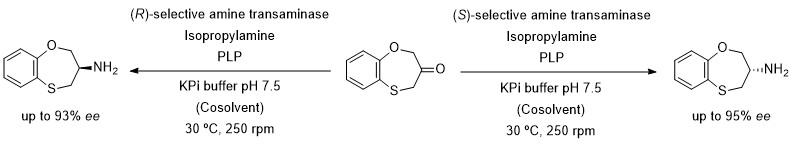
Development of Biotransamination Reactions towards the 3,4-Dihydro-2H-1,5-benzoxathiepin-3-amine Enantiomers

 and
and
Abstract
:
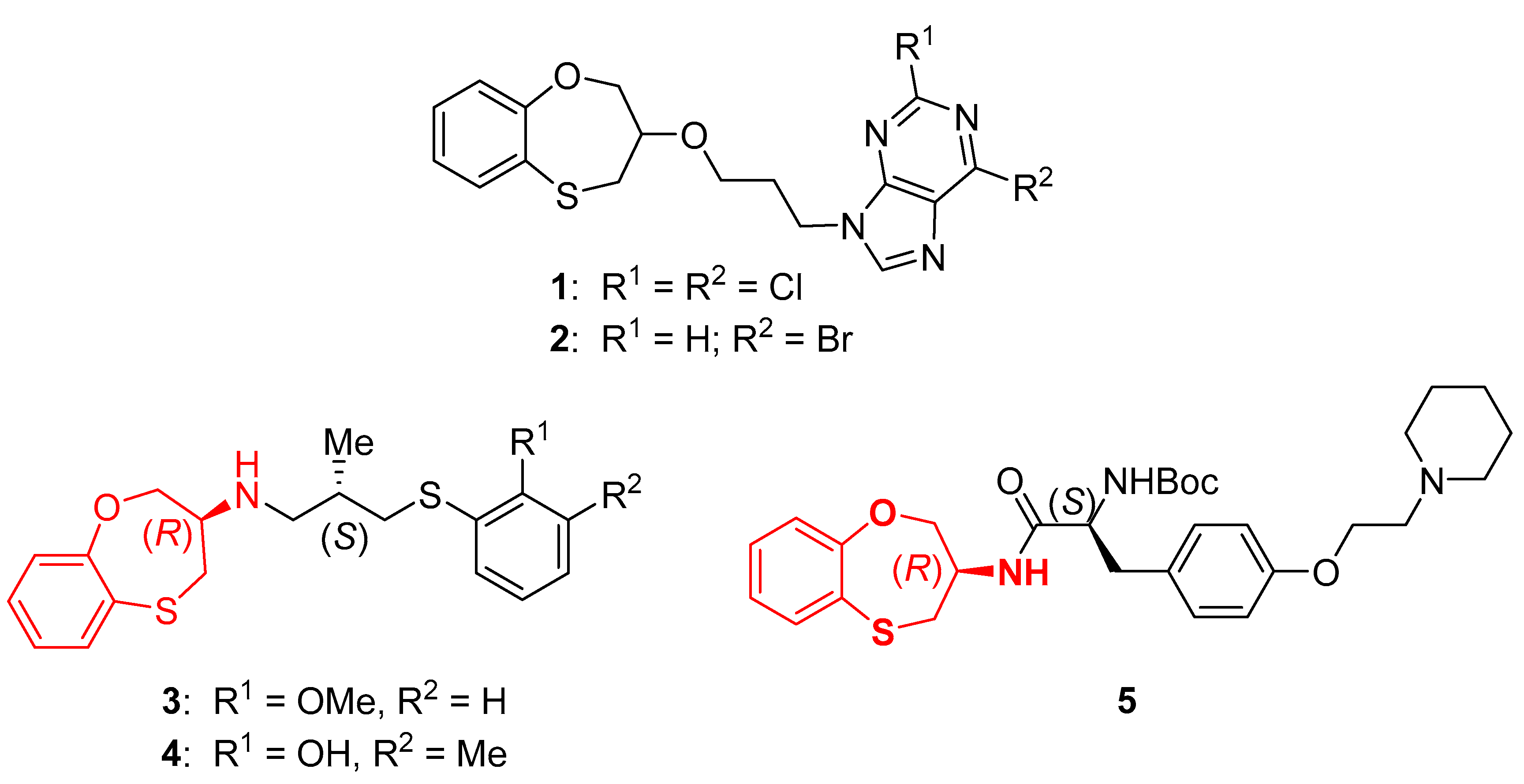
1. Introduction
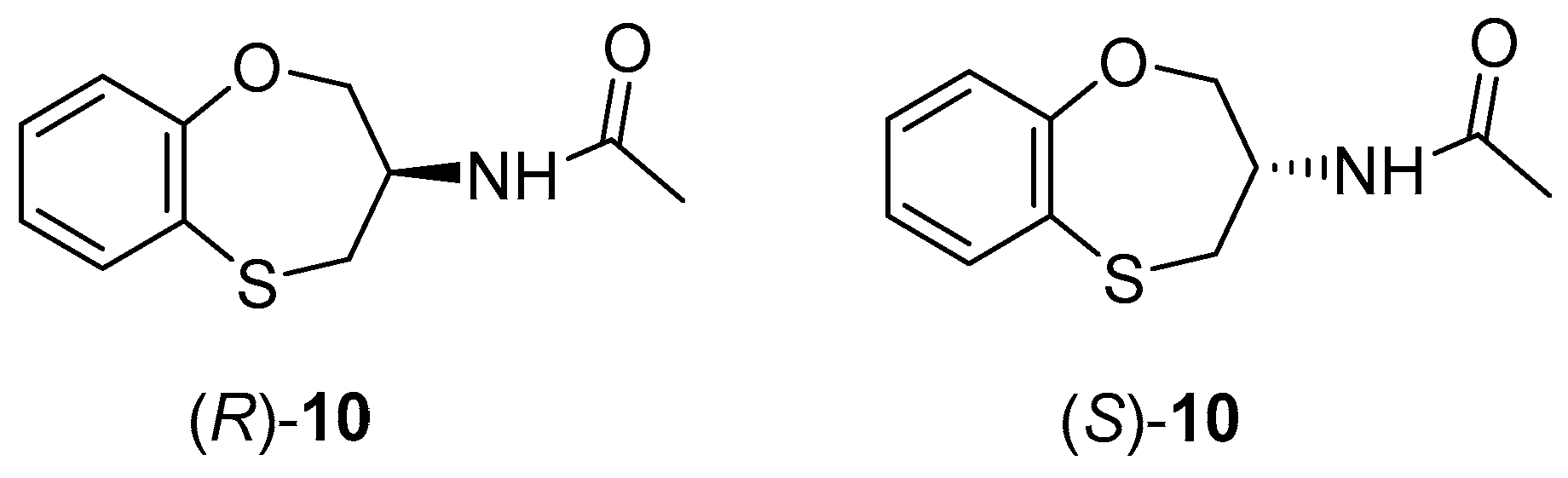
2. Results and Discussion
3. Materials and Methods
3.1. General Materials and Methods
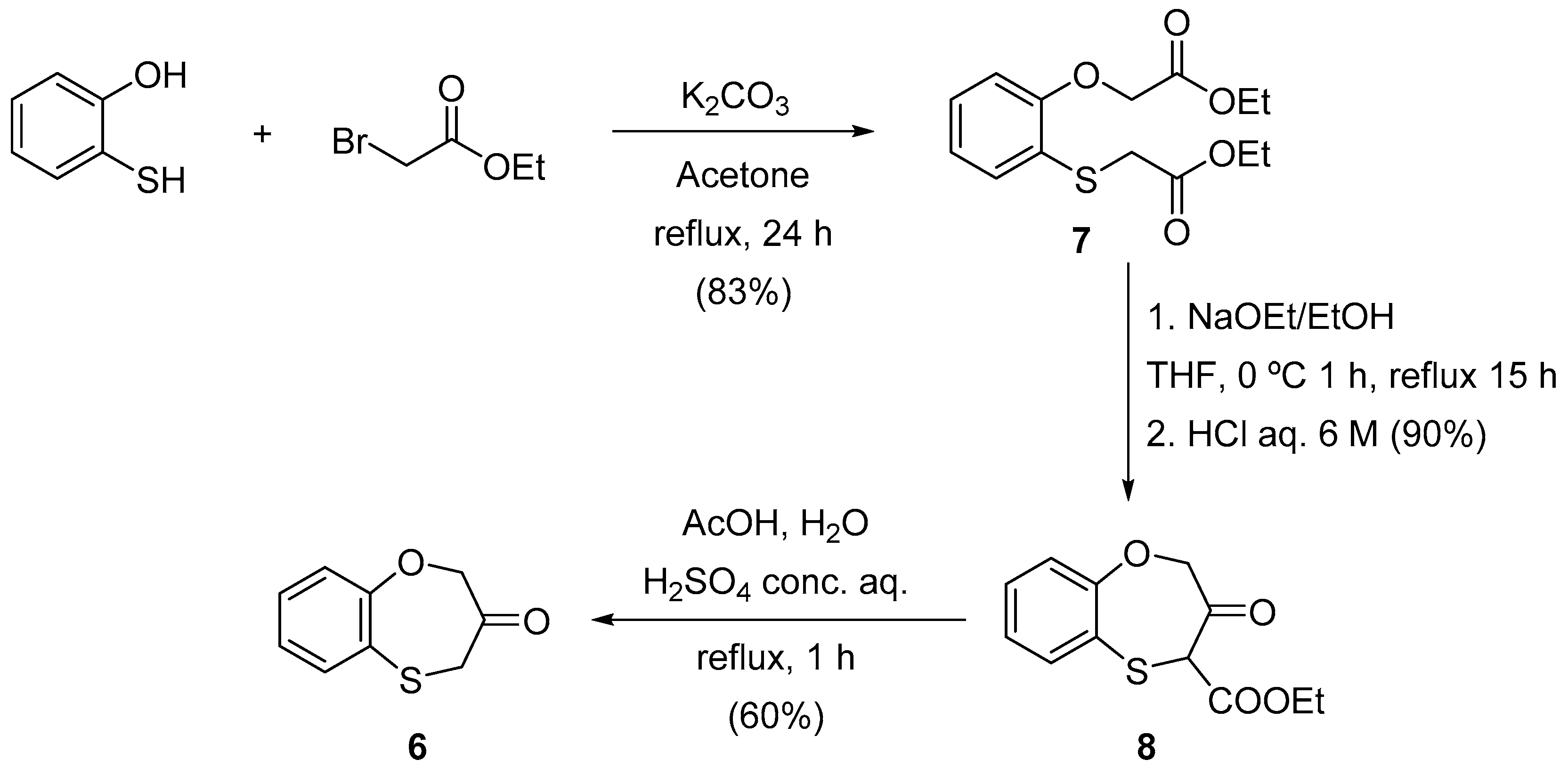
3.2. General Procedure for the Synthesis of Ethyl 2-Ethoxycarbonylmethylthiophenoxyacetate (7)
3.3. General Procedure for the Synthesis of Ethyl 3-Oxo-3,4-dihydro-2H-1,5-benzoxathiepin-4-carboxylate (8)
3.4. General Procedure for the Synthesis 3,4-Dihydro-2H-1,5-benzoxathiepin-3-one (6)
3.5. General Procedure for the Biotransamination of 6 Using ATAs Overexpressed in Escherichia coli
3.6. General Procedure for the Biotransamination of 6 Using Commercial ATAs
3.7. Preparative Biotransamination of 6 under Optimised Conditions
3.8. Computational Methods
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kimatrai, M.; Conejo-García, A.; Ramírez, A.; Andreolli, E.; García, M.Á.; Aránega, A.; Marchal, J.A.; Campos, J.M. Synthesis and Anticancer Activity of the (R,S)-Benzofused 1,5-Oxathiepin Moiety Tethered to Purines through Alkylidenoxy Linkers. ChemMedChem 2011, 6, 1854–1859. [Google Scholar] [CrossRef] [PubMed]
- Le Grand, B.; Pignier, C.; Létienne, R.; Cuisiat, F.; Rolland, F.; Mas, A.; Vacher, B. Sodium late current blockers in ischemia reperfusion: Is the bullet magic? J. Med. Chem. 2008, 51, 3856–3866. [Google Scholar] [CrossRef] [PubMed]
- Le Grand, B.; Pignier, C.; Létienne, R.; Cuisiat, F.; Rolland, F.; Mas, A.; Borras, M.; Vacher, B. Na+ currents in cardioprotection: Better to be late. J. Med. Chem. 2009, 52, 4149–4160. [Google Scholar] [CrossRef] [PubMed]
- Mahfoudh, N.; Marín-Ramos, N.I.; Gil, A.M.; Jiménez, A.I.; Choquesillo-Lazarte, D.; Kawano, D.F.; Campos, J.M.; Cativiela, C. Cysteine-based 3-substituted 1,5-benzoxathiepin derivatives: Two new classes of anti-proliferative agents. Arab. J. Chem. 2018, 11, 426–441. [Google Scholar] [CrossRef]
