Strategy for Modifying Layered Perovskites toward Efficient Solar Light-Driven Photocatalysts for Removal of Chlorinated Pollutants

 , ,
, ,
Abstract
:
1. Introduction
2. Results and Discussion
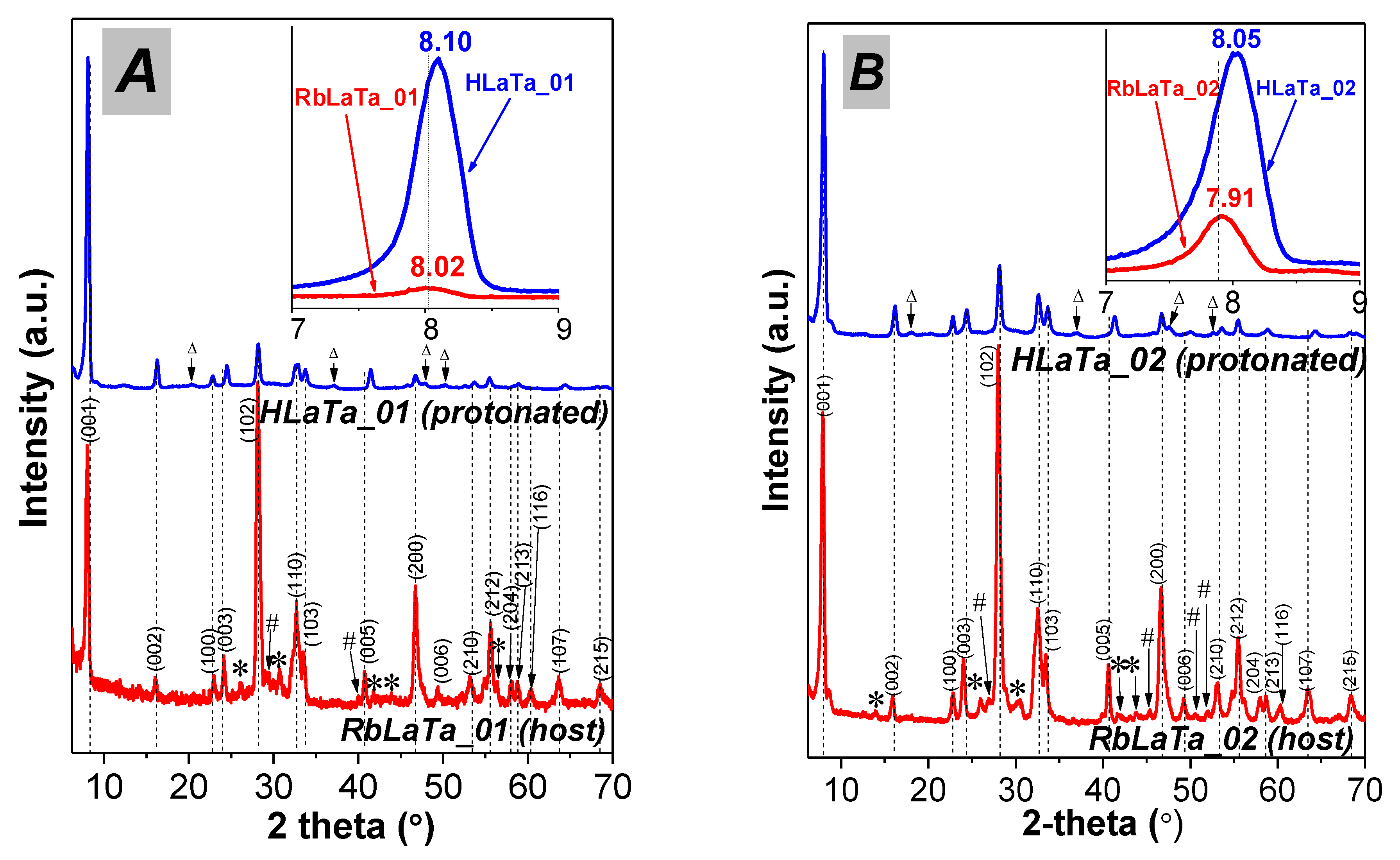
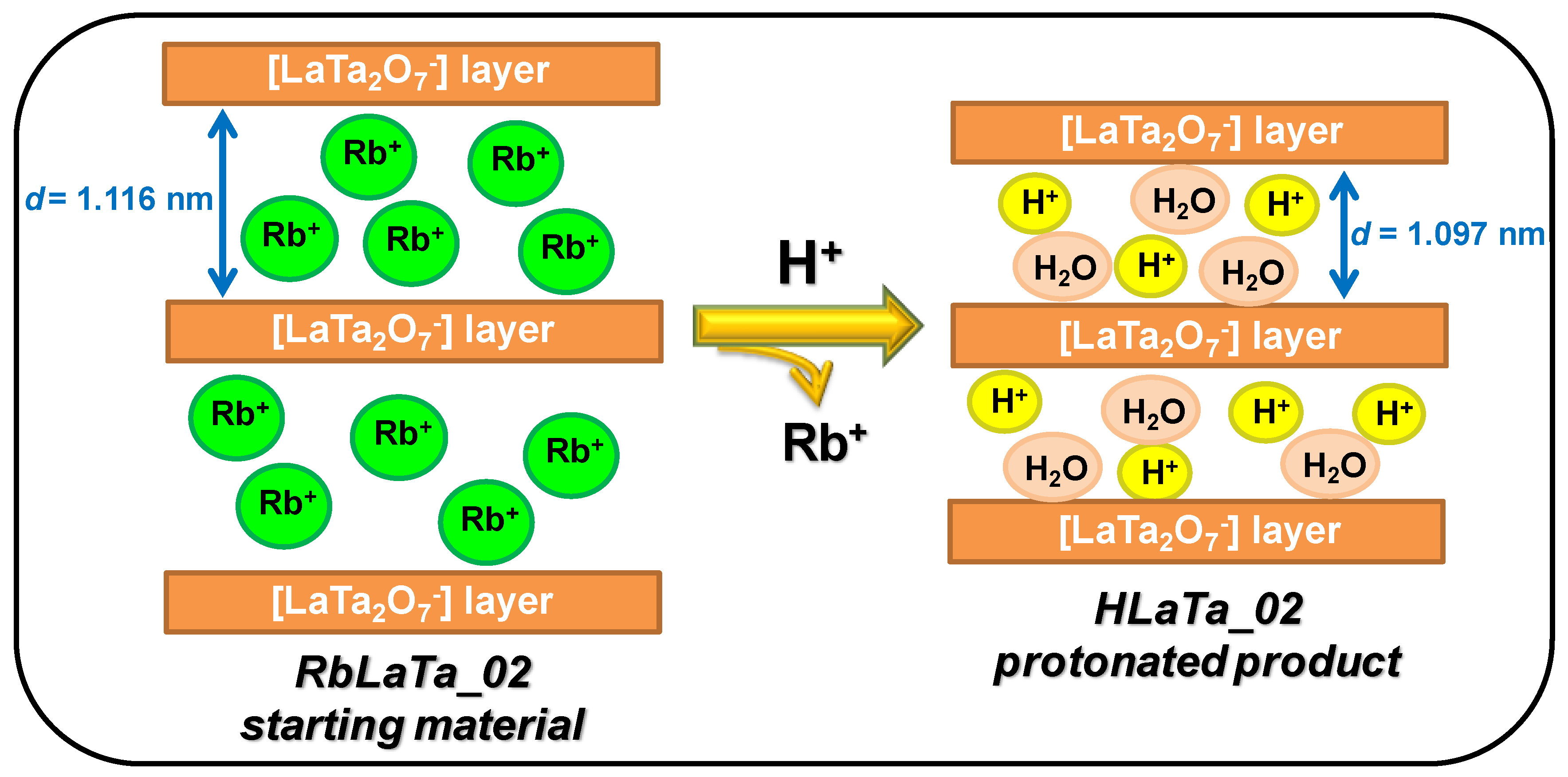
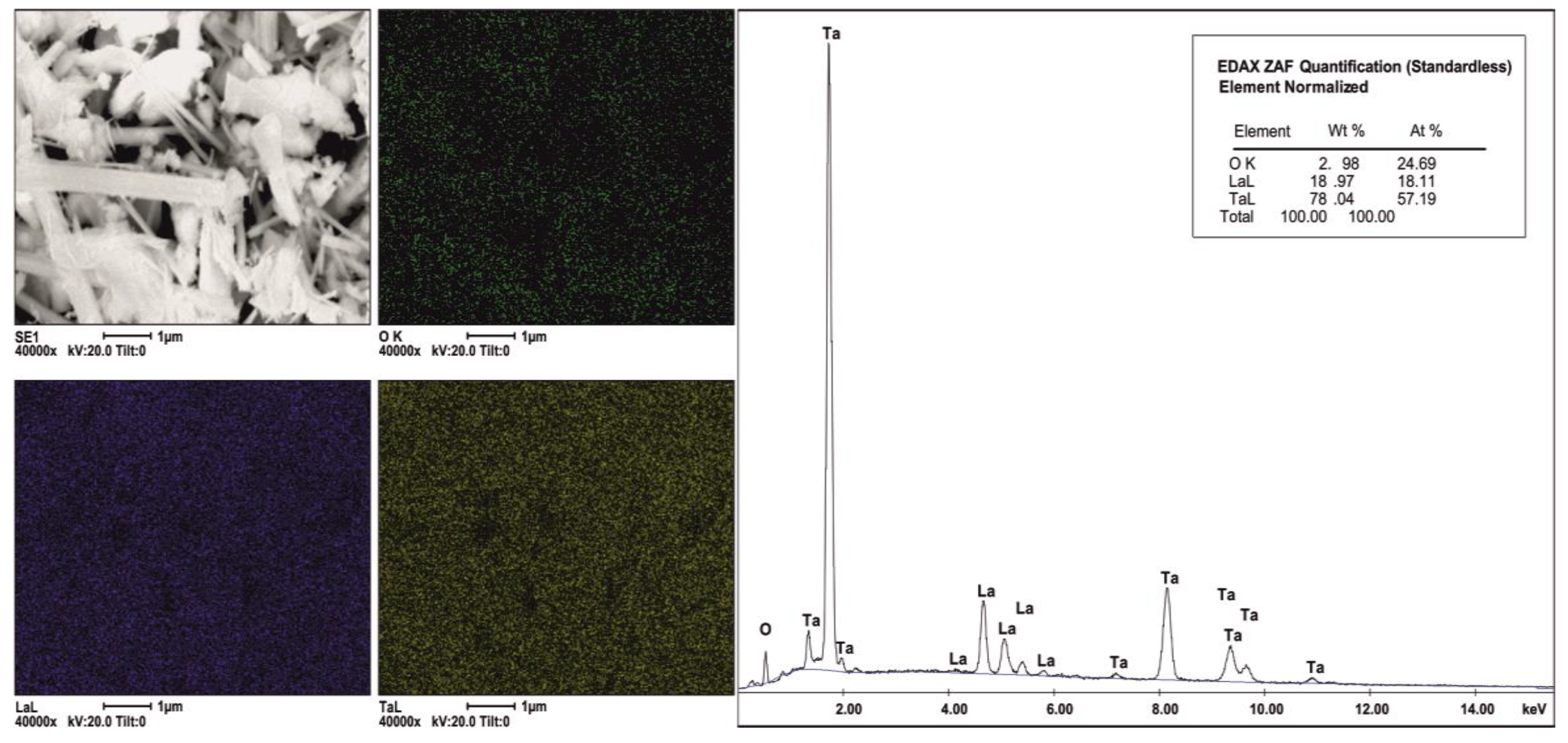
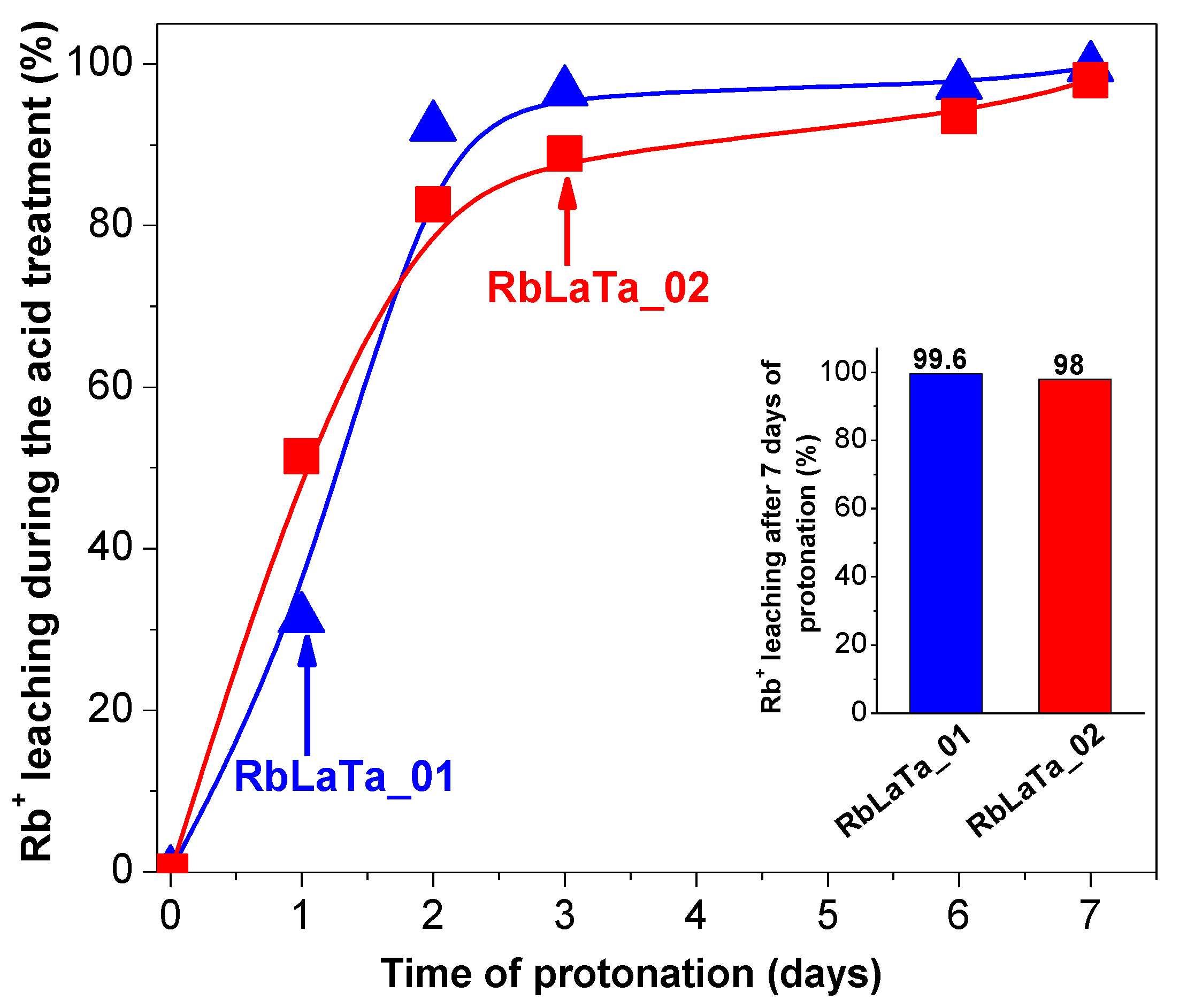
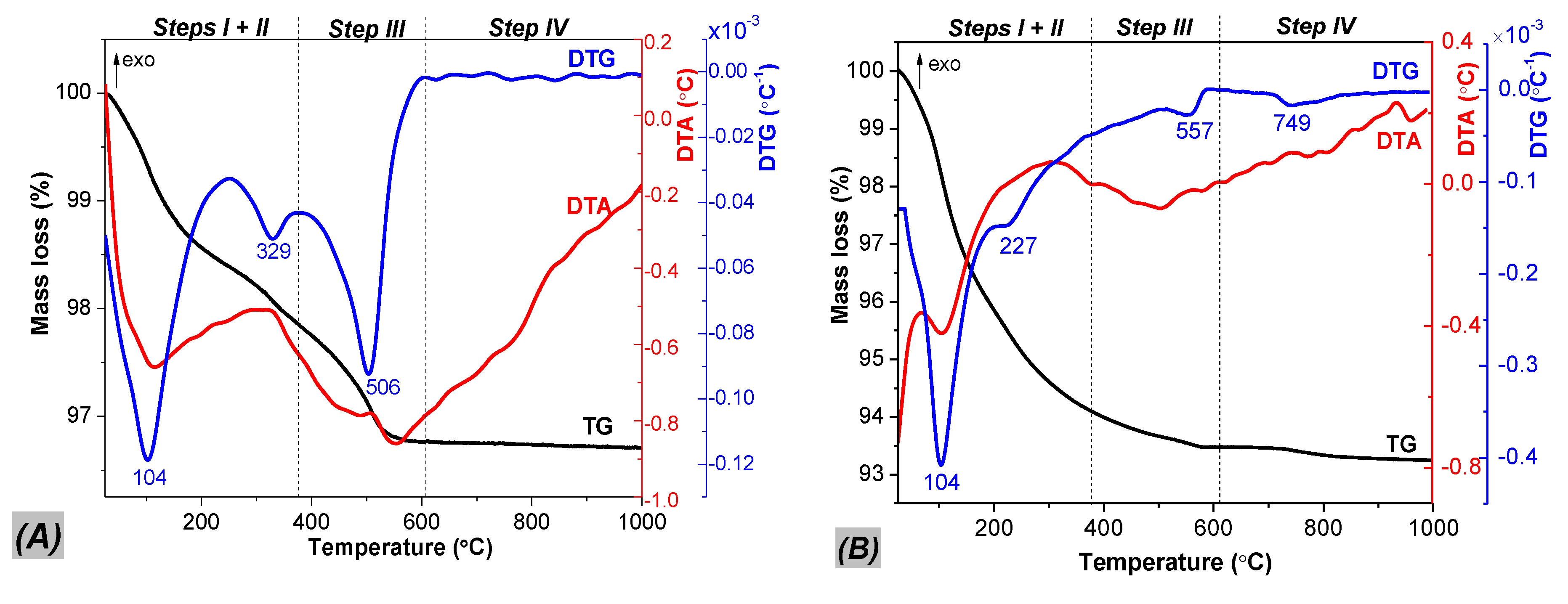
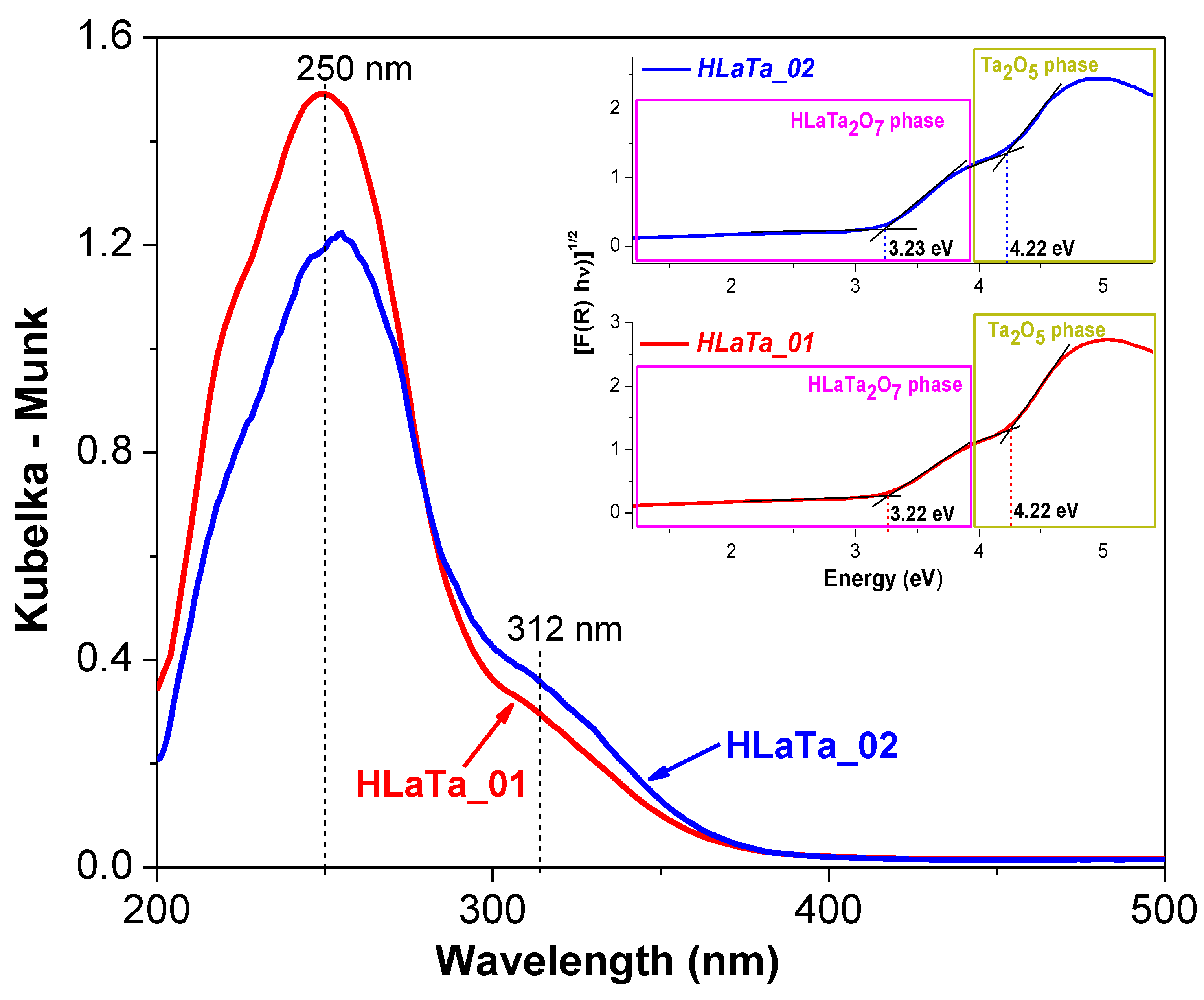
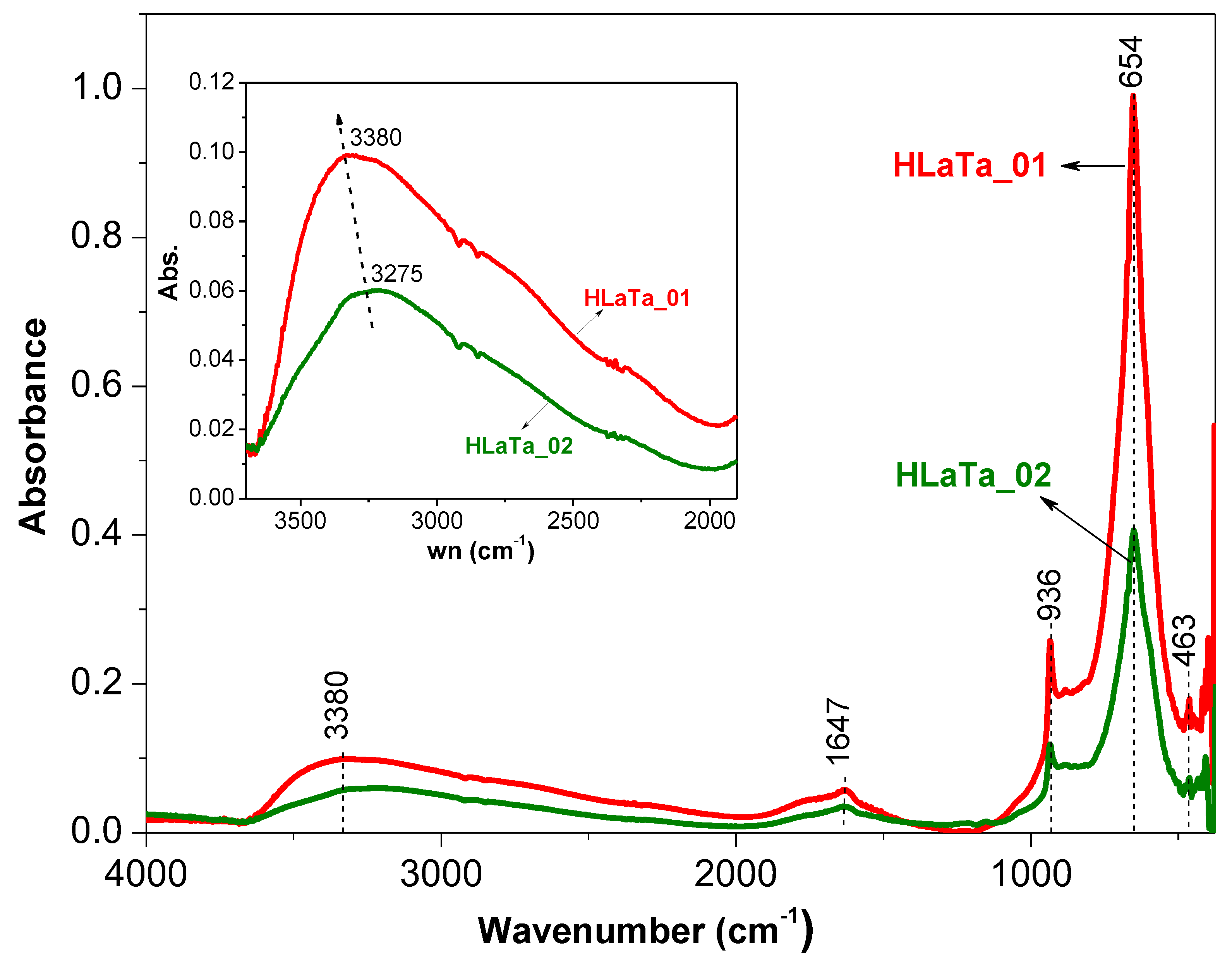
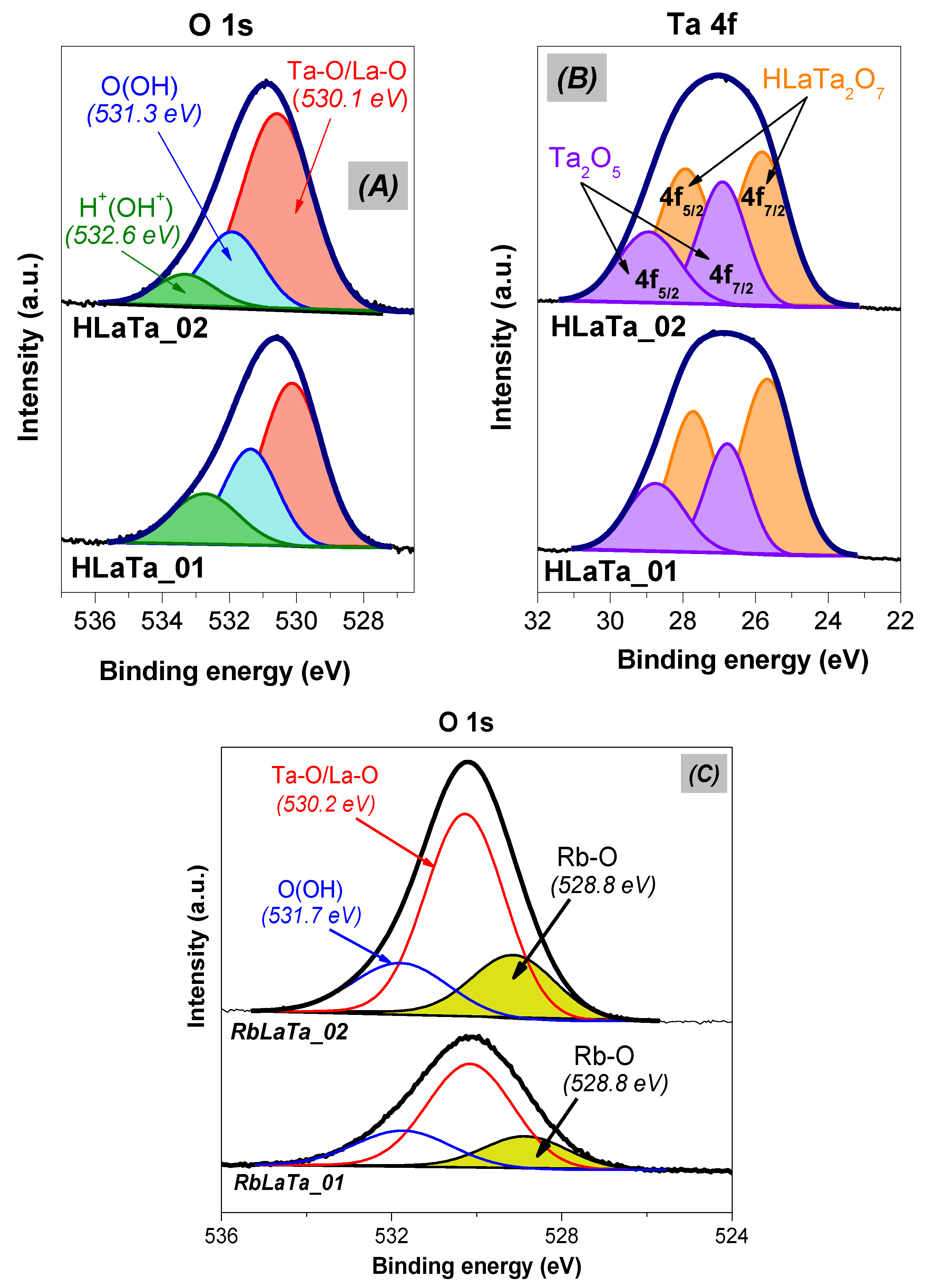
2.1. Phase Composition, Morphology and Structure of Ion Exchanged Layered Perovskites
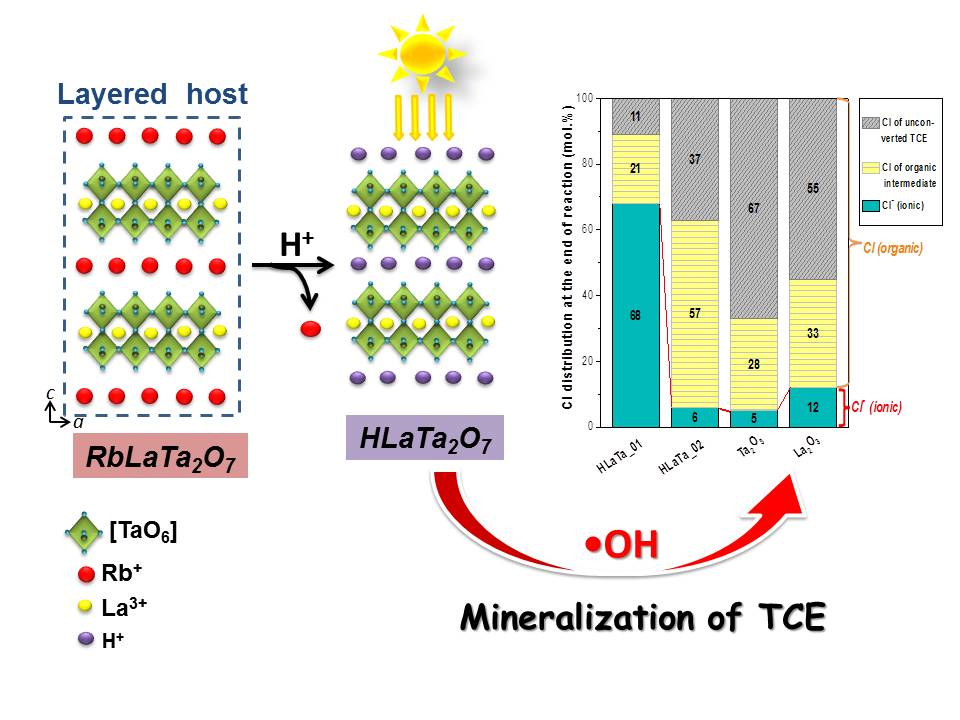
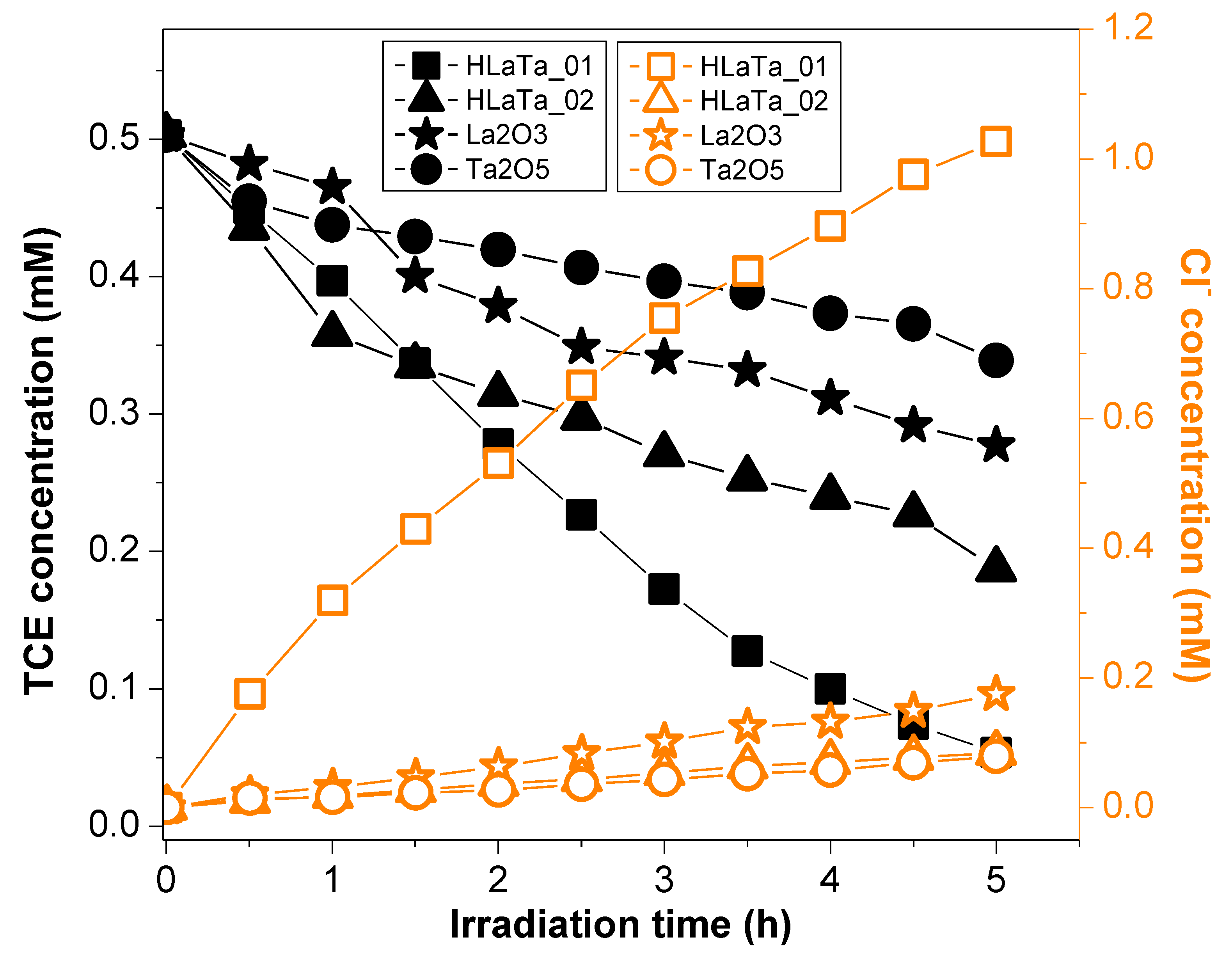
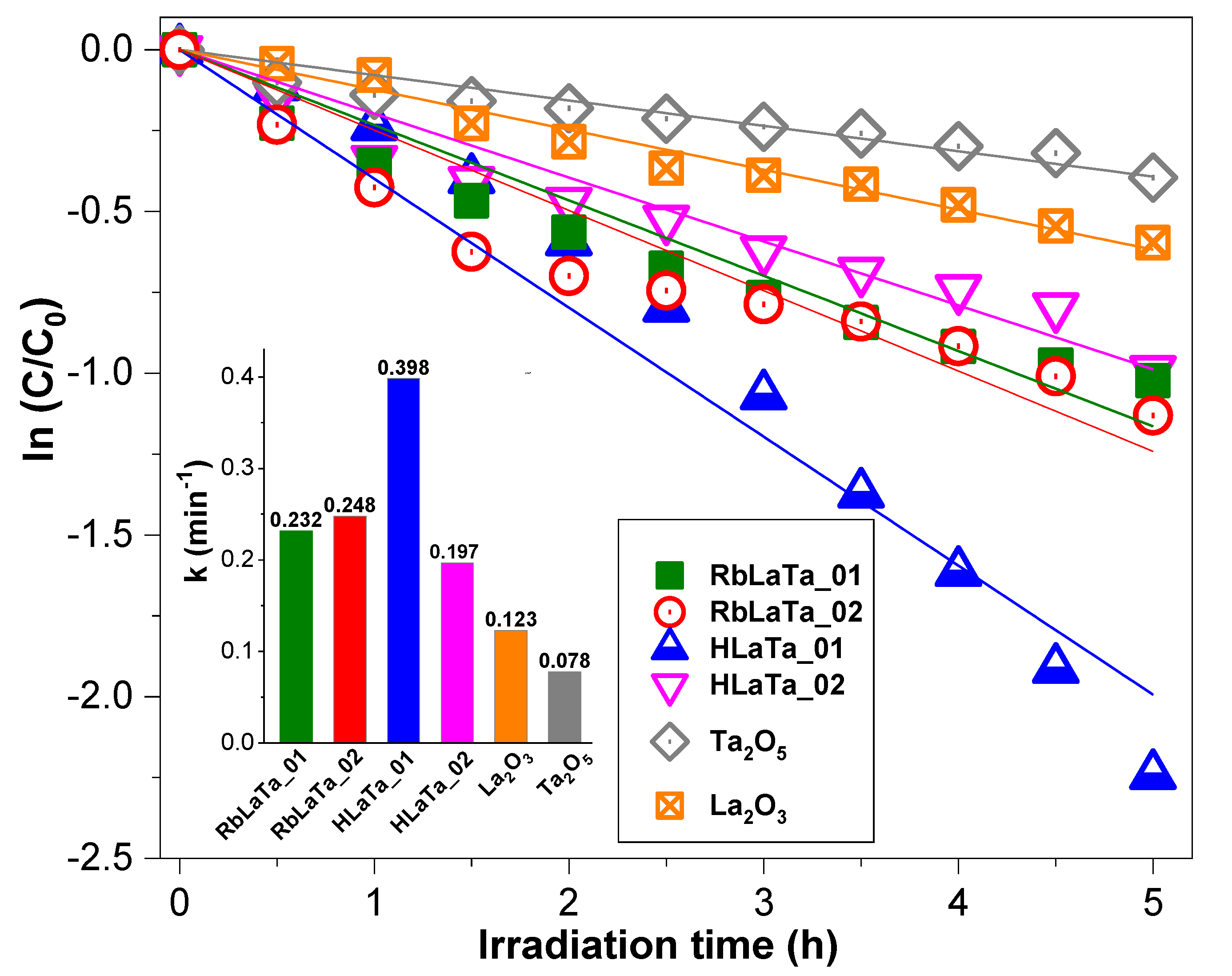
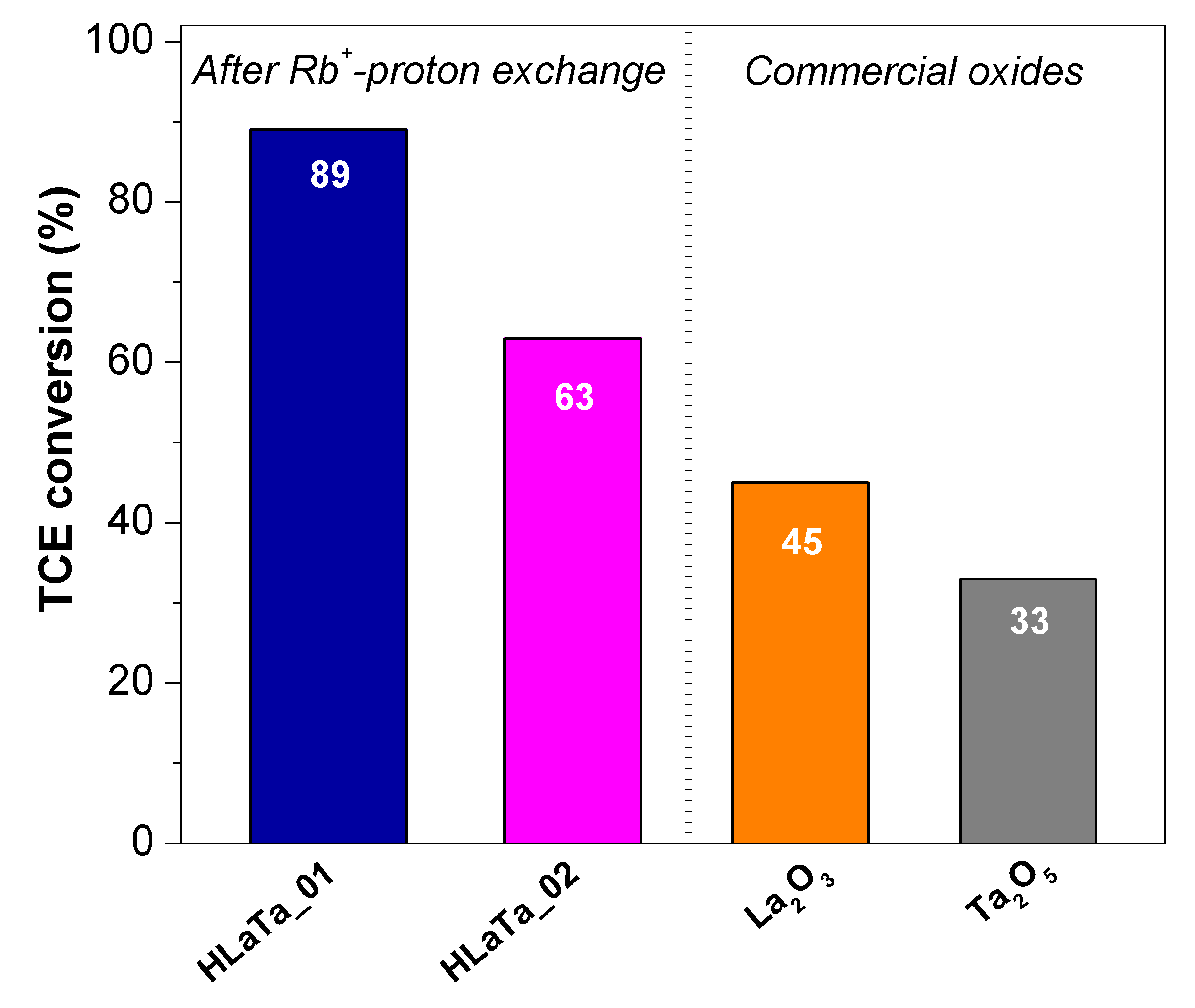
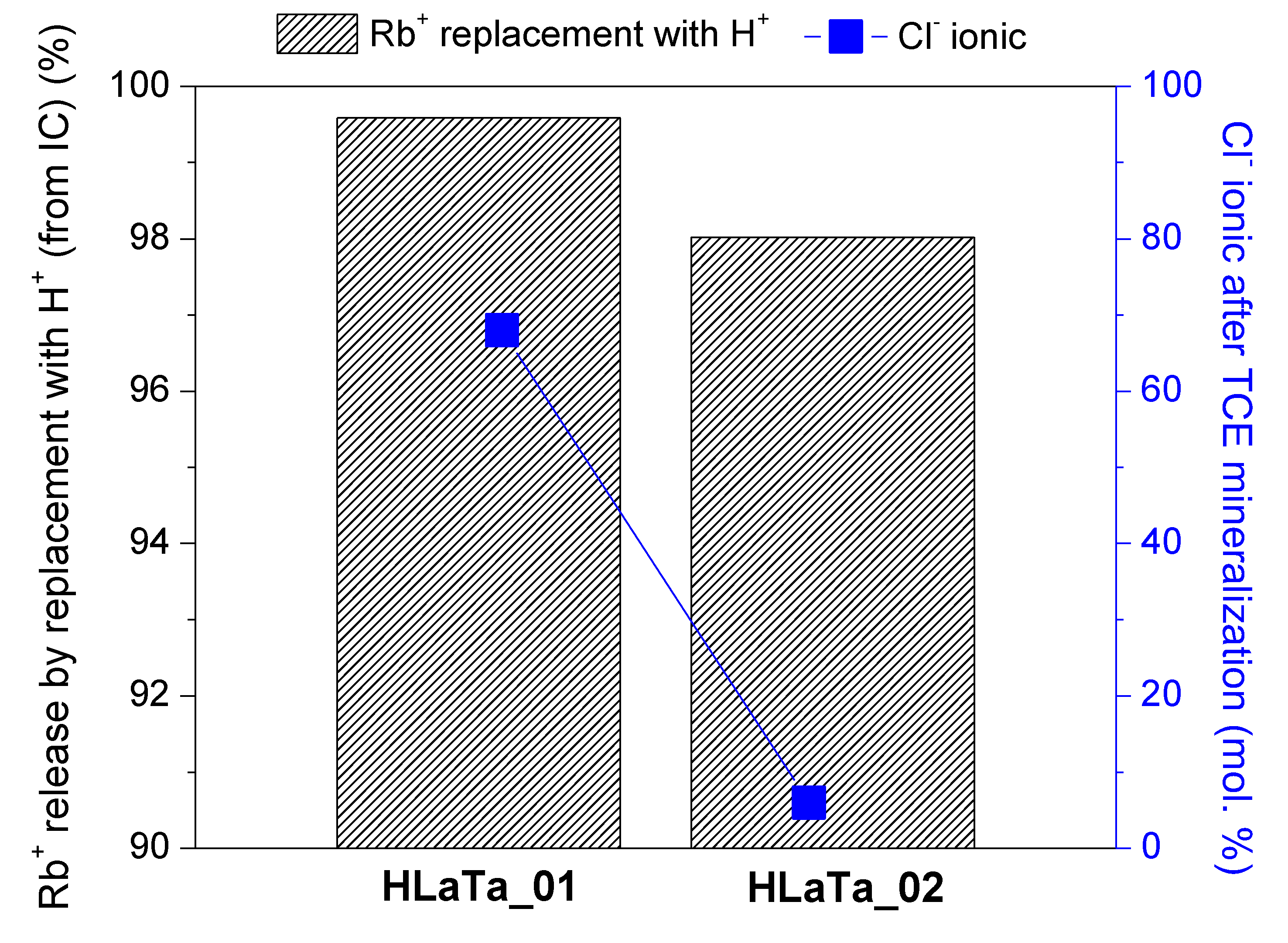
2.2. Photocatalytic Degradation of TCE over Rb+-Proton Exchanged Layered Perovskites
3. Materials and Methods
3.1. Synthesis of RbLaTa2O7 Starting Material
3.2. Preparation of Protonated HLaTa2O7 Compound
3.3. Samples Characterization
3.4. Photocatalytic Degradation of Trichloroethylene (TCE)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Osada, M.; Sasaki, T. Two-dimensional dielectric nanosheets: Novel nanoelectronics from nanocrystal building blocks. Adv. Mater. 2012, 24, 210–228. [Google Scholar] [CrossRef] [PubMed]
- Compton, O.C.; Osterloh, F.E. Niobate nanosheets as catalysts for photochemical water splitting into hydrogen and hydrogen peroxide. J. Phys. Chem. C 2009, 113, 479–485. [Google Scholar] [CrossRef] [Green Version]
- Rodionov, I.A.; Zvereva, I.A. Photocatalytic activity of layered perovskite-like oxides in practically valuable chemical reactions. Russ. Chem. Rev. 2016, 85, 248–279. [Google Scholar] [CrossRef]
- Pena, M.A.; Fierro, J.L.G. Chemical structures and performance of perovskite oxides. Chem. Rev. 2001, 101, 1981–2017. [Google Scholar] [CrossRef]
- Liu, G.; Zhen, C.; Kang, Y.; Wang, L.; Cheng, H.M. Unique physicochemical properties of two-dimensional light absorbers faciliating photocatalysis. Chem. Soc. Rev. 2018, 47, 6410–6444. [Google Scholar] [CrossRef]
- Tahara, S.; Takeda, Y.; Sugahara, Y. Preparation of organic−inorganic hybrids possessing nanosheets with perovskite-related structures via exfoliation during a sol−gel process. Chem. Mater. 2005, 17, 6198–6204. [Google Scholar] [CrossRef]
