
PRMT1 Regulates EGFR and Wnt Signaling Pathways and Is a Promising Target for Combinatorial Treatment of Breast Cancer

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Human Samples
2.2. Immunohistochemistry (IHC)
2.3. Cell Culture, RNA Interference, Antibodies, Small-Molecule Inhibitors, and Primers
2.4. Cell Proliferation Assay
2.5. Apoptosis Assays
2.6. Colony Formation Assay
2.7. Soft Agar Assay
2.8. Real-Time—Quantitative PCR Assay (RT-qPCR)
2.9. β-Catenin-Activated Reporter (BAR) Luciferase Assay
2.10. Chromatin Immunoprecipitation (ChIP)
2.11. Transcriptomic Analysis of PRMT1-Depleted Cells
2.12. GSK3368715 Treatment in Mice
2.13. Drug Combinations
2.14. Statistical Analysis
3. Results
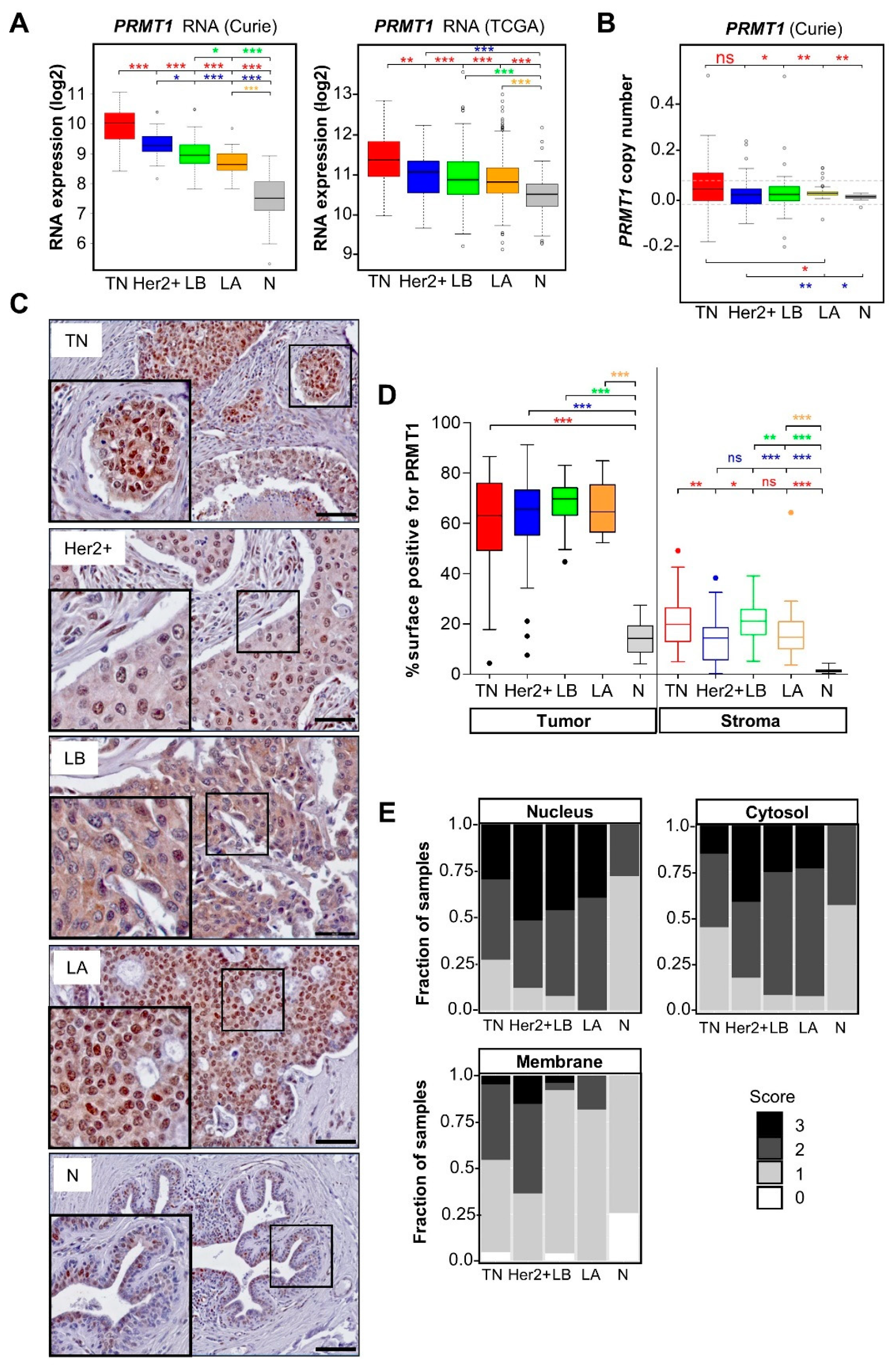
3.1. PRMT1 Is Overexpressed in All the Breast Cancer Subtypes Compared to Normal Breast Tissue
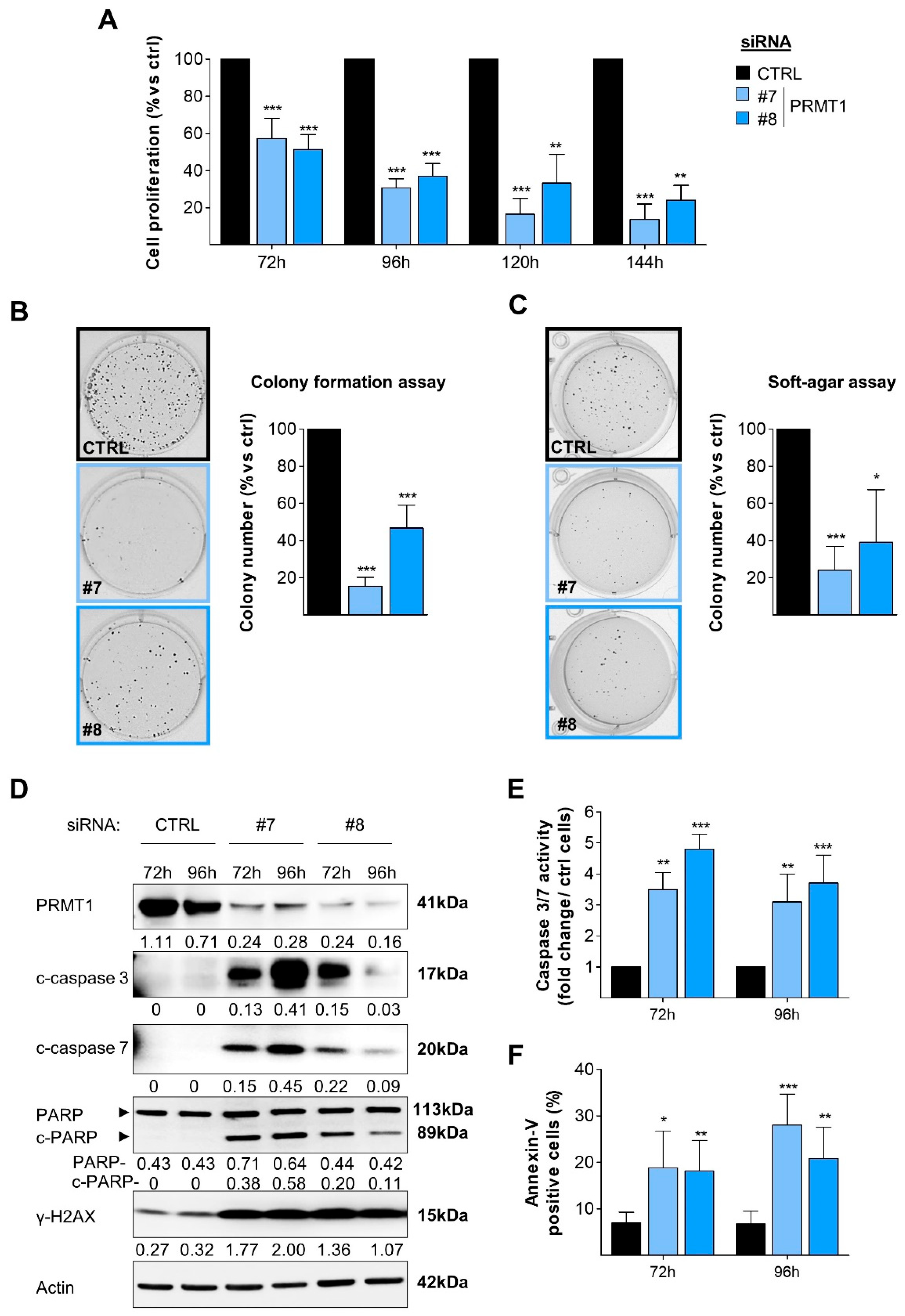
3.2. RNAi-Mediated Depletion of PRMT1 Decreases BC Cell Viability, Clonogenicity and Induces DNA Damage and Apoptosis
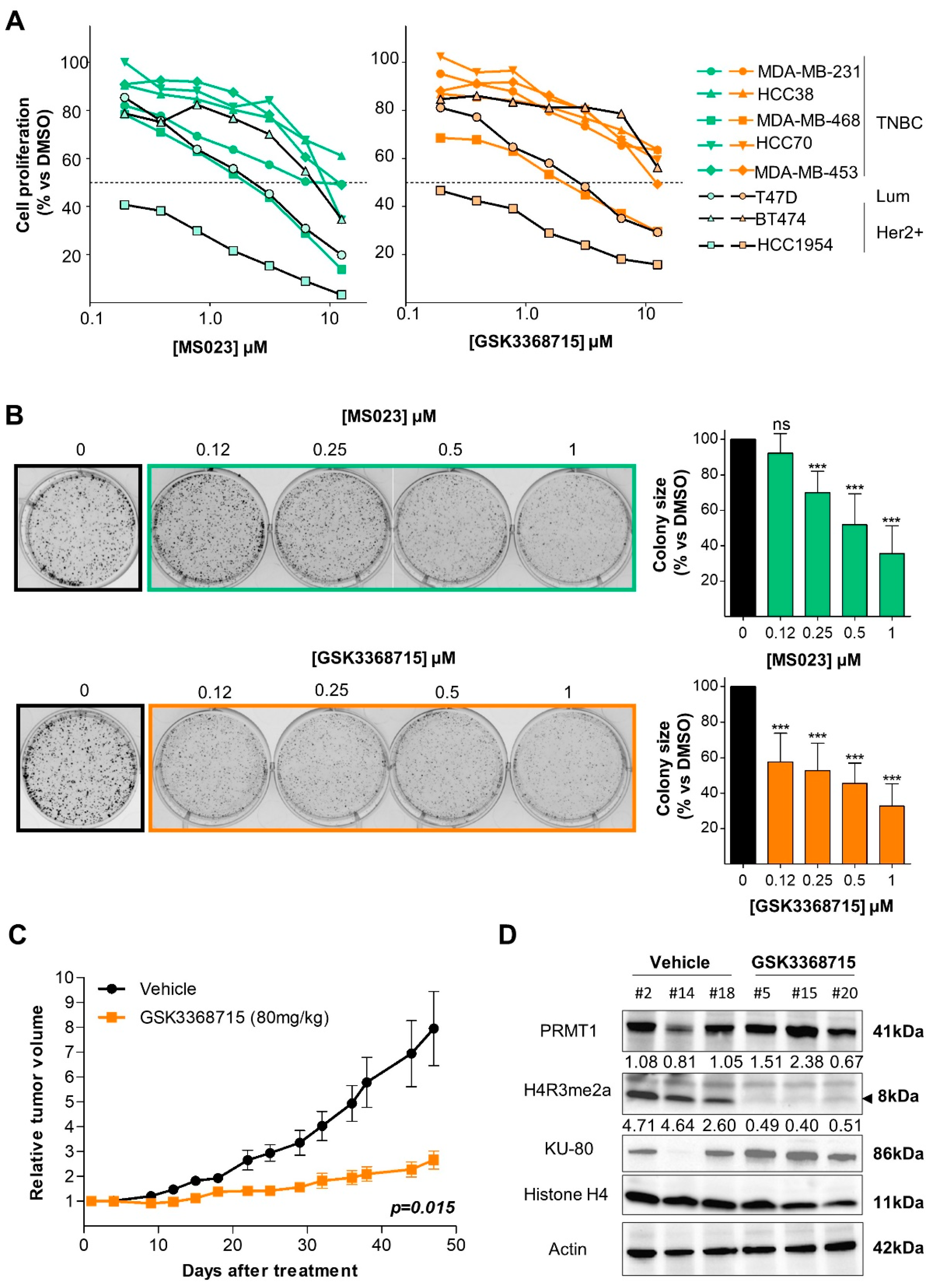
3.3. Type I PRMT Inhibitors Reduce BC Cell Growth
3.4. Type I PRMT Inhibition Slows Tumor Growth in a TNBC Xenograft Model
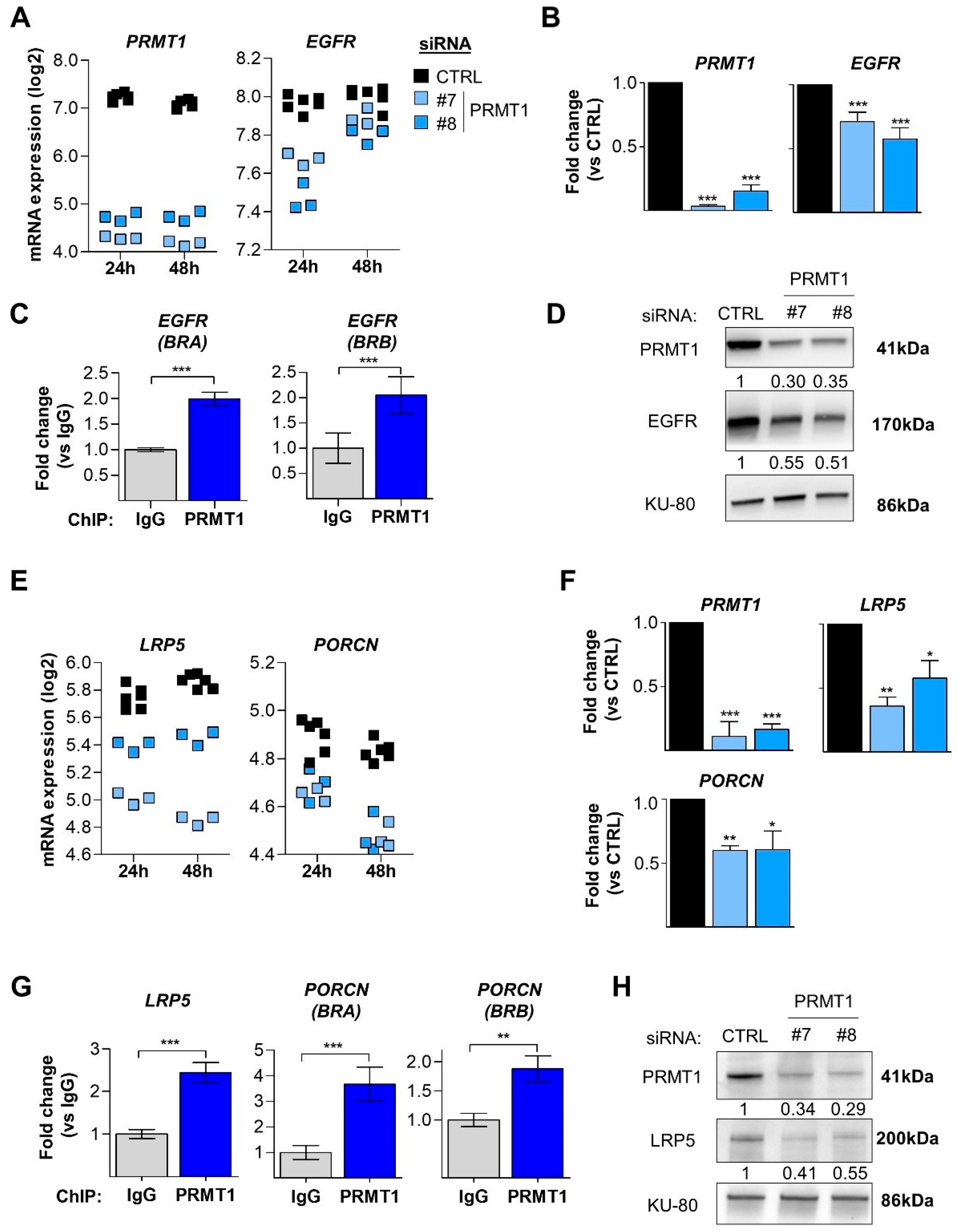
3.5. PRMT1 Regulates the EGFR and Wnt Signaling Pathways at the Transcriptomic Level
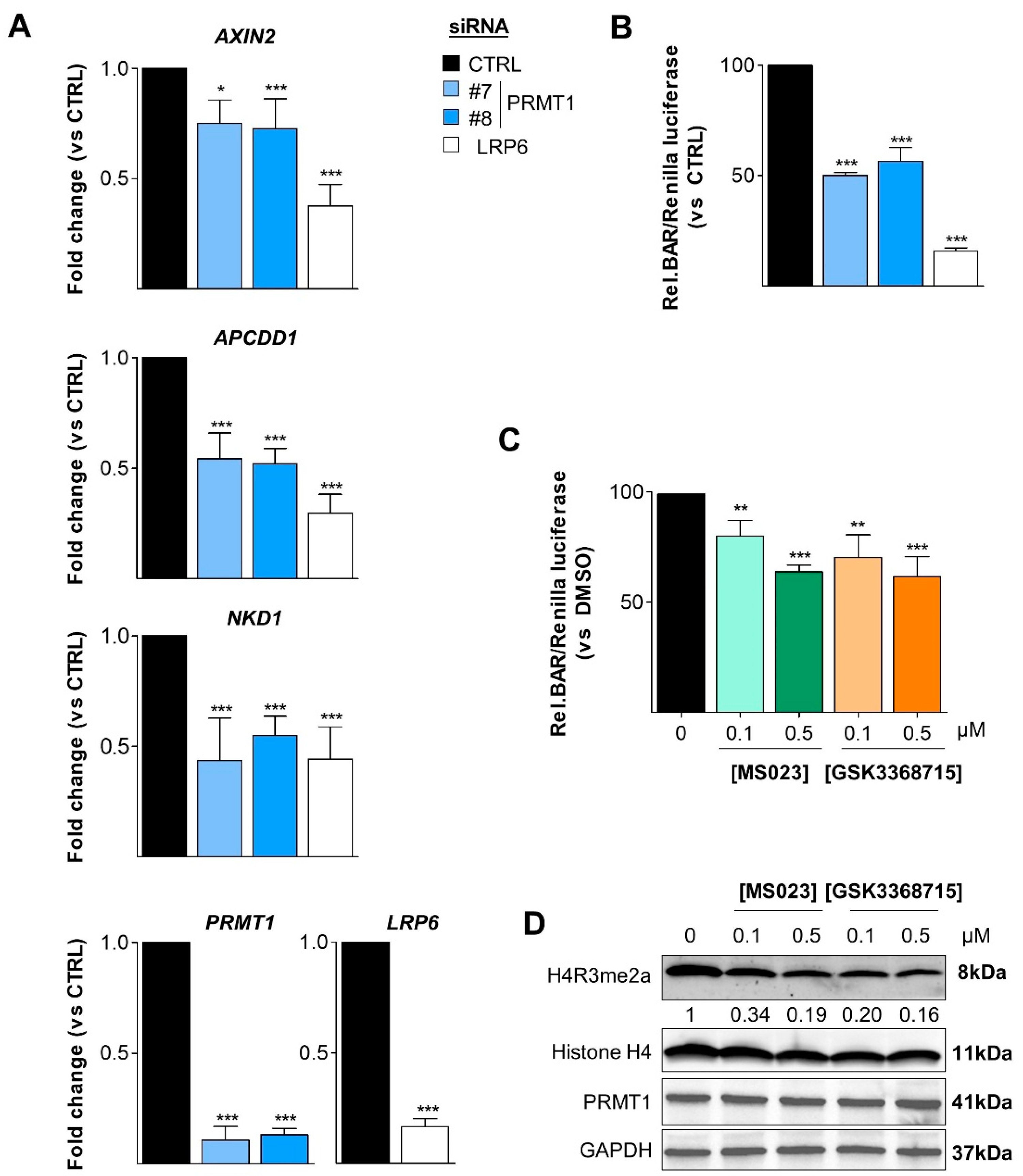
3.6. PRMT1 Activates the Canonical Wnt Signaling Pathway
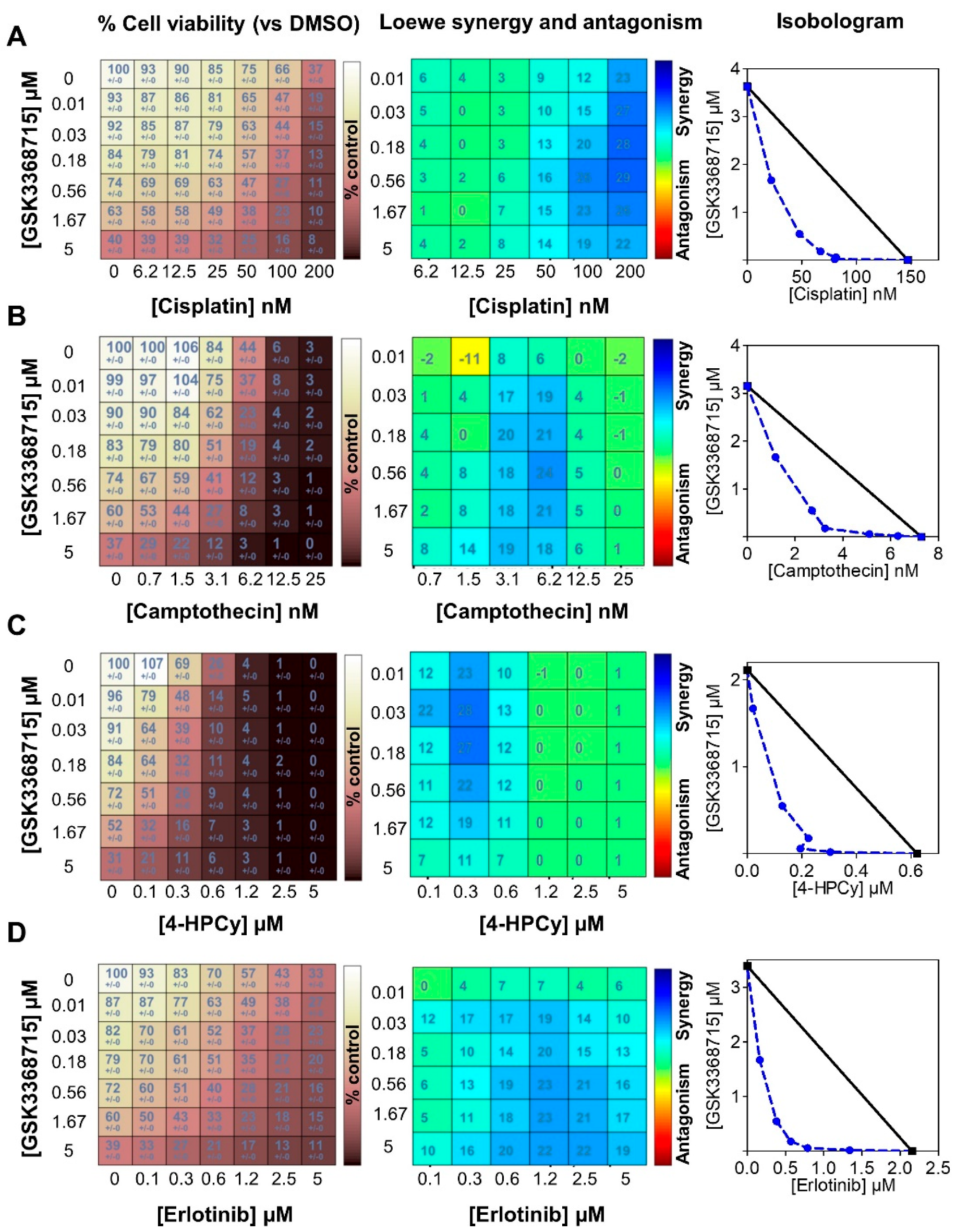
3.7. Type I PRMT Inhibitors Show Synergistic Interactions with Erlotinib or Chemotherapies
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Loibl, S.; Poortmans, P.; Morrow, M.; Denkert, C.; Curigliano, G. Breast cancer. Lancet 2021, 397, 1750–1769. [Google Scholar] [CrossRef]
- Manjunath, M.; Choudhary, B. Triple-negative breast cancer: A run-through of features, classification and current therapies. Oncol. Lett. 2021, 22, 512. [Google Scholar] [CrossRef]
- You, K.S.; Yi, Y.W.; Cho, J.; Park, J.S.; Seong, Y.S. Potentiating Therapeutic Effects of Epidermal Growth Factor Receptor Inhibition in Triple-Negative Breast Cancer. Pharmaceuticals 2021, 14, 589. [Google Scholar] [CrossRef]
- Pohl, S.G.; Brook, N.; Agostino, M.; Arfuso, F.; Kumar, A.P.; Dharmarajan, A. Wnt signaling in triple-negative breast cancer. Oncogenesis 2017, 6, e310. [Google Scholar] [CrossRef] [Green Version]
- Merikhian, P.; Eisavand, M.R.; Farahmand, L. Triple-negative breast cancer: Understanding Wnt signaling in drug resistance. Cancer Cell Int. 2021, 21, 419. [Google Scholar] [CrossRef]