- Vacher, V.; Brunel, Y.; Castan, C.F. An Improved Process for the Preparation of Benzoxathiepines and Their Intermediates. FR 2868779 A120051014, 14 October 2005. [Google Scholar]
- Hudlicky, T.; Reed, J.W. Applications of biotransformations and biocatalysis to complexity generation in organic synthesis. Chem. Soc. Rev. 2009, 38, 3117–3132. [Google Scholar] [CrossRef] [PubMed]
- Clouthier, C.M.; Pelletier, J.M. Expanding the organic toolbox: A guide to integrating biocatalysis in synthesis. Chem. Soc. Rev. 2012, 41, 1585–1605. [Google Scholar] [CrossRef] [PubMed]
- Milner, S.E.; Maguire, A.R. Recent trends in whole cell and isolated enzymes in enantioselective synthesis. Arkivoc 2012, 321–382. [Google Scholar]
- Torrelo, G.; Hanefeld, U.; Hollmann, F. Biocatalysis. Catal. Lett. 2015, 145, 309–345. [Google Scholar] [CrossRef]
- Albarrán-Velo, J.; González-Martínez, D.; Gotor-Fernández, V. Stereoselective Biocatalysis. A mature technology for the asymmetric synthesis of pharmaceutical building blocks. Biocatal. Biotransf. 2018, 36, 102–130. [Google Scholar] [CrossRef]
- Höhne, M.; Bornscheuer, U.T. Biocatalytic Routes to Optically Active Amines. ChemCatChem 2009, 1, 42–51. [Google Scholar] [CrossRef]
- Kroutil, W.; Fischereder, E.-M.; Fuchs, C.S.; Lechner, H.; Mutti, F.G.; Pressnitz, D.; Rajagopalan, A.; Sattler, J.H.; Simon, R.C.; Siirola, E. Asymmetric preparation of prim-, sec-, and tert-amines employing selected biocatalysts. Org. Process Res. Dev. 2013, 17, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Kohls, H.; Steffen-Munsberg, F.; Höhne, M. Recent achievements in developing the biocatalytic toolbox for chiral amine synthesis. Curr. Opin. Chem. Biol. 2014, 19, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Schrittwieser, J.H.; Velikogne, S.; Kroutil, W. Biocatalytic imine reduction and reductive amination of ketones. Adv. Synth. Catal. 2015, 357, 1655–1685. [Google Scholar] [CrossRef]
- Gamenara, D.; Domínguez de María, P. Enantioselective imine reduction catalyzed by imine reductases and artificial metalloenzymes. Org. Biomol. Chem. 2014, 12, 2989–2992. [Google Scholar] [CrossRef] [PubMed]