- Cattaneo, A.S.; Ferrara, C.; Marculescu, A.M.; Giannici, F.; Martorana, A.; Mustarelli, P.; Tealdi, C. Solid-state NMR characterization of the structure and thermal stability of hybrid organic–inorganic compounds based on a HLaNb2O7 Dion–Jacobson layered perovskite. Phys. Chem. Chem. Phys. 2016, 18, 21903–21912. [Google Scholar] [CrossRef] [Green Version]
- Raciulete, M.; Papa, F.; Kawamoto, D.; Munteanu, C.; Culita, D.C.; Atkinson, I.; Negrila, C.; Bratan, V.; Pandele-Cusu, J.; Balint, I. Particularities of trichloroethylene photocatalytic degradation over crystalline RbLaTa2O7 nanowire bundles grown by solid-state synthesis route. J. Environ. Chem. Eng. 2019, 7, 102789. [Google Scholar] [CrossRef]
- Tsunoda, Y.; Sugimoto, W.; Sugahara, Y. Intercalation behavior of n-alkylamines into a protonated form of a layered perovskite derived from Aurivillius phase Bi2SrTa2O9. Chem. Mater. 2003, 15, 632–635. [Google Scholar] [CrossRef]
- Thomas, C.I.; Karppinen, M. Intercalation of primary alcohols into layered titanoniobates. Inorg. Chem. 2017, 56, 9132–9138. [Google Scholar] [CrossRef] [Green Version]
- Sugaya, T.; Ozaki, M.; Guegan, R.; Idota, N.; Sugahara, Y. Surface modification of layered perovskite nanosheets with a phosphorus coupling reagent in a biphasic system. Langmuir 2019, 35, 6594–6601. [Google Scholar] [CrossRef] [PubMed]
- Silyukov, O.I.; Kurnosenko, S.A.; Zvereva, I.A. Intercalation of methylamine into the protonated forms of layered perovskite-like oxides HLnTiO4 (Ln = La and Nd). Glass Phys. Chem. 2018, 44, 428–432. [Google Scholar] [CrossRef]
- Ma, R.; Sasaki, T. Nanosheets of oxides and hydroxides: Ultimate 2D charge-bearing functional crystallites. Adv. Mater. 2010, 22, 5082–5104. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Watanabe, M. Osmotic swelling to exfoliation. Exceptionally high degrees of hydration of a layered titanate. J. Am. Chem. Soc. 1998, 120, 4682–4689. [Google Scholar] [CrossRef]
- Wang, T.H.; Draskovic, T.I.; Henderson, C.N.; Mallouk, T.E. Synthesis, exfoliation, and electronic/protonic conductivity of the Dion-Jacobson phase layer perovskite HLa2Ti2TaO10∙nH2O. Chem. Mater. 2014, 26, 898–906. [Google Scholar]
