green Version]
- Bouwes Bavinck, J.N.; Feltkamp, M.C.W.; Green, A.C.; Fiocco, M.; Euvrard, S.; Harwood, C.A.; Nasir, S.; Thomson, J.; Proby, C.M.; Naldi, L.; et al. Human papillomavirus and posttransplantation cutaneous squamous cell carcinoma: A multicenter, prospective cohort study. Am. J. Transplant. 2018, 18, 1220–1230. [Google Scholar] [CrossRef]
- Chahoud, J.; Semaan, A.; Chen, Y.; Cao, M.; Rieber, A.G.; Rady, P.; Tyring, S.K. Association Between β-Genus Human Papillomavirus and Cutaneous Squamous Cell Carcinoma in Immunocompetent Individuals—A Meta-analysis. JAMA Dermatol. 2016, 152, 1354–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karagas, M.R.; Nelson, H.H.; Sehr, P.; Waterboer, T.; Stukel, T.A.; Andrew, A.; Green, A.C.; Bouwes Bavinck, J.N.; Perry, A.; Spencer, S.; et al. Human Papillomavirus Infection and Incidence of Squamous Cell and Basal Cell Carcinomas of the Skin. JNCI J. Natl. Cancer Inst. 2006, 98, 389–395. [Google Scholar] [CrossRef]
- Andersson, K.; Waterboer, T.; Kirnbauer, R.; Slupetzky, K.; Iftner, T.; de Villiers, E.-M.; Forslund, O.; Pawlita, M.; Dillner, J. Seroreactivity to Cutaneous Human Papillomaviruses among Patients with Nonmelanoma Skin Cancer or Benign Skin Lesions. Cancer Epidemiol. Biomark. Prev. 2008, 17, 189–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iannacone, M.R.; Gheit, T.; Waterboer, T.; Giuliano, A.R.; Messina, J.L.; Fenske, N.A.; Cherpelis, B.S.; Sondak, V.K.; Roetzheim, R.G.; Ferrer-Gil, S.; et al. Case–Control Study of Cutaneous Human Papillomavirus Infection in Basal Cell Carcinoma of the Skin. J. Investig. Dermatol. 2013, 133, 1512–1520. [Google Scholar] [CrossRef] [Green Version]
- Howley, P.M.; Pfister, H.J. Beta genus papillomaviruses and skin cancer. Virology 2015, 479–480, 290–296. [Google Scholar] [CrossRef] [Green Version]
- Weissenborn, S.J.; Nindl, I.; Purdie, K.; Harwood, C.; Proby, C.; Breuer, J.; Majewski, S.; Pfister, H.; Wieland, U. Human papillomavirus-DNA loads in actinic keratoses exceed those in non-melanoma skin cancers. J. Investig. Dermatol. 2005, 125, 93–97. [Google Scholar] [CrossRef] [Green Version]
- Conforti, C.; Paolini, F.; Venuti, A.; Dianzani, C.; Zalaudek, I. The detection rate of human papillomavirus in well-differentiated squamous cell carcinoma and keratoacanthoma: Is there new evidence for a viral pathogenesis of keratoacanthoma? Br. J. Dermatol. 2019, 181, 1309–1311. [Google Scholar] [CrossRef]
- Viarisio, D.; Decker, K.M.; Aengeneyndt, B.; Flechtenmacher, C.; Gissmann, L.; Tommasino, M. Human papillomavirus type 38 E6 and E7 act as tumour promoters during chemically induced skin carcinogenesis. J. Gen. Virol. 2013, 94, 749–752. [Google Scholar] [CrossRef] [PubMed]
- Wallace, N.A.; Robinson, K.; Howie, H.L.; Galloway, D.A. HPV 5 and 8 E6 abrogate ATR activity resulting in increased persistence of UVB induced DNA damage. PLoS Pathog. 2012, 8, e1002807. [Google Scholar] [CrossRef] [Green Version]
- Cornet, I.; Bouvard, V.; Campo, M.S.; Thomas, M.; Banks, L.; Gissmann, L.; Lamartine, J.; Sylla, B.S.; Accardi, R.; Tommasino, M. Comparative Analysis of Transforming Properties of E6 and E7 from Different Beta Human Papillomavirus Types. J. Virol. 2012, 86, 2366–2370. [Google Scholar] [CrossRef] [Green Version]
- Caldeira, S.; Zehbe, I.; Accardi, R.; Malanchi, I.; Dong, W.; Giarrè, M.; de Villiers, E.-M.; Filotico, R.; Boukamp, P.; Tommasino, M. The E6 and E7 Proteins of the Cutaneous Human Papillomavirus Type 38 Display Transforming Properties. J. Virol. 2003, 77, 2195–2206. [Google Scholar] [CrossRef] [Green Version]