- Maubant, S.; Tahtouh, T.; Brisson, A.; Maire, V.; Nemati, F.; Tesson, B.; Ye, M.; Rigaill, G.; Noizet, M.; Dumont, A.; et al. LRP5 regulates the expression of STK40, a new potential target in triple-negative breast cancers. Oncotarget 2018, 9, 22586–22604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, V.I.; Godoy, J.A.; Inestrosa, N.C. Modulating Wnt signaling at the root: Porcupine and Wnt acylation. Pharmacol. Ther. 2019, 198, 34–45. [Google Scholar] [CrossRef]
- Xu, J.; Richard, S. Cellular pathways influenced by protein arginine methylation: Implications for cancer. Mol. Cell 2021, 81, 4357–4368. [Google Scholar] [CrossRef] [PubMed]
- Jarrold, J.; Davies, C.C. PRMTs and Arginine Methylation: Cancer’s Best-Kept Secret? Trends Mol. Med. 2019, 25, 993–1009. [Google Scholar] [CrossRef]
- Wu, Q.; Schapira, M.; Arrowsmith, C.H.; Barsyte-Lovejoy, D. Protein arginine methylation: From enigmatic functions to therapeutic targeting. Nat. Rev. Drug Discov. 2021, 20, 509–530. [Google Scholar] [CrossRef] [PubMed]
- Guccione, E.; Richard, S. The regulation, functions and clinical relevance of arginine methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 642–657. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Bedford, M.T. Protein arginine methyltransferases and cancer. Nat. Rev. Cancer 2013, 13, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Suresh, S.; Huard, S.; Dubois, T. CARM1/PRMT4: Making Its Mark beyond Its Function as a Transcriptional Coactivator. Trends Cell Biol. 2021, 31, 402–417. [Google Scholar] [CrossRef] [PubMed]
- Thiebaut, C.; Eve, L.; Poulard, C.; Le Romancer, M. Structure, Activity, and Function of PRMT1. Life 2021, 11, 1147. [Google Scholar] [CrossRef]
- Morettin, A.; Baldwin, R.M.; Cote, J. Arginine methyltransferases as novel therapeutic targets for breast cancer. Mutagenesis 2015, 30, 177–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lattouf, H.; Kassem, L.; Jacquemetton, J.; Choucair, A.; Poulard, C.; Tredan, O.; Corbo, L.; Diab-Assaf, M.; Hussein, N.; Treilleux, I.; et al. LKB1 regulates PRMT5 activity in breast cancer. Int. J. Cancer 2019, 144, 595–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinet, M.; Suresh, S.; Maire, V.; Monchecourt, C.; Nemati, F.; Lesage, L.; Pierre, F.; Ye, M.; Lescure, A.; Brisson, A.; et al. Protein arginine methyltransferase 5: A novel therapeutic target for triple-negative breast cancers. Cancer Med. 2019, 8, 2414–2428. [Google Scholar] [CrossRef] [Green Version]