- Takeda, Y.; Momma, T.; Osaka, T.; Kuroda, K.; Sugahara, Y. Organic derivatives of the layered perovskite HLaNb2O7∙xH2O with polyether chains on the interlayer surface: Characterization, intercalation of LiClO4 and ionic conductivity. J. Mater. Chem. 2008, 18, 3581–3587. [Google Scholar] [CrossRef]
- Parida, R.K.; Pattanayak, D.K.; Mohanty, B.; Parida, B.N. Optical and transport properties of double perovskite strontium bismuth vanadate. J. Mol. Struct. 2020, 1205, 127607. [Google Scholar] [CrossRef]
- Qiu, X.; Miyauchi, M.; Sunada, K.; Minoshima, M.; Liu, M.; Lu, Y.; Li, D.; Shimodaira, Y.; Hosogi, Y.; Kuroda, Y.; et al. Hybrid CuxO/TiO2 nanocomposites as risk-reduction materials in indoor environments. ACS Nano 2012, 6, 1609–1618. [Google Scholar] [CrossRef]
- Raciulete, M.; Kachina, A.; Puzenat, E.; Afanasiev, P. Preparation of nanodispersed titania using stabilized ammonium nitrate melts. J. Solid State Chem. 2010, 183, 2438–2444. [Google Scholar] [CrossRef]
- Huang, J.; Ma, R.; Ebina, Y.; Fukuda, K.; Takada, K.; Sasaki, T. Layer-by-layer assembly of TaO3 nanosheet/polycationcomposite nanostructures: Multilayer film, hollow sphere, and its photocatalytic activity for hydrogen evolution. Chem. Mater. 2010, 22, 2582–2587. [Google Scholar] [CrossRef]
- Raciulete, M.; Papa, F.; Culita, D.C.; Munteanu, C.; Atkinson, I.; Bratan, V.; Pandele-Cusu, J.; State, R.; Balint, I. Impact of RbLaTa2O7 layered perovskite synthesis conditions on their activity for photocatalytic abatement of trichloroethylene. Rev. Roum. Chim. 2018, 63, 821–828. [Google Scholar]
- Gadea, R.; Ahemed, J.; Yanapu, K.L.; Abate, S.Y.; Tao, Y.T.; Pola, S. Photodegradation of organic dyes and industrial wastewater in the presence of layer-type perovskite materials under visible light irradiation. J. Environ. Chem. Eng. 2018, 6, 4504–4513. [Google Scholar] [CrossRef]
- Ren, H.; Yu, S.; Chao, L.; Xia, Y.; Sun, Y.; Zuo, S.; Li, F.; Niu, T.; Yang, Y.; Ju, H.; et al. Efficient and stable Ruddlesden–Popper perovskite solar cell with tailored interlayer molecular interaction. Nat. Photonics 2020, 14, 154–163. [Google Scholar] [CrossRef]
- Huang, Y.; Wei, Y.; Fan, L.; Huang, M.; Lin, J.; Wu, J. Photocatalytic activities of HLaNb2O7 prepared by polymerized complex method. Int. J. Hydrogen Energy 2009, 34, 5318–5325. [Google Scholar] [CrossRef]
- Machida, M.; Yabunaka, J.; Kijima, T. Synthesis and photocatalytic property of layered perovskite tantalates, RbLnTa2O7 (Ln = La, Pr, Nd, and Sm). Chem. Mater. 2000, 12, 812–817. [Google Scholar] [CrossRef]