- Brimer, N.; Lyons, C.; Wallberg, A.E.; Vande Pol, S.B. Cutaneous papillomavirus E6 oncoproteins associate with MAML1 to repress transactivation and NOTCH signaling. Oncogene 2012, 31, 4639–4646. [Google Scholar] [CrossRef] [Green Version]
- Meyers, J.M.; Uberoi, A.; Grace, M.; Lambert, P.F.; Munger, K. Cutaneous HPV8 and MmuPV1 E6 Proteins Target the NOTCH and TGF-β Tumor Suppressors to Inhibit Differentiation and Sustain Keratinocyte Proliferation. PLoS Pathog. 2017, 13, e1006171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, M.J.A.; White, E.A.; Sowa, M.E.; Harper, J.W.; Aster, J.C.; Howley, P.M. Cutaneous β-human papillomavirus E6 proteins bind Mastermind-like coactivators and repress Notch signaling. Proc. Natl. Acad. Sci. USA 2012, 109, E1473–E1480. [Google Scholar] [CrossRef] [Green Version]
- Rozenblatt-Rosen, O.; Deo, R.C.; Padi, M.; Adelmant, G.; Calderwood, M.A.; Rolland, T.; Grace, M.; Dricot, A.; Askenazi, M.; Tavares, M.; et al. Interpreting cancer genomes using systematic host network perturbations by tumour virus proteins. Nature 2012, 487, 491–495. [Google Scholar] [CrossRef]
- Olivero, C.; Lanfredini, S.; Borgogna, C.; Gariglio, M.; Patel, G.K. HPV-Induced Field Cancerisation: Transformation of Adult Tissue Stem Cell Into Cancer Stem Cell. Front. Microbiol. 2018, 9, 546. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Harris, C.C. p53: Traffic cop at the crossroads of DNA repair and recombination. Nat. Rev. Mol. Cell Biol. 2005, 6, 44–55. [Google Scholar] [CrossRef]
- Kraemer, K.H.; Tamura, D.; Khan, S.G.; Digiovanna, J.J. Burning issues in the diagnosis of xeroderma pigmentosum. Br. J. Dermatol. 2013, 169, 1176. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, H.; English, D.; Randell, P.L.; Nakazawa, K.; Martel, N.; Armstrong, B.K.; Yamasaki, H. UV and skin cancer: Specific p53 gene mutation in normal skin as a biologically relevant exposure measurement. Proc. Natl. Acad. Sci. USA 1994, 91, 360–364. [Google Scholar] [CrossRef] [Green Version]
- Jonason, A.S.; Kunala, S.; Price, G.J.; Restifo, R.J.; Spinelli, H.M.; Persing, J.A.; Leffell, D.J.; Tarone, R.E.; Brash, D.E. Frequent clones of p53-mutated keratinocytes in normal human skin. Proc. Natl. Acad. Sci. USA 1996, 93, 14025–14029. [Google Scholar] [CrossRef] [Green Version]
- Ren, Z.P.; Hedrum, A.; Pontén, F.; Nistér, M.; Ahmadian, A.; Lundeberg, J.; Uhlén, M.; Pontén, J. Human epidermal cancer and accompanying precursors have identical p53 mutations different from p53 mutations in adjacent areas of clonally expanded non-neoplastic keratinocytes. Oncogene 1996, 12, 765–773. [Google Scholar]
- Ziegler, A.; Jonason, A.S.; Leffell, D.J.; Simon, J.A.; Sharma, H.W.; Kimmelman, J.; Remington, L.; Jacks, T.; Brash, D.E. Sunburn and p53 in the onset of skin cancer. Nature 1994, 372, 773–776. [Google Scholar] [CrossRef] [PubMed]
- Durinck, S.; Ho, C.; Wang, N.J.; Liao, W.; Jakkula, L.R.; Collisson, E.A.; Pons, J.; Chan, S.-W.; Lam, E.T.; Chu, C.; et al. Temporal Dissection of Tumorigenesis in Primary Cancers. Cancer Discov. 2011, 1, 137–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- South, A.P.; Purdie, K.J.; Watt, S.A.; Haldenby, S.; den Breems, N.Y.; Dimon, M.; Arron, S.T.; Kluk, M.J.; Aster, J.C.; McHugh, A.; et al. NOTCH1 Mutations Occur Early during Cutaneous Squamous Cell Carcinogenesis. J. Investig. Dermatol. 2014, 134, 2630–2638. [Google Scholar] [CrossRef] [Green Version]
- Di Nardo, L.; Pellegrini, C.; Di Stefani, A.; Del Regno, L.; Sollena, P.; Piccerillo, A.; Longo, C.; Garbe, C.; Fargnoli, M.C.; Peris, K. Molecular genetics of cutaneous squamous cell carcinoma: Perspective for treatment strategies. J. Eur. Acad. Dermatol. Venereol. 2020, 34, 932–941. [Google Scholar] [CrossRef]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Hruban, R.H.; Goggins, M.; Parsons, J.; Kern, S.E. Progression model for pancreatic cancer. Clin. Cancer Res. 2000, 6, 2969–2972. [Google Scholar] [PubMed]