- Eram, M.S.; Shen, Y.; Szewczyk, M.; Wu, H.; Senisterra, G.; Li, F.; Butler, K.V.; Kaniskan, H.U.; Speed, B.A.; Dela Sena, C.; et al. A Potent, Selective, and Cell-Active Inhibitor of Human Type I Protein Arginine Methyltransferases. ACS Chem. Biol. 2016, 11, 772–781. [Google Scholar] [CrossRef] [Green Version]
- Fedoriw, A.; Rajapurkar, S.R.; O’Brien, S.; Gerhart, S.V.; Mitchell, L.H.; Adams, N.D.; Rioux, N.; Lingaraj, T.; Ribich, S.A.; Pappalardi, M.B.; et al. Anti-tumor Activity of the Type I PRMT Inhibitor, GSK3368715, Synergizes with PRMT5 Inhibition through MTAP Loss. Cancer Cell 2019, 36, 100–114.e25. [Google Scholar] [CrossRef]
- Liao, H.W.; Hsu, J.M.; Xia, W.; Wang, H.L.; Wang, Y.N.; Chang, W.C.; Arold, S.T.; Chou, C.K.; Tsou, P.H.; Yamaguchi, H.; et al. PRMT1-mediated methylation of the EGF receptor regulates signaling and cetuximab response. J. Clin. Investig. 2015, 125, 4529–4543. [Google Scholar] [CrossRef] [Green Version]
- Nakai, K.; Xia, W.; Liao, H.W.; Saito, M.; Hung, M.C.; Yamaguchi, H. The role of PRMT1 in EGFR methylation and signaling in MDA-MB-468 triple-negative breast cancer cells. Breast Cancer 2018, 25, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.M.; Chen, C.T.; Chou, C.K.; Kuo, H.P.; Li, L.Y.; Lin, C.Y.; Lee, H.J.; Wang, Y.N.; Liu, M.; Liao, H.W.; et al. Crosstalk between Arg 1175 methylation and Tyr 1173 phosphorylation negatively modulates EGFR-mediated ERK activation. Nat. Cell Biol. 2011, 13, 174–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, B.; Gui, T.; Zeng, X.; Deng, Y.; Wang, Z.; Wang, Y.; Yang, D.; Li, Q.; Xu, P.; Hu, R.; et al. PRMT1-mediated H4R3me2a recruits SMARCA4 to promote colorectal cancer progression by enhancing EGFR signaling. Genome Med. 2021, 13, 58. [Google Scholar] [CrossRef] [PubMed]
- Takai, H.; Masuda, K.; Sato, T.; Sakaguchi, Y.; Suzuki, T.; Suzuki, T.; Koyama-Nasu, R.; Nasu-Nishimura, Y.; Katou, Y.; Ogawa, H.; et al. 5-Hydroxymethylcytosine plays a critical role in glioblastomagenesis by recruiting the CHTOP-methylosome complex. Cell Rep. 2014, 9, 48–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bikkavilli, R.K.; Malbon, C.C. Wnt3a-stimulated LRP6 phosphorylation is dependent upon arginine methylation of G3BP2. J. Cell Sci. 2012, 125, 2446–2456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bikkavilli, R.K.; Malbon, C.C. Arginine methylation of G3BP1 in response to Wnt3a regulates beta-catenin mRNA. J. Cell Sci. 2011, 124, 2310–2320. [Google Scholar] [CrossRef] [Green Version]