- Machida, M.; Miyazaki, K.; Matsushima, S.; Arai, M. Photocatalytic properties of layered perovskite tantalates, MLnTa2O7 (M = Cs, Rb, Na, and H; Ln = La, Pr, Nd, and Sm). J. Mater. Chem. 2003, 13, 1433–1437. [Google Scholar] [CrossRef]
- Gömpel, D.; Tahir, M.N.; Panthöfer, M.; Mugnaioli, E.; Brandscheid, R.; Kolbb, U.; Tremel, W. Facile Hydrothermal Synthesis of Crystalline Ta2O5 Nanorods, MTaO3 (M = H, Na, K, Rb) Nanoparticles, and Their Photocatalytic Behaviour. J. Mater. Chem. A 2014, 2, 8033–8040. [Google Scholar] [CrossRef]
- Yoshimura, J.; Ebina, Y.; Kondo, J.; Domen, K.; Tanaka, A. Visible-light induced photocatalytic behavior of a layered perovskite type niobate, RbPb2Nb3O10. J. Phys. Chem. 1993, 97, 1970–1973. [Google Scholar] [CrossRef]
- Domen, K.; Yoshimura, J.; Sekine, T.; Tanaka, A.; Onishi, T. A novel series of photocatalysts with an ion-exchangeable layered structure of niobate. Catal. Lett. 1990, 4, 339–343. [Google Scholar] [CrossRef]
- Palacin, M.R.; Lira, M.; Garcia, J.L.; Caldes, M.T.; Casan-Pastor, N.; Fuertes, A.; Gomez-Romero, P. Synthesis, deintercalation and transport properties of a mixed-valence derivate of the layered oxide HLaNb2O7. Mater. Res. Bull. 1996, 31, 217–225. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chaleogenides. Acta Crystallogr. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Rodionov, I.A.; Maksimova, E.A.; Pozhidaev, A.Y.; Kurnosenko, S.A.; Silyukov, O.I.; Zvereva, I.A. Layered titanate H2Nd2Ti3O10 Intercalated with n-Butylamine: A New Highly Efficient Hybrid Photocatalyst for Hydrogen Production from Aqueous Solutions of Alcohols. Front. Chem. 2019, 7, 863. [Google Scholar] [CrossRef] [PubMed]
- Osada, M.; Sasaki, T. Nanoarchitectonics in dielectric/ferroelectric layered perovskites: From bulk 3D systems to 2D nanosheets. Dalton Trans. 2018, 47, 2841–2851. [Google Scholar] [CrossRef] [PubMed]
- Tahara, S.; Shimada, A.; Kumada, N.; Sugahara, Y. Characterization of Bi5Nb3O15 by refinement of neutron diffraction pattern, acid treatment and reaction of the acid-treated product with n-alkylamines. J. Solid State Chem. 2007, 180, 2517–2524. [Google Scholar] [CrossRef]