- Olivier, M.; Langerød, A.; Carrieri, P.; Bergh, J.; Klaar, S.; Eyfjord, J.; Theillet, C.; Rodriguez, C.; Lidereau, R.; Bièche, I.; et al. The clinical value of somatic TP53 gene mutations in 1794 patients with breast cancer. Clin. Cancer Res. 2006, 12, 1157–1167. [Google Scholar] [CrossRef] [Green Version]
- Berg, R.J.; van Kranen, H.J.; Rebel, H.G.; de Vries, A.; van Vloten, W.A.; Van Kreijl, C.F.; van der Leun, J.C.; de Gruijl, F.R. Early p53 alterations in mouse skin carcinogenesis by UVB radiation: Immunohistochemical detection of mutant p53 protein in clusters of preneoplastic epidermal cells. Proc. Natl. Acad. Sci. USA 1996, 93, 274–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Tron, V.; Ho, V. Induction of squamous cell carcinoma in p53-deficient mice after ultraviolet irradiation. J. Investig. Dermatol. 1998, 110, 72–75. [Google Scholar] [CrossRef] [Green Version]
- Mudgil, A.V.; Segal, N.; Andriani, F.; Wang, Y.; Fusenig, N.E.; Garlick, J.A. Ultraviolet B irradiation induces expansion of intraepithelial tumor cells in a tissue model of early cancer progression. J. Investig. Dermatol. 2003, 121, 191–197. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.Y.; Hanna, G.J.; Laga, A.C.; Haddad, R.I.; Lorch, J.H.; Hammerman, P.S. Genomic analysis of metastatic cutaneous squamous cell carcinoma. Clin. Cancer Res. 2015, 21, 1447–1456. [Google Scholar] [CrossRef] [Green Version]
- Artavanis-Tsakonas, S.; Rand, M.D.; Lake, R.J. Notch Signaling: Cell Fate Control and Signal Integration in Development. Science 1999, 284, 770–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watt, F.M.; Estrach, S.; Ambler, C.A. Epidermal Notch signalling: Differentiation, cancer and adhesion. Curr. Opin. Cell Biol. 2008, 20, 171–179. [Google Scholar] [CrossRef] [Green Version]
- Nowell, C.S.; Radtke, F. Notch as a tumour suppressor. Nat. Rev. Cancer 2017, 17, 145–159. [Google Scholar] [CrossRef]
- Balint, K.; Xiao, M.; Pinnix, C.C.; Soma, A.; Veres, I.; Juhasz, I.; Brown, E.J.; Capobianco, A.J.; Herlyn, M.; Liu, Z.J. Activation of Notch1 signaling is required for beta-catenin-mediated human primary melanoma progression. J. Clin. Investig. 2005, 115, 3166–3176. [Google Scholar] [CrossRef]
- Weng, A.P.; Ferrando, A.A.; Lee, W.; Morris, J.P.t.; Silverman, L.B.; Sanchez-Irizarry, C.; Blacklow, S.C.; Look, A.T.; Aster, J.C. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004, 306, 269–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolas, M.; Wolfer, A.; Raj, K.; Kummer, J.A.; Mill, P.; van Noort, M.; Hui, C.-c.; Clevers, H.; Dotto, G.P.; Radtke, F. Notch1 functions as a tumor suppressor in mouse skin. Nat. Genet. 2003, 33, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Pickering, C.R.; Zhou, J.H.; Lee, J.J.; Drummond, J.A.; Peng, S.A.; Saade, R.E.; Tsai, K.Y.; Curry, J.L.; Tetzlaff, M.T.; Lai, S.Y. Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin. Cancer Res. 2014, 20, 6582–6592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefort, K.; Mandinova, A.; Ostano, P.; Kolev, V.; Calpini, V.; Kolfschoten, I.; Devgan, V.; Lieb, J.; Raffoul, W.; Hohl, D.; et al. Notch1 is a p53 target gene involved in human keratinocyte tumor suppression through negative regulation of ROCK1/2 and MRCKalpha kinases. Genes Dev 2007, 21, 562–577. [Google Scholar] [CrossRef] [Green Version]
- Proweller, A.; Tu, L.; Lepore, J.J.; Cheng, L.; Lu, M.M.; Seykora, J.; Millar, S.E.; Pear, W.S.; Parmacek, M.S. Impaired Notch Signaling Promotes De novo Squamous Cell Carcinoma Formation. Cancer Res. 2006, 66, 7438–7444. [Google Scholar] [CrossRef] [Green Version]