- Cha, B.; Kim, W.; Kim, Y.K.; Hwang, B.N.; Park, S.Y.; Yoon, J.W.; Park, W.S.; Cho, J.W.; Bedford, M.T.; Jho, E.H. Methylation by protein arginine methyltransferase 1 increases stability of Axin, a negative regulator of Wnt signaling. Oncogene 2011, 30, 2379–2389. [Google Scholar] [CrossRef] [Green Version]
- Bikkavilli, R.K.; Avasarala, S.; Vanscoyk, M.; Sechler, M.; Kelley, N.; Malbon, C.C.; Winn, R.A. Dishevelled3 is a novel arginine methyl transferase substrate. Sci. Rep. 2012, 2, 805. [Google Scholar] [CrossRef] [Green Version]
- Poulard, C.; Corbo, L.; Le Romancer, M. Protein arginine methylation/demethylation and cancer. Oncotarget 2016, 7, 67532–67550. [Google Scholar] [CrossRef] [Green Version]
- Maire, V.; Nemati, F.; Richardson, M.; Vincent-Salomon, A.; Tesson, B.; Rigaill, G.; Gravier, E.; Marty-Prouvost, B.; De Koning, L.; Lang, G.; et al. Polo-like kinase 1: A potential therapeutic option in combination with conventional chemotherapy for the management of patients with triple-negative breast cancer. Cancer Res. 2013, 73, 813–823. [Google Scholar] [CrossRef] [Green Version]
- Maire, V.; Baldeyron, C.; Richardson, M.; Tesson, B.; Vincent-Salomon, A.; Gravier, E.; Marty-Prouvost, B.; De Koning, L.; Rigaill, G.; Dumont, A.; et al. TTK/hMPS1 is an attractive therapeutic target for triple-negative breast cancer. PLoS ONE 2013, 8, e63712. [Google Scholar] [CrossRef]
- Marty, B.; Maire, V.; Gravier, E.; Rigaill, G.; Vincent-Salomon, A.; Kappler, M.; Lebigot, I.; Djelti, F.; Tourdes, A.; Gestraud, P.; et al. Frequent PTEN genomic alterations and activated phosphatidylinositol 3-kinase pathway in basal-like breast cancer cells. Breast Cancer Res. 2008, 10, R101. [Google Scholar] [CrossRef] [Green Version]
- Bao, X.; Siprashvili, Z.; Zarnegar, B.J.; Shenoy, R.M.; Rios, E.J.; Nady, N.; Qu, K.; Mah, A.; Webster, D.E.; Rubin, A.J.; et al. CSNK1a1 Regulates PRMT1 to Maintain the Progenitor State in Self-Renewing Somatic Tissue. Dev. Cell 2017, 43, 227–239.e5. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Di Veroli, G.Y.; Fornari, C.; Wang, D.; Mollard, S.; Bramhall, J.L.; Richards, F.M.; Jodrell, D.I. Combenefit: An interactive platform for the analysis and visualization of drug combinations. Bioinformatics 2016, 32, 2866–2868. [Google Scholar] [CrossRef]