- Colomban, P.; Zaafrani, O.; Slodczyk, A. Proton content and nature in perovskite ceramic membranes for medium temperature fuel cells and electrolysers. Membranes 2012, 2, 493–509. [Google Scholar] [CrossRef] [Green Version]
- Bhuvanesh, N.S.P.; Crosnier-Lopez, M.P.; Duroy, H.; Fourquet, J.L. Synthesis, characterization and dehydration study of H2A0.5nBnO3n+1xH2O (n= 2 and 3, A= Ca, Sr and B= Nb, Ta) compounds obtained by ion-exchange from the layered Li2A0.5nBnO3n+1 perovskite materials. J. Mater. Chem. 2000, 10, 1685–1692. [Google Scholar] [CrossRef]
- Utkina, T.; Chislov, M.; Silyukov, O.; Burovikhina, A.; Zvereva, I.A. TG and DSC investigation of water intercalation and protonation processes in perovskite-like layered structure of titanate K2Nd2Ti3O10. J. Therm. Anal. Calorim. 2016, 125, 281–287. [Google Scholar] [CrossRef]
- Hermann, A.T.; Wiley, J.B. Thermal stability of Dion–Jacobson mixed-metal-niobate double-layered perovskites. Mater. Res. Bull. 2009, 44, 1046–1050. [Google Scholar] [CrossRef]
- Silyukov, O.; Chislov, M.; Burovikhina, A.; Utkina, T.; Zvereva, I. Thermogravimetry study of ion exchange and hydration in layered oxide materials. J. Therm. Anal. Calorim. 2012, 110, 187–192. [Google Scholar] [CrossRef]
- Nishimoto, S.; Matsuda, M.; Miyake, M. Novel protonated and hydrated n = 1 Ruddlesden–Popper phases, HxNa1−xLaTiO4∙yH2O, formed by ion-exchange/intercalation reaction. J. Solid State Chem. 2005, 178, 811–818. [Google Scholar] [CrossRef]
- Suram, S.K.; Newhouse, P.F.; Gregoire, J.M. High throughput light absorber discovery, part 1: An algorithm for automated Tauc analysis. ACS Comb. Sci. 2016, 18, 673–681. [Google Scholar] [CrossRef]
- Lee, J.; Lu, W.; Kioupakis, E. Electronic properties of tantalum pentoxide polymorphs from first-principles calculations. Appl. Phys. Lett. 2014, 105, 202108. [Google Scholar] [CrossRef]
- Baqe, A.A.; Matori, K.A.; Al-Hada, N.M.; Shaari, A.H.; Kamari, H.M.; Saion, E.; Chyi, J.L.Y.; Abdullah, C.A.C. Synthesis and characterization of binary (CuO)0.6(CeO2)0.4 nanoparticles via a simple heat treatment method. Res. Phys. 2018, 9, 471–478. [Google Scholar]
- Wang, Y.; Wang, C.; Wang, L.; Hao, Q.; Zhu, X.; Chen, X.; Tang, K. Preparation of interlayer surface tailored protonated double-layered perovskite H2CaTa2O7 with n-alcohols and their photocatalytic activity. RSC Adv. 2014, 4, 4047–4054. [Google Scholar] [CrossRef]