- Grogan, G.; Turner, N.J. InspIRED by nature: NADPH-dependent imine reductases (IREDs) as catalysts for the preparation of chiral amines. Chem. Eur. J. 2016, 22, 1900–1907. [Google Scholar] [CrossRef] [PubMed]
- Mangas-Sánchez, J.; France, S.P.; Montgomery, S.L.; Aleku, G.A.; Man, H.; Sharma, M.; Ramsden, J.I.; Grogan, G.; Turner, N.J. Imine reductases (IREDs). Curr. Opin. Chem. Biol. 2017, 37, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Tufvesson, P.; Lima-Ramos, J.; Jensen, J.S.; Al-Haque, N.; Neto, W.; Woodley, J.M. Process Considerations for the Asymmetric Synthesis of Chiral Amines Using Transaminases. Biotechnol. Bioeng. 2011, 108, 1479–1493. [Google Scholar] [CrossRef] [PubMed]
- Mathew, S.; Yun, H. ω-Transaminases for the production of optically pure amines and unnatural amino acids. ACS Catal. 2012, 2, 993–1001. [Google Scholar] [CrossRef]
- Simon, R.C.; Richter, N.; Busto, E.; Kroutil, W. Recent developments of cascade reactions involving ω-transaminases. ACS Catal. 2014, 4, 129–143. [Google Scholar] [CrossRef]
- Fuchs, M.; Farnberger, J.E.; Kroutil, W. The industrial age of biocatalytic transamination. Eur. J. Org. Chem. 2015, 6965–6982. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Berglund, P. Transaminase biocatalysis: Optimization and application. Green Chem. 2017, 19, 333–360. [Google Scholar] [CrossRef]
- Patil, M.D.; Grogan, G.; Bommarius, A.; Yun, H. Recent advances in ω-transaminase-mediated biocatalysis for the enantioselective synthesis of chiral amines. Catalysts 2018, 8, 254. [Google Scholar] [CrossRef]
- Koszelewski, D.; Lavandera, I.; Clay, D.; Rozzell, D.; Kroutil, W. Asymmetric synthesis of optically pure pharmacologically relevant amines Employing ω-transaminases. Adv. Synth. Catal. 2008, 350, 2761–2766. [Google Scholar] [CrossRef]
- Höhne, M.; Kühl, S.; Robins, K.; Bornscheuer, U.T. Efficient asymmetric synthesis of chiral amines by combining transaminase and pyruvate decarboxylase. ChemBioChem 2008, 9, 363–365. [Google Scholar] [CrossRef] [PubMed]