- Gyorffy, B.; Lanczky, A.; Eklund, A.C.; Denkert, C.; Budczies, J.; Li, Q.; Szallasi, Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1809 patients. Breast Cancer Res. Treat. 2010, 123, 725–731. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.M.; Sun, W.Z.; Fan, X.Z.; Xu, Y.L.; Cheng, M.B.; Zhang, Y. Methylation of C/EBPalpha by PRMT1 Inhibits Its Tumor-Suppressive Function in Breast Cancer. Cancer Res. 2019, 79, 2865–2877. [Google Scholar] [CrossRef] [Green Version]
- Giuliani, V.; Miller, M.A.; Liu, C.Y.; Hartono, S.R.; Class, C.A.; Bristow, C.A.; Suzuki, E.; Sanz, L.A.; Gao, G.; Gay, J.P.; et al. PRMT1-dependent regulation of RNA metabolism and DNA damage response sustains pancreatic ductal adenocarcinoma. Nat. Commun. 2021, 12, 4626. [Google Scholar] [CrossRef]
- Malbeteau, L.; Poulard, C.; Languilaire, C.; Mikaelian, I.; Flamant, F.; Le Romancer, M.; Corbo, L. PRMT1 Is Critical for the Transcriptional Activity and the Stability of the Progesterone Receptor. iScience 2020, 23, 101236. [Google Scholar] [CrossRef]
- Maubant, S.; Tesson, B.; Maire, V.; Ye, M.; Rigaill, G.; Gentien, D.; Cruzalegui, F.; Tucker, G.C.; Roman-Roman, S.; Dubois, T. Transcriptome analysis of Wnt3a-treated triple-negative breast cancer cells. PLoS ONE 2015, 10, e0122333. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Wennerberg, K.; Aittokallio, T. What is synergy? The Saariselka agreement revisited. Front. Pharmacol. 2015, 6, 181. [Google Scholar] [CrossRef] [PubMed]
- Yoshimatsu, M.; Toyokawa, G.; Hayami, S.; Unoki, M.; Tsunoda, T.; Field, H.I.; Kelly, J.D.; Neal, D.E.; Maehara, Y.; Ponder, B.A.; et al. Dysregulation of PRMT1 and PRMT6, Type I arginine methyltransferases, is involved in various types of human cancers. Int. J. Cancer 2011, 128, 562–573. [Google Scholar] [CrossRef]
- Goulet, I.; Gauvin, G.; Boisvenue, S.; Cote, J. Alternative splicing yields protein arginine methyltransferase 1 isoforms with distinct activity, substrate specificity, and subcellular localization. J. Biol. Chem. 2007, 282, 33009–33021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathioudaki, K.; Scorilas, A.; Ardavanis, A.; Lymberi, P.; Tsiambas, E.; Devetzi, M.; Apostolaki, A.; Talieri, M. Clinical evaluation of PRMT1 gene expression in breast cancer. Tumour Biol. 2011, 32, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, R.M.; Morettin, A.; Cote, J. Role of PRMTs in cancer: Could minor isoforms be leaving a mark? World J. Biol. Chem. 2014, 5, 115–129. [Google Scholar] [CrossRef]