- Coates, J. Interpretation of Infrared Spectra, a Practical Approach. In Encyclopedia of Analytical Chemistry; Meyers, R.A., Ed.; John Wiley & Sons Ltd.: Chichester, UK, 2000. [Google Scholar]
- Sasaki, T.; Kooli, F.; Iida, M.; Michiue, Y.; Takenouchi, S.; Yajima, Y.; Izumi, F.; Chakoumakos, B.C.; Watanabe, M. A mixed alkali metal titanate with the lepidocrocite-like layered structure. Preparation, crystal structure, protonic form, and acid-base intercalation properties. Chem. Mater. 1998, 10, 4123–4128. [Google Scholar] [CrossRef]
- Yuan, H.; Besselink, R.; Liao, Z.; ten Elshof, J.E. The swelling transition of lepidocrocite-type protonated layered titanates into anatase under hydrothermal treatment. Sci. Rep. 2015, 4, 4584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahalingam, T.; Selvakumar, C.; Ranjith Kumar, E.; Venkatachalam, T. Structural, optical, morphological and thermal properties of TiO2–Al and TiO2–Al2O3 composite powders by ball milling. Phys. Lett. A 2017, 381, 1815–1819. [Google Scholar] [CrossRef]
- Wang, Y.; Zhu, D.; Xu, X. Zr-doped mesoporous Ta3N5 microspheres for efficient photocatalytic water oxidation. ACS Appl. Mater. Interfaces 2016, 8, 35407–35418. [Google Scholar] [CrossRef]
- Cordero-Garcia, A.; Turnes Palomino, G.; Hinojosa-Reyes, L.; Guzmán-Mar, J.L.; Maya-Tevino, L.; Hernandez-Ramirez, A. Photocatalytic behaviour of WO3/TiO2-N for diclofenac degradation using simulated solar radiation as an activation source. Environ. Sci. Pollut. Res. 2017, 24, 4613–4624. [Google Scholar] [CrossRef]
- Loh, J.Y.Y.; Kherani, N.P. X-ray photospectroscopy and electronic studies of reactor parameters on photocatalytic hydrogenation of carbon dioxide by defect-laden indium oxide hydroxide nanorods. Molecules 2019, 24, 3818. [Google Scholar] [CrossRef] [Green Version]
- Joo, J.C.; Ahn, C.H.; Jang, D.G.; Yoon, Y.H.; Kim, K.J.; Campos, L.; Ahn, H. Photocatalytic degradation of trichloroethylene in aqueous phase using nano-ZNO/laponite composites. J. Hazard. Mater. 2013, 263, 569–574. [Google Scholar] [CrossRef]
- Awaludin, Z.; Safuan, M.; Okajima, T.; Ohsaka, T. Investigating the physical and electrochemical effects of cathodic polarization treatment on TaOx. J. Mater. Chem. A 2015, 3, 16791–16800. [Google Scholar] [CrossRef]