- Mutti, F.G.; Fuchs, C.S.; Pressnitz, D.; Sattler, J.H.; Kroutil, W. Stereoselectivity of four (R)-selective transaminases for the asymmetric amination of ketones. Adv. Synth. Catal. 2011, 353, 3227–3233. [Google Scholar] [CrossRef]
- Truppo, M.; Janey, J.M.; Grau, B.; Morley, K.; Pollack, S.; Hughes, G.; Davies, I. Asymmetric, biocatalytic labeled compound synthesis using transaminases. Catal. Sci. Technol. 2012, 2, 1556–1559. [Google Scholar] [CrossRef]
- Pressnitz, D.; Fuchs, C.S.; Sattler, J.H.; Knaus, T.; Macheroux, P.; Mutti, F.G.; Kroutil, W. Asymmetric amination of tetralone and chromanone derivatives employing ω-transaminases. ACS Catal. 2013, 3, 555–559. [Google Scholar] [CrossRef]
- Park, E.-S.; Malik, M.S.; Dong, J.-Y.; Shin, J.-S. One-pot production of enantiopure alkylamines and arylalkylamines of opposite chirality catalyzed by ω-transaminase. ChemCatChem 2013, 5, 1734–1738. [Google Scholar] [CrossRef]
- Richter, N.; Simon, R.C.; Lechner, H.; Kroutil, W.; Ward, J.M.; Hailes, H.C. ω-Transaminases for the amination of functionalised cyclic ketones. Org. Biomol. Chem. 2015, 13, 8843–8851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Montero, L.; Gotor, V.; Gotor-Fernández, V.; Lavandera, I. But-2-ene-1,4-diamine and but-2-ene-1,4-diol as donors for thermodynamically favored transaminase- and alcohol dehydrogenase-catalyzed processes. Adv. Synth. Catal. 2016, 358, 1618–1624. [Google Scholar] [CrossRef]
- Gundersen, M.T.; Tufvesson, P.; Rackham, E.J.; Lloyd, R.C.; Woodley, J.M. A rapid selection procedure for simple commercial implementation of ω-transaminase reactions. Org. Process Res. Dev. 2016, 20, 602–608. [Google Scholar] [CrossRef] [Green Version]
- Siirola, E.; Mutti, F.G.; Grischek, B.; Hoefler, S.F.; Fabian, W.M.F.; Grogan, G.; Kroutil, W. Asymmetric synthesis of 3-substituted cyclohexylamine derivatives from prochiral diketones via three biocatalytic steps. Adv. Synth. Catal. 2013, 355, 1703–1708. [Google Scholar] [CrossRef]