- Demehri, S.; Turkoz, A.; Kopan, R. Epidermal Notch1 Loss Promotes Skin Tumorigenesis by Impacting the Stromal Microenvironment. Cancer Cell 2009, 16, 55–66. [Google Scholar] [CrossRef] [Green Version]
- Extance, A. Alzheimer’s failure raises questions about disease-modifying strategies. Nat. Rev. Drug Discov. 2010, 9, 749–751. [Google Scholar] [CrossRef]
- Brown, V.L.; Harwood, C.A.; Crook, T.; Cronin, J.G.; Kelsell, D.P.; Proby, C.M. p16INK4a and p14ARF Tumor Suppressor Genes Are Commonly Inactivated in Cutaneous Squamous Cell Carcinoma. J. Investig. Dermatol. 2004, 122, 1284–1292. [Google Scholar] [CrossRef] [Green Version]
- Inman, G.J.; Wang, J.; Nagano, A.; Alexandrov, L.B.; Purdie, K.J.; Taylor, R.G.; Sherwood, V.; Thomson, J.; Hogan, S.; Spender, L.C.; et al. The genomic landscape of cutaneous SCC reveals drivers and a novel azathioprine associated mutational signature. Nat. Commun. 2018, 9, 3667. [Google Scholar] [CrossRef]
- Ryan, M.B.; Corcoran, R.B. Therapeutic strategies to target RAS-mutant cancers. Nat. Rev. Clin. Oncol. 2018, 15, 709–720. [Google Scholar] [CrossRef]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K pathway in human disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Arcy, M.E.; Pfeiffer, R.M.; Rivera, D.R.; Hess, G.P.; Cahoon, E.K.; Arron, S.T.; Brownell, I.; Cowen, E.W.; Israni, A.K.; Triplette, M.A.; et al. Voriconazole and the Risk of Keratinocyte Carcinomas among Lung Transplant Recipients in the United States. JAMA Dermatol. 2020, 156, 772–779. [Google Scholar] [CrossRef] [PubMed]
- Dajee, M.; Lazarov, M.; Zhang, J.Y.; Cai, T.; Green, C.L.; Russell, A.J.; Marinkovich, M.P.; Tao, S.; Lin, Q.; Kubo, Y.; et al. NF-κB blockade and oncogenic Ras trigger invasive human epidermal neoplasia. Nature 2003, 421, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Ridky, T.W.; Chow, J.M.; Wong, D.J.; Khavari, P.A. Invasive three-dimensional organotypic neoplasia from multiple normal human epithelia. Nat. Med. 2010, 16, 1450–1455. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howard, B.D.; Tessman, I. Identification of the altered bases in mutated single-stranded DNA: II. In vivo mutagenesis by 5-bromodeoxyuridine and 2-aminopurine. J. Mol. Biol. 1964, 9, 364–371. [Google Scholar] [CrossRef]
- Setlow, R.; Carrier, W. Pyrimidine dimers in ultraviolet-irradiated DNA’s. J. Mol. Biol. 1966, 17, 237–254. [Google Scholar] [CrossRef]
- Cho, R.J.; Alexandrov, L.B.; den Breems, N.Y.; Atanasova, V.S.; Farshchian, M.; Purdom, E.; Nguyen, T.N.; Coarfa, C.; Rajapakshe, K.; Prisco, M.; et al. APOBEC mutation drives early-onset squamous cell carcinomas in recessive dystrophic epidermolysis bullosa. Sci. Transl. Med. 2018, 10, eaas9668. [Google Scholar] [CrossRef] [Green Version]
- Haisma, M.S.; Plaat, B.E.C.; Bijl, H.P.; Roodenburg, J.L.N.; Diercks, G.F.H.; Romeijn, T.R.; Terra, J.B. Multivariate analysis of potential risk factors for lymph node metastasis in patients with cutaneous squamous cell carcinoma of the head and neck. J. Am. Acad. Dermatol. 2016, 75, 722–730. [Google Scholar] [CrossRef]
- Koyfman, S.A.; Cooper, J.S.; Beitler, J.J.; Busse, P.M.; Jones, C.U.; McDonald, M.W.; Quon, H.; Ridge, J.A.; Saba, N.F.; Salama, J.K.; et al. ACR Appropriateness Criteria(®) Aggressive Nonmelanomatous Skin Cancer of the Head and Neck. Head Neck 2016, 38, 175–182. [Google Scholar] [CrossRef] [Green Version]
- National Comprehensive Cancer Network. Squamous Cell Skin Cancer. (Version 1). 2021. Available online: https://www.nccn.org/professionals/physician_gls/pdf/squamous.pdf (accessed on 5 April 2021).