- Wang, J.; Wang, C.; Xu, P.; Li, X.; Lu, Y.; Jin, D.; Yin, X.; Jiang, H.; Huang, J.; Xiong, H.; et al. PRMT1 is a novel molecular therapeutic target for clear cell renal cell carcinoma. Theranostics 2021, 11, 5387–5403. [Google Scholar] [CrossRef]
- Filipovic, J.; Bosic, M.; Cirovic, S.; Zivotic, M.; Dunderovic, D.; Dordevic, D.; Zivkovic-Perisic, S.; Lipkovski, A.; Markovic-Lipkovski, J. PRMT1 expression in renal cell tumors- application in differential diagnosis and prognostic relevance. Diagn Pathol. 2019, 14, 120. [Google Scholar] [CrossRef] [PubMed]
- Repenning, A.; Happel, D.; Bouchard, C.; Meixner, M.; Verel-Yilmaz, Y.; Raifer, H.; Holembowski, L.; Krause, E.; Kremmer, E.; Feederle, R.; et al. PRMT1 promotes the tumor suppressor function of p14(ARF) and is indicative for pancreatic cancer prognosis. EMBO J. 2021, 40, e106777. [Google Scholar] [CrossRef]
- Choucair, A.; Pham, T.H.; Omarjee, S.; Jacquemetton, J.; Kassem, L.; Tredan, O.; Rambaud, J.; Marangoni, E.; Corbo, L.; Treilleux, I.; et al. The arginine methyltransferase PRMT1 regulates IGF-1 signaling in breast cancer. Oncogene 2019, 38, 4015–4027. [Google Scholar] [CrossRef]
- Gao, Y.; Zhao, Y.; Zhang, J.; Lu, Y.; Liu, X.; Geng, P.; Huang, B.; Zhang, Y.; Lu, J. The dual function of PRMT1 in modulating epithelial-mesenchymal transition and cellular senescence in breast cancer cells through regulation of ZEB1. Sci. Rep. 2016, 6, 19874. [Google Scholar] [CrossRef]
- Liu, L.M.; Tang, Q.; Hu, X.; Zhao, J.J.; Zhang, Y.; Ying, G.G.; Zhang, F. Arginine Methyltransferase PRMT1 Regulates p53 Activity in Breast Cancer. Life 2021, 11, 789. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, R.M.; Morettin, A.; Paris, G.; Goulet, I.; Cote, J. Alternatively spliced protein arginine methyltransferase 1 isoform PRMT1v2 promotes the survival and invasiveness of breast cancer cells. Cell Cycle 2012, 11, 4597–4612. [Google Scholar] [CrossRef] [Green Version]
- Yin, X.K.; Wang, Y.L.; Wang, F.; Feng, W.X.; Bai, S.M.; Zhao, W.W.; Feng, L.L.; Wei, M.B.; Qin, C.L.; Wang, F.; et al. PRMT1 enhances oncogenic arginine methylation of NONO in colorectal cancer. Oncogene 2021, 40, 1375–1389. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.P.; Jiang, Y.B.; Zhong, C.Q.; Ma, N.; Zhang, E.B.; Zhang, F.; Li, J.J.; Deng, Y.Z.; Wang, K.; Xie, D.; et al. PRMT1 Promoted HCC Growth and Metastasis In Vitro and In Vivo via Activating the STAT3 Signalling Pathway. Cell Physiol. Biochem. 2018, 47, 1643–1654. [Google Scholar] [CrossRef]