- Sethulakshmi, N.; Unnimaya, A.N.; Al-Omari, I.A.; Al-Harthi, S.; Sagar, S.; Thomas, S.; Srinivasan, G.; Anantharaman, M.R. On magnetic ordering in heavily sodium substituted hole doped lanthanum manganites. J. Magn. Magn. Mater. 2015, 391, 75–82. [Google Scholar] [CrossRef]
- Min, X.; Fangn, M.; Huang, Z.; Liu, Y.; Tang, C.; Qian, T.; Wu, X. Synthesis and luminescence properties of nitrided lanthanum magnesium hexaluminate LaMgAl11O19 phosphors. Ceram. Int. 2014, 40, 4535–4539. [Google Scholar] [CrossRef]
- Huang, B.; Lei, C.; Wei, C.; Zeng, G. Chlorinated volatile organic compounds (Cl-VOCs) in environment-sources, potential human health impacts, and current remediation technologies. Environ. Int. 2014, 71, 118–138. [Google Scholar] [CrossRef]
- Lee, H.I.; Kim, J.H.; Lee, H.S.; Lee, W.D. Purification of toxic compounds in water and treatment of polymeric materials. In Environmentally Benign Photocatalysts: Nanostructure Science and Technology; Anpo, M., Kamat, P.V., Eds.; Springer: New York, NY, USA, 2010; p. 350. ISBN 978-0-387-48441-9. [Google Scholar]
- Pruden, A.L.; Ollis, D.F. Photoassisted heterogeneous catalysis: The degradation of trichloroethyelene in water. J. Catal. 1983, 82, 404–417. [Google Scholar] [CrossRef]
- Phillips, L.A.; Raupp, G.B. Infrared spectroscopic investigation of gas solid heterogeneous photocatalytic oxidation of trichloroethylene. J. Mol. Catal. 1992, 77, 297–311. [Google Scholar] [CrossRef]
- Fan, J.; Yates, J.T. Mechanism of photooxidation of trichloroethylene on TiO2: Detection of intermediates by infrared spectroscopy. J. Am. Chem. Soc. 1996, 118, 4686–4692. [Google Scholar] [CrossRef]
- Reza, K.M.; Kurny, A.; Gulshan, F. Parameters affecting the photocatalytic degradation of dyes using TiO2: A review. Appl. Water Sci. 2017, 7, 1569–1578. [Google Scholar] [CrossRef] [Green Version]
- Kopidakis, N.; Benkstein, K.D.; van de Lagemaat, J.; Frank, A.J. Temperature dependence of the electron diffusion coefficient in electrolyte-filled TiO2 nanoparticle films: Evidence against multiple trapping in exponential conduction-band tails. Phys. Rev. B 2006, 73, 045326. [Google Scholar] [CrossRef] [Green Version]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Reaction Temperature /Synthesis Time | D(102) 1 (nm) | Lattice Parameters (nm) | Interlayer Spacing 2 (nm) | [Ref.] |
|---|---|---|---|---|---|