- Tauber, K.; Fuchs, M.; Sattler, J.H.; Pitzer, J.; Pressnitz, D.; Koszelewski, D.; Faber, K.; Pfeffer, J.; Haas, T.; Kroutil, W. Artificial multi-enzyme networks for the asymmetric amination of sec- alcohols. Chem. Eur. J. 2013, 19, 4030–4035. [Google Scholar] [CrossRef] [PubMed]
- Skalden, L.; Peters, C.; Dickerhoff, J.; Nobili, A.; Joosten, H.-J.; Weisz, K.; Höhne, M.; Bornscheuer, U.T. Two subtle amino acid changes in a transaminase substantially enhance or invert enantiopreference in cascade syntheses. ChemBioChem 2015, 15, 1041–1045. [Google Scholar] [CrossRef] [PubMed]
- Monti, D.; Forchin, M.C.; Crotti, M.; Parmeggiani, F.; Gatti, F.G.; Brenna, E.; Riva, S. Cascade coupling of ene-reductases and ω-Transaminases for the stereoselective synthesis of diastereomerically enriched amines. ChemCatChem 2015, 7, 3106–3109. [Google Scholar] [CrossRef]
- Skalden, L.; Peters, C.; Ratz, L.; Bornscheuer, U.T. Synthesis of (1R,3R)-1-amino-3-methylcyclon-n-hexane by an enzyme cascade reaction. Tetrahedron 2016, 72, 7207–7211. [Google Scholar] [CrossRef]
- Liardo, E.; Ríos-Lombardía, N.; Morís, F.; Rebolledo, F.; González-Sabín, J. Hybrid organo- and biocatalytic process for the asymmetric transformation of alcohols into amines in aqueous medium. ACS Catal. 2017, 7, 4768–4774. [Google Scholar] [CrossRef]
- Molinaro, C.; Bulger, P.G.; Lee, E.E.; Kosjek, B.; Lau, S.; Gauvreau, D.; Howard, M.E.; Wallace, D.J.; O’Shea, P.D. CRTH2 antagonist MK-7246: A synthetic evolution from discovery through development. J. Org. Chem. 2012, 77, 2299–2309. [Google Scholar] [CrossRef] [PubMed]
- Richter, N.; Simon, R.C.; Kroutil, W.; Ward, J.M.; Hailes, H.C. Synthesis of pharmaceutically relevant 17-α-amino steroids using an ω-transaminase. Chem. Commun. 2014, 50, 6098–6100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limanto, J.; Ashley, E.R.; Yin, J.; Beutner, G.L.; Grau, B.T.; Kassim, A.M.; Kim, M.M.; Klapars, A.; Liu, Z.; Strotman, H.R.; Truppo, M.D. A highly efficient asymmetric synthesis of Vernakalant. Org. Lett. 2014, 16, 2716–2719. [Google Scholar] [CrossRef] [PubMed]