- Chuang, C.Y.; Chang, C.P.; Lee, Y.J.; Lin, W.L.; Chang, W.W.; Wu, J.S.; Cheng, Y.W.; Lee, H.; Li, C. PRMT1 expression is elevated in head and neck cancer and inhibition of protein arginine methylation by adenosine dialdehyde or PRMT1 knockdown downregulates proliferation and migration of oral cancer cells. Oncol. Rep. 2017, 38, 1115–1123. [Google Scholar] [CrossRef]
- Musiani, D.; Giambruno, R.; Massignani, E.; Ippolito, M.R.; Maniaci, M.; Jammula, S.; Manganaro, D.; Cuomo, A.; Nicosia, L.; Pasini, D.; et al. PRMT1 Is Recruited via DNA-PK to Chromatin Where It Sustains the Senescence-Associated Secretory Phenotype in Response to Cisplatin. Cell Rep. 2020, 30, 1208–1222.e9. [Google Scholar] [CrossRef] [Green Version]
- Lim, Y.; Lee, J.Y.; Ha, S.J.; Yu, S.; Shin, J.K.; Kim, H.C. Proteome-wide identification of arginine methylation in colorectal cancer tissues from patients. Proteome Sci. 2020, 18, 6. [Google Scholar] [CrossRef]
- Hsu, W.J.; Chen, C.H.; Chang, Y.C.; Cheng, C.H.; Tsa, I.Y.; Lin, C.W. PRMT1 Confers Resistance to Olaparib via Modulating MYC Signaling in Triple-Negative Breast Cancer. J. Pers. Med. 2021, 11, 1009. [Google Scholar] [CrossRef]
- Dominici, C.; Sgarioto, N.; Yu, Z.; Sesma-Sanz, L.; Masson, J.Y.; Richard, S.; Raynal, N.J. Synergistic effects of type I PRMT and PARP inhibitors against non-small cell lung cancer cells. Clin. Epigenetics 2021, 13, 54. [Google Scholar] [CrossRef] [PubMed]
- Fong, J.Y.; Pignata, L.; Goy, P.A.; Kawabata, K.C.; Lee, S.C.; Koh, C.M.; Musiani, D.; Massignani, E.; Kotini, A.G.; Penson, A.; et al. Therapeutic Targeting of RNA Splicing Catalysis through Inhibition of Protein Arginine Methylation. Cancer Cell 2019, 36, 194–209.e9. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Zhang, L.; Villarreal, O.D.; He, W.; Su, D.; Bedford, E.; Moh, P.; Shen, J.; Shi, X.; Bedford, M.T.; et al. PRMT1 loss sensitizes cells to PRMT5 inhibition. Nucleic Acids Res. 2019, 47, 5038–5048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; He, X.; Lin, Y.C.; Dong, H.; Zhang, L.; Chen, X.; Wang, Z.; Shen, Y.; Li, M.; Wang, H.; et al. Targeting PRMT1-mediated FLT3 methylation disrupts maintenance of MLL-rearranged acute lymphoblastic leukemia. Blood 2019, 134, 1257–1268. [Google Scholar] [CrossRef]