- Cooper, J.S.; Pajak, T.F.; Forastiere, A.A.; Jacobs, J.; Campbell, B.H.; Saxman, S.B.; Kish, J.A.; Kim, H.E.; Cmelak, A.J.; Rotman, M. Postoperative concurrent radiotherapy and chemotherapy for high-risk squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2004, 350, 1937–1944. [Google Scholar] [CrossRef] [Green Version]
- Bernier, J.; Domenge, C.; Ozsahin, M.; Matuszewska, K.; Lefèbvre, J.-L.; Greiner, R.H.; Giralt, J.; Maingon, P.; Rolland, F.; Bolla, M. Postoperative irradiation with or without concomitant chemotherapy for locally advanced head and neck cancer. N. Engl. J. Med. 2004, 350, 1945–1952. [Google Scholar] [CrossRef] [Green Version]
- Denis, F.; Garaud, P.; Bardet, E.; Alfonsi, M.; Sire, C.; Germain, T.; Bergerot, P.; Rhein, B.; Tortochaux, J.; Calais, G. Final results of the 94-01 French Head and Neck Oncology and Radiotherapy Group randomized trial comparing radiotherapy alone with concomitant radiochemotherapy in advanced-stage oropharynx carcinoma. J. Clin. Oncol. 2004, 22, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Bourhis, J.; Sire, C.; Graff, P.; Grégoire, V.; Maingon, P.; Calais, G.; Gery, B.; Martin, L.; Alfonsi, M.; Desprez, P.; et al. Concomitant chemoradiotherapy versus acceleration of radiotherapy with or without concomitant chemotherapy in locally advanced head and neck carcinoma (GORTEC 99-02): An open-label phase 3 randomised trial. Lancet Oncol. 2012, 13, 145–153. [Google Scholar] [CrossRef]
- Porceddu, S.V.; Bressel, M.; Poulsen, M.G.; Stoneley, A.; Veness, M.J.; Kenny, L.M.; Wratten, C.; Corry, J.; Cooper, S.; Fogarty, G.B.; et al. Postoperative Concurrent Chemoradiotherapy Versus Postoperative Radiotherapy in High-Risk Cutaneous Squamous Cell Carcinoma of the Head and Neck: The Randomized Phase III TROG 05.01 Trial. J. Clin. Oncol. 2018, 36, 1275–1283. [Google Scholar] [CrossRef]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef] [Green Version]
- McGranahan, N.; Furness, A.J.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Blake, S.J.; Yong, M.C.; Harjunpää, H.; Ngiow, S.F.; Takeda, K.; Young, A.; O’Donnell, J.S.; Allen, S.; Smyth, M.J.; et al. Improved Efficacy of Neoadjuvant Compared to Adjuvant Immunotherapy to Eradicate Metastatic Disease. Cancer Discov 2016, 6, 1382–1399. [Google Scholar] [CrossRef] [Green Version]
- Ferrarotto, R.; Amit, M.; Nagarajan, P.; Rubin, M.L.; Yuan, Y.; Bell, D.; El-Naggar, A.K.; Johnson, J.M.; Morrison, W.H.; Rosenthal, D.I.; et al. Pilot Phase II Trial of Neoadjuvant Immunotherapy in Locoregionally Advanced, Resectable Cutaneous Squamous Cell Carcinoma of the Head and Neck. Clin. Cancer Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Neoadjuvant Atezolizumab in Surgically Resectable Advanced Cutaneous Squamous Cell Carcinoma. U.S. National Library of Medicine. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT04710498 (accessed on 15 June 2021).
- Neo-adjuvant Nivolumab or Nivolumab With Ipilimumab in Advanced Cutaneous Squamous Cell Carcinoma Prior to Surgery (MATISSE)—NCT04620200. U.S. National Library of Medicine. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT04620200 (accessed on 1 August 2021).