- Weiβ, M. S.; Pavlidis, I.V.; Spurr, P.; Hanlon, S.P.; Wirz, B.; Iding, H.; Bornscheuer, U.T. Protein-engineering of an amine transaminase for the stereoselective synthesis of a pharmaceutically relevant bicyclic amine. Org. Biomol. Chem. 2016, 14, 10249–10254. [Google Scholar] [Green Version]
- Feng, Y.; Luo, Z.; Sun, G.; Chen, M.; Lai, J.; Lin, W.; Goldmann, S.; Zhang, L.; Wang, Z. Development of an Efficient and Scalable Biocatalytic Route to (3R)-3-Aminoazepane: A Pharmaceutically Important Intermediate. Org. Process Res. Dev. 2017, 21, 648–654. [Google Scholar] [CrossRef]
- Sugihara, H.; Mabuchi, H.; Kawamatsu, Y. 1,5-Benzoxathiepin derivatives, I. Synthesis and reaction of 1,5-benzoxathiepin derivatives. Chem. Pharm. Bull. 1987, 35, 1919–1929. [Google Scholar] [CrossRef] [PubMed]
- López-Iglesias, M.; González-Martínez, D.; Gotor, V.; Busto, E.; Kroutil, W.; Gotor-Fernández, V. Biocatalytic Transamination for the Asymmetric Synthesis of Pyridylalkylamines. Structural and Activity Features in the Reactivity of Transaminases. ACS Catal. 2016, 6, 4003–4009. [Google Scholar] [CrossRef] [Green Version]
- López-Iglesias, M.; González-Martínez, D.; Rodríguez-Mata, M.; Gotor, V.; Busto, E.; Kroutil, W.; Gotor-Fernández, V. Asymmetric Biocatalytic Synthesis of Fluorinated Pyridines through Transesterification or Transamination: Computational Insights into the Reactivity of Transaminases. Adv. Synth. Catal. 2017, 359, 279–291. [Google Scholar] [CrossRef] [Green Version]
- Cassimjee, K.E.; Branneby, C.; Abedi, V.; Wells, A.; Berglund, P. Transaminations with isopropyl amine: Equilibrium displacement with yeast alcohol dehydrogenase coupled to in situ cofactor regeneration. Chem. Commun. 2010, 46, 5569–5571. [Google Scholar] [CrossRef] [PubMed]
- Green, A.P.; Turner, N.J.; O’Reilly, E. Chiral Amine Synthesis Using ω-Transaminases: An Amine Donor that Displaces Equilibria and Enables High-Throughput Screening. Angew. Chem. Int. Ed. 2014, 53, 10714–10717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomm, A.; Lewis, W.; Green, A.P.; O’Reilly, E. A New Generation of Smart Amine Donors for Transaminase-Mediated Biotransformations. Chem. Eur. J. 2016, 22, 12692–12695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payer, S.E.; Schrittwieser, J.H.; Kroutil, W. Vicinal Diamines as Smart Cosubstrates in the Transaminase-Catalyzed Asymmetric Amination of Ketones. Eur. J. Org. Chem. 2017, 2553–2559. [Google Scholar] [CrossRef]