- He, X.; Zhu, Y.; Lin, Y.C.; Li, M.; Du, J.; Dong, H.; Sun, J.; Zhu, L.; Wang, H.; Ding, Z.; et al. PRMT1-mediated FLT3 arginine methylation promotes maintenance of FLT3-ITD(+) acute myeloid leukemia. Blood 2019, 134, 548–560. [Google Scholar] [CrossRef]
- Lu, S.X.; De Neef, E.; Thomas, J.D.; Sabio, E.; Rousseau, B.; Gigoux, M.; Knorr, D.A.; Greenbaum, B.; Elhanati, Y.; Hogg, S.J.; et al. Pharmacologic modulation of RNA splicing enhances anti-tumor immunity. Cell 2021, 184, 4032–4047.e31. [Google Scholar] [CrossRef]
- Zheng, N.N.; Zhou, M.; Sun, F.; Huai, M.X.; Zhang, Y.; Qu, C.Y.; Shen, F.; Xu, L.M. Combining protein arginine methyltransferase inhibitor and anti-programmed death-ligand-1 inhibits pancreatic cancer progression. World J. Gastroenterol. 2020, 26, 3737–3749. [Google Scholar] [CrossRef]
- Hu, G.; Yan, C.; Xie, P.; Cao, Y.; Shao, J.; Ge, J. PRMT2 accelerates tumorigenesis of hepatocellular carcinoma by activating Bcl2 via histone H3R8 methylation. Exp. Cell Res. 2020, 394, 112152. [Google Scholar] [CrossRef]
- Plotnikov, A.; Kozer, N.; Cohen, G.; Carvalho, S.; Duberstein, S.; Almog, O.; Solmesky, L.J.; Shurrush, K.A.; Babaev, I.; Benjamin, S.; et al. PRMT1 inhibition induces differentiation of colon cancer cells. Sci. Rep. 2020, 10, 20030. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suresh, S.; Huard, S.; Brisson, A.; Némati, F.; Dakroub, R.; Poulard, C.; Ye, M.; Martel, E.; Reyes, C.; Silvestre, D.C.; et al. PRMT1 Regulates EGFR and Wnt Signaling Pathways and Is a Promising Target for Combinatorial Treatment of Breast Cancer. Cancers 2022, 14, 306. https://doi.org/10.3390/cancers14020306
Suresh S, Huard S, Brisson A, Némati F, Dakroub R, Poulard C, Ye M, Martel E, Reyes C, Silvestre DC, et al. PRMT1 Regulates EGFR and Wnt Signaling Pathways and Is a Promising Target for Combinatorial Treatment of Breast Cancer. Cancers. 2022; 14(2):306. https://doi.org/10.3390/cancers14020306
Chicago/Turabian StyleSuresh, Samyuktha, Solène Huard, Amélie Brisson, Fariba Némati, Rayan Dakroub, Coralie Poulard, Mengliang Ye, Elise Martel, Cécile Reyes, David C. Silvestre, and et al. 2022. "PRMT1 Regulates EGFR and Wnt Signaling Pathways and Is a Promising Target for Combinatorial Treatment of Breast Cancer" Cancers 14, no. 2: 306. https://doi.org/10.3390/cancers14020306