- Rischin, D.; Fury, M.G.; Lowy, I.; Stankevich, E.; Han, H.; Porceddu, S. A phase III, randomized, double-blind study of adjuvant cemiplimab versus placebo post-surgery and radiation therapy (RT) in patients (pts) with high-risk cutaneous squamous cell carcinoma (CSCC). J. Clin. Oncol. 2020, 38, TPS10084. [Google Scholar] [CrossRef]
- Geiger, J.L.; Daniels, G.A.; Cohen, E.E.W.; Ge, J.Y.; Gumuscu, B.; Swaby, R.F.; Chang, A.L.S. KEYNOTE-630: Phase 3 study of adjuvant pembrolizumab versus placebo in patients with high-risk, locally advanced cutaneous squamous cell carcinoma. J. Clin. Oncol. 2019, 37, TPS9597. [Google Scholar] [CrossRef]
- Jarkowski, A., 3rd; Hare, R.; Loud, P.; Skitzki, J.J.; Kane, J.M., 3rd; May, K.S.; Zeitouni, N.C.; Nestico, J.; Vona, K.L.; Groman, A.; et al. Systemic Therapy in Advanced Cutaneous Squamous Cell Carcinoma (CSCC): The Roswell Park Experience and a Review of the Literature. Am. J. Clin. Oncol. 2016, 39, 545–548. [Google Scholar] [CrossRef]
- Migden, M.R.; Rischin, D.; Schmults, C.D.; Guminski, A.; Hauschild, A.; Lewis, K.D.; Chung, C.H.; Hernandez-Aya, L.; Lim, A.M.; Chang, A.L.S.; et al. PD-1 Blockade with Cemiplimab in Advanced Cutaneous Squamous-Cell Carcinoma. N. Engl. J. Med. 2018, 379, 341–351. [Google Scholar] [CrossRef] [Green Version]
- Rischin, D.; Khushalani, N.I.; Schmults, C.D.; Guminski, A.D.; Chang, A.L.S.; Lewis, K.D.; Lim, A.M.L.; Hernandez-Aya, L.F.; Hughes, B.G.M.; Schadendorf, D.; et al. Phase II study of cemiplimab in patients (pts) with advanced cutaneous squamous cell carcinoma (CSCC): Longer follow-up. J. Clin. Oncol. 2020, 38, 10018. [Google Scholar] [CrossRef]
- Rischin, D.; Migden, M.R.; Lim, A.M.; Schmults, C.D.; Khushalani, N.I.; Hughes, B.G.M.; Schadendorf, D.; Dunn, L.A.; Hernandez-Aya, L.; Chang, A.L.S.; et al. Phase 2 study of cemiplimab in patients with metastatic cutaneous squamous cell carcinoma: Primary analysis of fixed-dosing, long-term outcome of weight-based dosing. J. Immunother. Cancer 2020, 8, e000775. [Google Scholar] [CrossRef]
- Rischin, D.; Khushalani, N.I.; Schmults, C.D.; Guminski, A.; Chang, A.L.S.; Lewis, K.D.; Lim, A.M.; Hernandez-Aya, L.; Hughes, B.G.M.; Schadendorf, D.; et al. Integrated analysis of a phase 2 study of cemiplimab in advanced cutaneous squamous cell carcinoma: Extended follow-up of outcomes and quality of life analysis. J. Immunother. Cancer 2021, 9, e002757. [Google Scholar] [CrossRef] [PubMed]
- Grob, J.J.; Gonzalez, R.; Basset-Seguin, N.; Vornicova, O.; Schachter, J.; Joshi, A.; Meyer, N.; Grange, F.; Piulats, J.M.; Bauman, J.R.; et al. Pembrolizumab Monotherapy for Recurrent or Metastatic Cutaneous Squamous Cell Carcinoma: A Single-Arm Phase II Trial (KEYNOTE-629). J. Clin. Oncol. 2020, 38, 2916–2925. [Google Scholar] [CrossRef] [PubMed]
- Maubec, E.; Boubaya, M.; Petrow, P.; Beylot-Barry, M.; Basset-Seguin, N.; Deschamps, L.; Grob, J.-J.; Dréno, B.; Scheer-Senyarich, I.; Bloch-Queyrat, C.; et al. Phase II Study of Pembrolizumab As First-Line, Single-Drug Therapy for Patients With Unresectable Cutaneous Squamous Cell Carcinomas. J. Clin. Oncol. 2020, 38, 3051–3061. [Google Scholar] [CrossRef] [PubMed]
- Munhoz, R.R.; Camargo, V.P.D.; Marta, G.N.; Martins, J.C.; Nardo, M.; Barbosa, C.C.; Souza, C.E.d.; Barbosa, I.; Ricci, H.; Mattos, M.R.d.; et al. CA209-9JC: A phase II study of first-line nivolumab (NIVO) in patients (pts) with locally advanced or metastatic cutaneous squamous cell carcinoma. J. Clin. Oncol. 2020, 38, 10044. [Google Scholar] [CrossRef]
- The UNSCARRed Study: UNresctable Squamous Cell Carcinoma Treated With Avelumab and Radical Radiotherapy (UNSCARRed). U.S. National Library of Medicine. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT03737721 (accessed on 14 July 2021).
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients with Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef]
- Puzanov, I.; Milhem, M.M.; Minor, D.; Hamid, O.; Li, A.; Chen, L.; Chastain, M.; Gorski, K.S.; Anderson, A.; Chou, J.; et al. Talimogene Laherparepvec in Combination with Ipilimumab in Previously Untreated, Unresectable Stage IIIB-IV Melanoma. J. Clin. Oncol. 2016, 34, 2619–2626. [Google Scholar] [CrossRef] [Green Version]
- Long, G.; Dummer, R.; Johnson, D.; Michielin, O.; Martin-Algarra, S.; Treichel, S.; Chan, E.; Diede, S.; Ribas, A. 429 Long-term analysis of MASTERKEY-265 phase 1b trial of talimogene laherparepvec (T-VEC) plus pembrolizumab in patients with unresectable stage IIIB-IVM1c melanoma. J. Immunother. Cancer 2020, 8, A261. [Google Scholar] [CrossRef]
- Study Evaluating Cemiplimab Alone and Combined With RP1 in Treating Advanced Squamous Skin Cancer (CERPASS) U.S National Library of Medicine. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT04050436 (accessed on 2 September 2021).