- Paul, C.E.; Rodríguez-Mata, M.; Busto, E.; Lavandera, I.; Gotor-Fernández, V.; Gotor, V.; García-Cerrada, S.; Mendiola, J.; de Frutos, Ó.; Collado, I. Transaminases applied to the synthesis of high added-value enantiopure amines. Org. Process Res. Dev. 2014, 18, 788–792. [Google Scholar] [CrossRef]
- Kaulman, U.; Smithies, K.; Smith, M.E.B.; Hailes, H.C.; Ward, J.M. Substrate spectrum of ω-transaminase from Chromobacterium violaceum DSM30191 and its potential for biocatalysis. Enzyme Microb. Technol. 2007, 41, 628–637. [Google Scholar] [CrossRef]
- Yamada, Y.; Iwasaki, A.; Kizaki, N.; Matsumoto, K.; Ikenaka, Y.; Ogura, M.; Hasegawa, J. Enzymic Preparation of Optically Active (R)-Amino Compounds with Transaminase of Arthrobacter. PCT Int. Appl. WO 9848030A1, 29 October 1998. [Google Scholar]
- Pannuri, S.; Kamat, S.V.; Garcia, A.R.M. Methods for Engineering Arthrobacter citreus ω-Transaminase Variants with Improved Thermostability for Use in Enantiomeric Enrichment and Stereoselective Synthesis. PCT Int. Appl. WO 2006063336A2, 15 June 2006. [Google Scholar]
- Savile, C.K.; Janey, J.M.; Mundorff, E.M.; Moore, J.C.; Tam, S.; Jarvis, W.R.; Colbeck, J.C.; Krebber, A.; Fleitz, F.J.; Brands, J.; et al. Biocatalytic Asymmetric Synthesis of Chiral Amines from Ketones Applied to Sitagliptin Manufacture. Science 2010, 329, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Koszelewski, D.; Göritzer, M.; Clay, D.; Seisser, B.; Kroutil, W. Synthesis of optically active amines employing recombinant ω-transaminases in E. coli cells. ChemCatChem 2010, 2, 73–77. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]




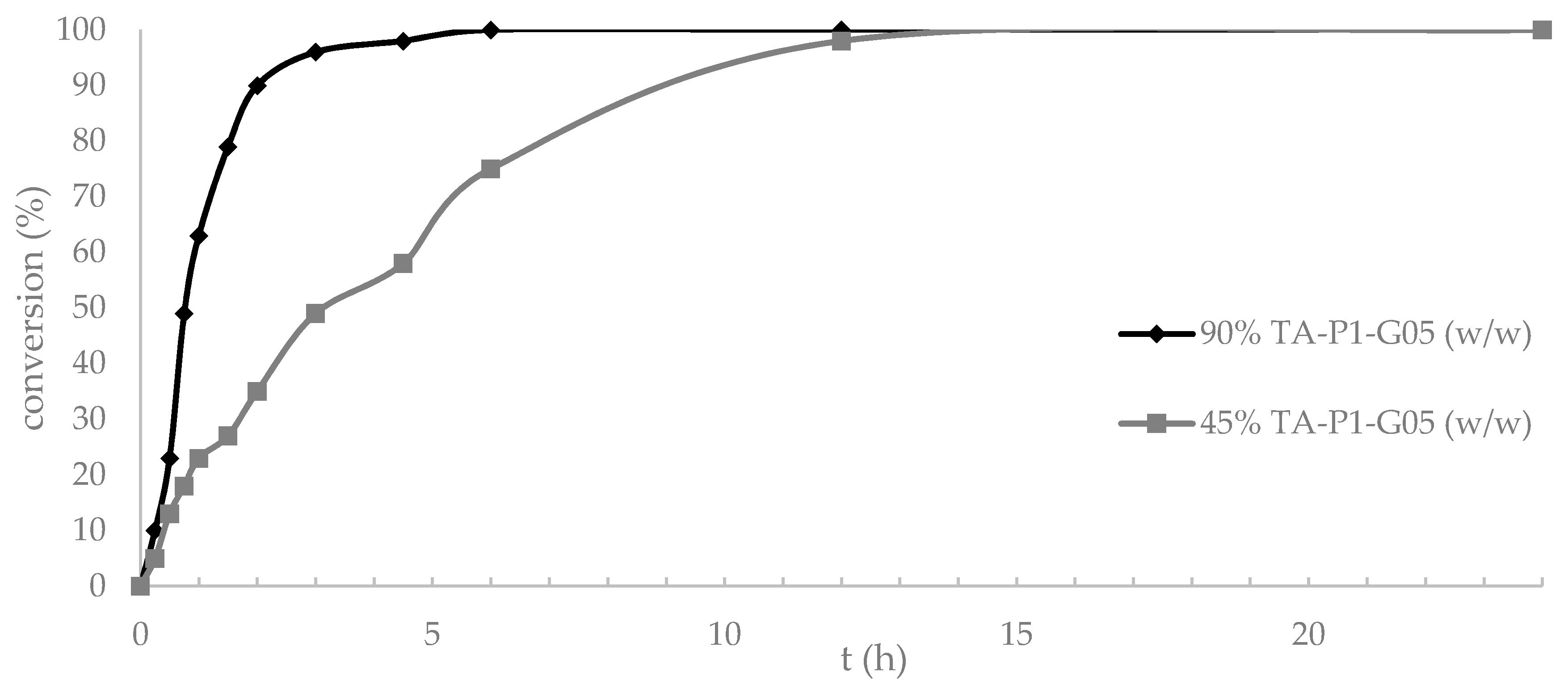
 ) 90% of enzyme loading (w/w) or (
) 90% of enzyme loading (w/w) or (  ) 45% of enzyme loading (w/w vs. 6).
) 90% of enzyme loading (w/w) or ( ) 45% of enzyme loading (w/w vs. 6).