| HLaTa_01 | Protonation at RT */7 days | 20.1 | a = 0.388 c = 1.076 | 1.092 | this work |
| HLaTa_02 | Protonation at RT */7 days | 19.2 | a = 0.388 c =1.077 | 1.097 | this work |
| RbLaTa_01 | 1200 °C/18 h | 18.3 | a = 0.387 c = 1.104 | 1.100 | [8] |
| RbLaTa_02 | 950 and 1200 °C/36 h | 18.2 | a = 0.388 c = 1.112 | 1.116 | [8] |
| Catalyst | Mass Loss (%) | Amount of H2O per Unit Formula (Moles) Steps I + II | Protonation Degree Calculated from Step III (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Step I 25–250 °C | Step II 250–380 °C | Steps I + II 25–380 °C | Step III 380–610 °C | Step IV 610–1000 °C | |||||
| th. | exp. | th. | exp. | ||||||
| HLaTa_01 | 1.61 (max. at 104 °C) | 0.55 (max. at 329 °C) | 2.80 | 2.16 | 1.46 | 1.07 (max. at 506 °C) | traces | 1 | 73 |
| HLaTa_02 | 4.11 (max. at 104 °C) | 1.73 (max. at 227 °C) | 4.90 | 5.84 | 1.46 | 0.67 (max. at 557 °C) | 0.22 (max. at 749 °C) | 2 | 46 |
| Catalyst | O (at. %) | La (at. %) | Ta (at. %) | La/Ta | Proportion of Phase Composition after Protonation (%) 1 | Amount of –OH Calculated from FTIR (μmol·g−1) 2 | SSA (m2·g−1) | |
|---|---|---|---|---|---|---|---|---|
| HLaTa2O7 | Ta2O5 | |||||||
| HLaTa_01 | 71.9 | 4.1 | 24.2 | 0.20 | 66 | 34 | 2876 | 8 |
| HLaTa_02 | 73.7 | 3.8 | 22.5 | 0.20 | 61 | 39 | 2198 | 6 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raciulete, M.; Papa, F.; Negrila, C.; Bratan, V.; Munteanu, C.; Pandele-Cusu, J.; Culita, D.C.; Atkinson, I.; Balint, I. Strategy for Modifying Layered Perovskites toward Efficient Solar Light-Driven Photocatalysts for Removal of Chlorinated Pollutants. Catalysts 2020, 10, 637. https://doi.org/10.3390/catal10060637
Raciulete M, Papa F, Negrila C, Bratan V, Munteanu C, Pandele-Cusu J, Culita DC, Atkinson I, Balint I. Strategy for Modifying Layered Perovskites toward Efficient Solar Light-Driven Photocatalysts for Removal of Chlorinated Pollutants. Catalysts. 2020; 10(6):637. https://doi.org/10.3390/catal10060637
Chicago/Turabian StyleRaciulete, Monica, Florica Papa, Catalin Negrila, Veronica Bratan, Cornel Munteanu, Jeanina Pandele-Cusu, Daniela C. Culita, Irina Atkinson, and Ioan Balint. 2020. "Strategy for Modifying Layered Perovskites toward Efficient Solar Light-Driven Photocatalysts for Removal of Chlorinated Pollutants" Catalysts 10, no. 6: 637. https://doi.org/10.3390/catal10060637