- Talimogene Laherparepvec and Panitumumab for the Treatment of Locally Advanced or Metastatic Squamous Cell Carcinoma of the Skin U.S National Library of Medicine. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT04163952 (accessed on 2 September 2021).
- Avelumab with or without Cetuximab in Treating Patients with Advanced Skin Squamous Cell Cancer. U.S. National Library of Medicine. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT03944941 (accessed on 14 July 2021).
- Salomon, D.S.; Brandt, R.; Ciardiello, F.; Normanno, N. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit. Rev. Oncol. Hematol. 1995, 19, 183–232. [Google Scholar] [CrossRef]
- Maubec, E.; Duvillard, P.; Velasco, V.; Crickx, B.; Avril, M.F. Immunohistochemical analysis of EGFR and HER-2 in patients with metastatic squamous cell carcinoma of the skin. Anticancer Res. 2005, 25, 1205–1210. [Google Scholar]
- Shimizu, T.; Izumi, H.; Oga, A.; Furumoto, H.; Murakami, T.; Ofuji, R.; Muto, M.; Sasaki, K. Epidermal Growth Factor Receptor Overexpression and Genetic Aberrations in Metastatic Squamous-Cell Carcinoma of the Skin. Dermatology 2001, 202, 203–206. [Google Scholar] [CrossRef]
- Maubec, E.; Petrow, P.; Scheer-Senyarich, I.; Duvillard, P.; Lacroix, L.; Gelly, J.; Certain, A.; Duval, X.; Crickx, B.; Buffard, V.; et al. Phase II study of cetuximab as first-line single-drug therapy in patients with unresectable squamous cell carcinoma of the skin. J. Clin. Oncol. 2011, 29, 3419–3426. [Google Scholar] [CrossRef]
- Foote, M.C.; McGrath, M.; Guminski, A.; Hughes, B.G.M.; Meakin, J.; Thomson, D.; Zarate, D.; Simpson, F.; Porceddu, S.V. Phase II study of single-agent panitumumab in patients with incurable cutaneous squamous cell carcinoma. Ann. Oncol. 2014, 25, 2047–2052. [Google Scholar] [CrossRef] [PubMed]
- Reigneau, M.; Robert, C.; Routier, E.; Mamelle, G.; Moya-Plana, A.; Tomasic, G.; Mateus, C. Efficacy of neoadjuvant cetuximab alone or with platinum salt for the treatment of unresectable advanced nonmetastatic cutaneous squamous cell carcinomas. Br. J. Dermatol. 2015, 173, 527–534. [Google Scholar] [CrossRef]
- William, W.N., Jr.; Feng, L.; Ferrarotto, R.; Ginsberg, L.; Kies, M.; Lippman, S.; Glisson, B.; Kim, E.S. Gefitinib for patients with incurable cutaneous squamous cell carcinoma: A single-arm phase II clinical trial. J. Am. Acad. Dermatol. 2017, 77, 1110–1113.e1112. [Google Scholar] [CrossRef]
- Gold, K.A.; Kies, M.S.; William, W.N.; Johnson, F.M.; Lee, J.J.; Glisson, B.S. Erlotinib in the treatment of recurrent or metastatic cutaneous squamous cell carcinoma: A single-arm phase 2 clinical trial. Cancer 2018, 124, 2169–2173. [Google Scholar] [CrossRef] [PubMed]
- Cavalieri, S.; Perrone, F.; Miceli, R.; Ascierto, P.A.; Locati, L.D.; Bergamini, C.; Granata, R.; Alfieri, S.; Resteghini, C.; Galbiati, D.; et al. Efficacy and safety of single-agent pan-human epidermal growth factor receptor (HER) inhibitor dacomitinib in locally advanced unresectable or metastatic skin squamous cell cancer. Eur. J. Cancer 2018, 97, 7–15. [Google Scholar] [CrossRef]
- Dunn, L.; Ho, A.L.; Eng, J.; Michel, L.S.; Fetten, J.V.; Warner, E.; Kriplani, A.; Zhi, W.I.; Ng, K.K.; Haque, S.; et al. A phase I/Ib study of lenvatinib and cetuximab in patients with recurrent/metastatic (R/M) head and neck squamous cell carcinoma (HNSCC). J. Clin. Oncol. 2020, 38, 6541. [Google Scholar] [CrossRef]
- Adelmann, C.H.; Truong, K.A.; Liang, R.J.; Bansal, V.; Gandee, L.; Saporito, R.C.; Lee, W.; Du, L.; Nicholas, C.; Napoli, M.; et al. MEK Is a Therapeutic and Chemopreventative Target in Squamous Cell Carcinoma. J. Investig. Dermatol. 2016, 136, 1920–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cobimetinib and Atezolizumab in Treating Participants with Advanced or Refractory Rare Tumors. U.S. National Library of Medicine. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT03108131 (accessed on 2 September 2021).