) 45% of enzyme loading (w/w vs. 6).
) 90% of enzyme loading (w/w) or ( ) 45% of enzyme loading (w/w vs. 6).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Enzyme | Conversion (%) b | ee (%) c |
|---|---|---|---|
| 1 | ATA-7 | <1 | n.d. |
| 2 | ATA-13 | 30 | n.d. |
| 3 | ATA-24 | 93 | <1 |
| 4 | ATA-25 | 96 | <1 |
| 5 | ATA-33 | >99 | <1 |
| 6 | ATA-113 | 13 | n.d. |
| 7 | ATA-117 | 2 | n.d. |
| 8 | ATA-200 | >99 | 85 (S) |
| 9 | ATA-217 | 6 | n.d. |
| 10 | ATA-234 | 4 | n.d. |
| 11 | ATA-237 | >99 | 41 (S) |
| 12 | ATA-238 | 4 | n.d. |
| 13 | ATA-251 | >99 | 72 (S) |
| 14 | ATA-254 | >99 | 56 (S) |
| 15 | ATA-256 | >99 | 63 (S) |
| 16 | ATA-260 | >99 | 79 (S) |
| 17 | ATA-301 | >99 | 7 (S) |
| 18 | ATA-303 | >99 | <1 |
| 19 | ATA-412 | >99 | 55 (S) |
| 20 | ATA-415 | >99 | <1 |
| 21 | TA-P1-A01 | >99 | 62 (R) |
| 22 | TA-P1-A06 | >99 | 50 (R) |
| 23 | TA-P1-B04 | >99 | 82 (R) |
| 24 | TA-P1-F03 | >99 | 90 (R) |
| 25 | TA-P1-F12 | >99 | 28 (R) |
| 26 | TA-P1-G05 | >99 | 93 (R) |
| 27 | TA-P1-G06 | >99 | 67 (R) |
| 28 | TA-P2-A01 | 4 | n.d. |
| 29 | TA-P2-A07 | 60 | 16 (S) |
| 30 | TA-P2-B01 | 99 | 19 (R) |
| Entry | Enzyme | Conversion (%) b | ee (%) c |
|---|---|---|---|
| 1 | ATA01 Aspergillus fumigatus | 9 | n.d. |
| 2 | ATA02 Gibberella zeae | <1 | n.d. |
| 3 | ATA03 Neosartorya fischeri | 29 | 90 (S) |
| 4 | ATA04 Aspergillius oryza | 2 | n.d. |
| 5 | ATA05 Aspergillius terreus | 8 | n.d. |
| 6 | ATA06 Penicillium chrysogenum | <1 | n.d. |
| 7 | ATA07 Mycobacterium vanbaalenii | 20 | 95 (S) |
| 8 | ATA08 Silicibacter pomeroyi | >99 | 91 (R) |

| Entry | Enzyme | [6] (mM) | Cosolvent b | T (°C) | t (h) | c (%) c |
|---|---|---|---|---|---|---|
| 1 | ATA03 Neosartorya fischeri | 20 | MeCN (5%) | 30 | 20 | 29 |
| 2 | ATA03 Neosartorya fischeri | 10 | none | 30 | 48 | 86 |
| 3 | ATA07 Mycobacterium vanbaalenii | 20 | MeCN (5%) | 30 | 20 | 20 |
| 4 | ATA07 Mycobacterium vanbaalenii | 10 | none | 30 | 20 | 73 |
| 5 | ATA07 Mycobacterium vanbaalenii | 20 | none | 45 | 48 | 43 |
| 6 d | ATA07 Mycobacterium vanbaalenii d | 20 | none | 30 | 65 | 91 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Martínez, D.; Fernández-Sáez, N.; Cativiela, C.; Campos, J.M.; Gotor-Fernández, V. Development of Biotransamination Reactions towards the 3,4-Dihydro-2H-1,5-benzoxathiepin-3-amine Enantiomers. Catalysts 2018, 8, 470. https://doi.org/10.3390/catal8100470
González-Martínez D, Fernández-Sáez N, Cativiela C, Campos JM, Gotor-Fernández V. Development of Biotransamination Reactions towards the 3,4-Dihydro-2H-1,5-benzoxathiepin-3-amine Enantiomers. Catalysts. 2018; 8(10):470. https://doi.org/10.3390/catal8100470
Chicago/Turabian StyleGonzález-Martínez, Daniel, Nerea Fernández-Sáez, Carlos Cativiela, Joaquín M. Campos, and Vicente Gotor-Fernández. 2018. "Development of Biotransamination Reactions towards the 3,4-Dihydro-2H-1,5-benzoxathiepin-3-amine Enantiomers" Catalysts 8, no. 10: 470. https://doi.org/10.3390/catal8100470