- Ding, L.T.; Zhao, P.; Yang, M.L.; Lv, G.Z.; Zhao, T.L. GDC-0084 inhibits cutaneous squamous cell carcinoma cell growth. Biochem. Biophys. Res. Commun. 2018, 503, 1941–1948. [Google Scholar] [CrossRef]
- Zou, Y.; Ge, M.; Wang, X. Targeting PI3K-AKT-mTOR by LY3023414 inhibits human skin squamous cell carcinoma cell growth in vitro and in vivo. Biochem. Biophys. Res. Commun. 2017, 490, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.C.; Varghese, A.M.; Hyman, D.M.; Bauer, T.M.; Pant, S.; Callies, S.; Lin, J.; Martinez, R.; Wickremsinhe, E.; Fink, A.; et al. A First-in-Human Phase 1 Study of LY3023414, an Oral PI3K/mTOR Dual Inhibitor, in Patients with Advanced Cancer. Clin. Cancer Res. 2018, 24, 3253–3262. [Google Scholar] [CrossRef] [Green Version]
- Wen, P.Y.; De Groot, J.F.; Battiste, J.D.; Goldlust, S.A.; Garner, J.S.; Simpson, J.A.; Kijlstra, J.; Olivero, A.; Cloughesy, T.F. Escalation portion of phase II study to evaluate the safety, pharmacokinetics, and clinical activity of the PI3K/mTOR inhibitor paxalisib (GDC-0084) in glioblastoma (GBM) with unmethylated O6-methylguanine-methyltransferase (MGMT) promotor status. J. Clin. Oncol. 2020, 38, 2550. [Google Scholar] [CrossRef]
- Manohar, S.; Thongprayoon, C.; Cheungpasitporn, W.; Markovic, S.N.; Herrmann, S.M. Systematic Review of the Safety of Immune Checkpoint Inhibitors among Kidney Transplant Patients. Kidney Int. Rep. 2019, 5, 149–158. [Google Scholar] [CrossRef] [Green Version]
- Lai, H.-C.; Lin, J.-F.; Hwang, T.I.S.; Liu, Y.-F.; Yang, A.-H.; Wu, C.-K. Programmed Cell Death 1 (PD-1) Inhibitors in Renal Transplant Patients with Advanced Cancer: A Double-Edged Sword? Int. J. Mol. Sci. 2019, 20, 2194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tacrolimus, Nivolumab, and Ipilimumab in Treating Kidney Transplant Recipients with Selected Unresectable or Metastatic Cancers. U.S National Library of Medicine. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT03816332 (accessed on 2 July 2021).
- Cemiplimab in AlloSCT/SOT Recipients with CSCC (CONTRAC) U.S. National Library of Medicine. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT04339062 (accessed on 2 July 2021).
- Cemiplimab-rwlc for Unresectable Locally Recurrent and/or Metastatic CSCC U.S National Library of Medicine. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT04242173 (accessed on 2 July 2021).
Figure 1.
Risk factors for CSCC include UV radiation exposure, immunosuppressive conditions and drugs, inflammations such as those from chronic wounds, and impaired DNA repair. * Infection with β-human papillomavirus and JAK inhibitors are also thought to increase the risk of CSCC, but clear evidence and the underlying biology are not as well established. Created using biorender.com (accessed on 1 September 2021).
Figure 1.
Risk factors for CSCC include UV radiation exposure, immunosuppressive conditions and drugs, inflammations such as those from chronic wounds, and impaired DNA repair. * Infection with β-human papillomavirus and JAK inhibitors are also thought to increase the risk of CSCC, but clear evidence and the underlying biology are not as well established. Created using biorender.com (accessed on 1 September 2021).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Thai, A.A.; Lim, A.M.; Solomon, B.J.; Rischin, D. Biology and Treatment Advances in Cutaneous Squamous Cell Carcinoma. Cancers 2021, 13, 5645. https://doi.org/10.3390/cancers13225645
AMA Style
Thai AA, Lim AM, Solomon BJ, Rischin D. Biology and Treatment Advances in Cutaneous Squamous Cell Carcinoma. Cancers. 2021; 13(22):5645. https://doi.org/10.3390/cancers13225645
Chicago/Turabian StyleThai, Alesha A., Annette M. Lim, Benjamin J. Solomon, and Danny Rischin. 2021. "Biology and Treatment Advances in Cutaneous Squamous Cell Carcinoma" Cancers 13, no. 22: 5645. https://doi.org/10.3390/cancers13225645
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.