The Relationship between Reaction Temperature and Carbon Deposition on Nickel Catalysts Based on Al2O3, ZrO2 or SiO2 Supports during the Biogas Dry Reforming Reaction



 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Characterization Results for Fresh Catalysts
2.2. Characterization Results for Spent Catalysts
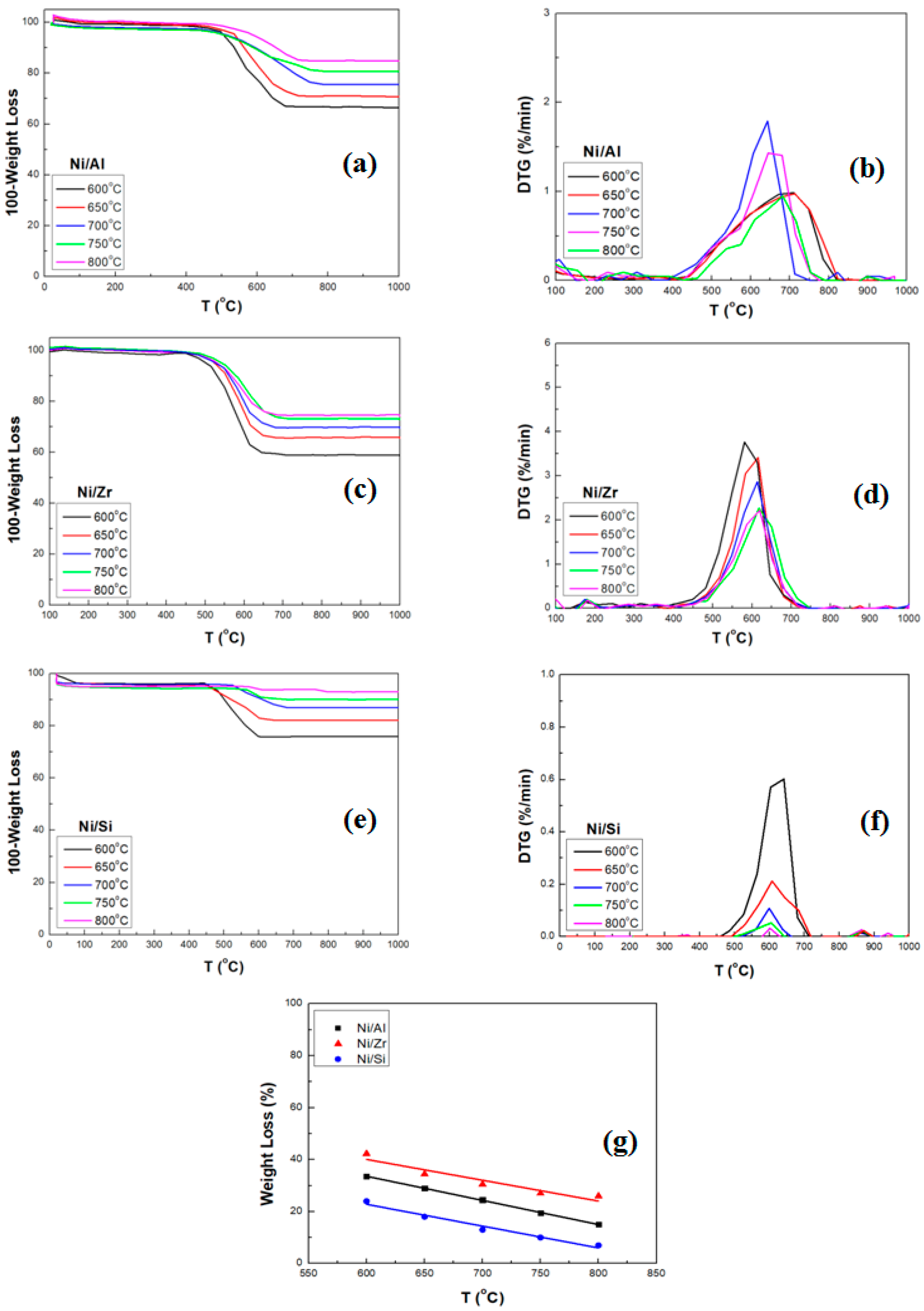
2.2.1. TGA/DTG Analysis
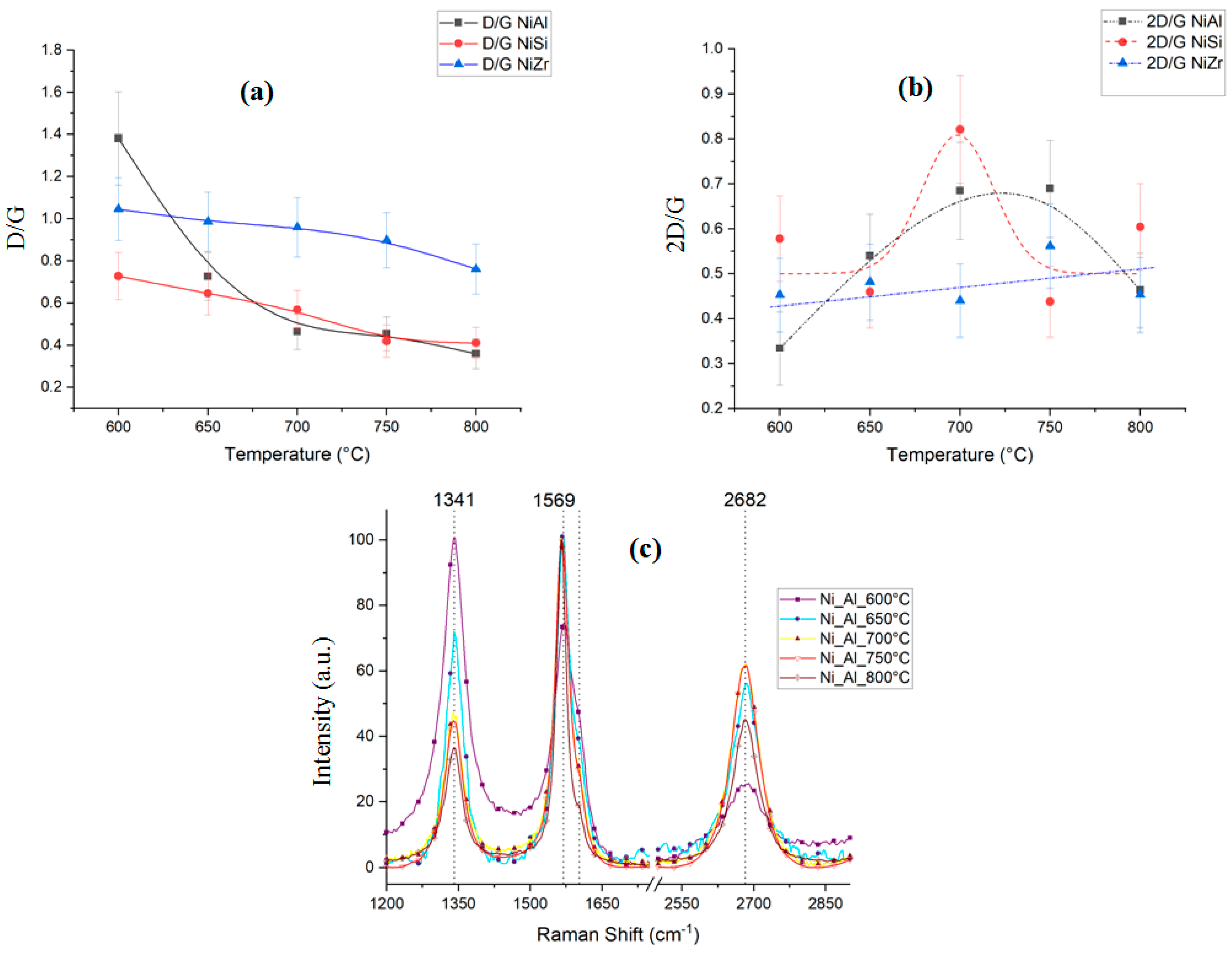
2.2.2. Raman Spectroscopy Analysis
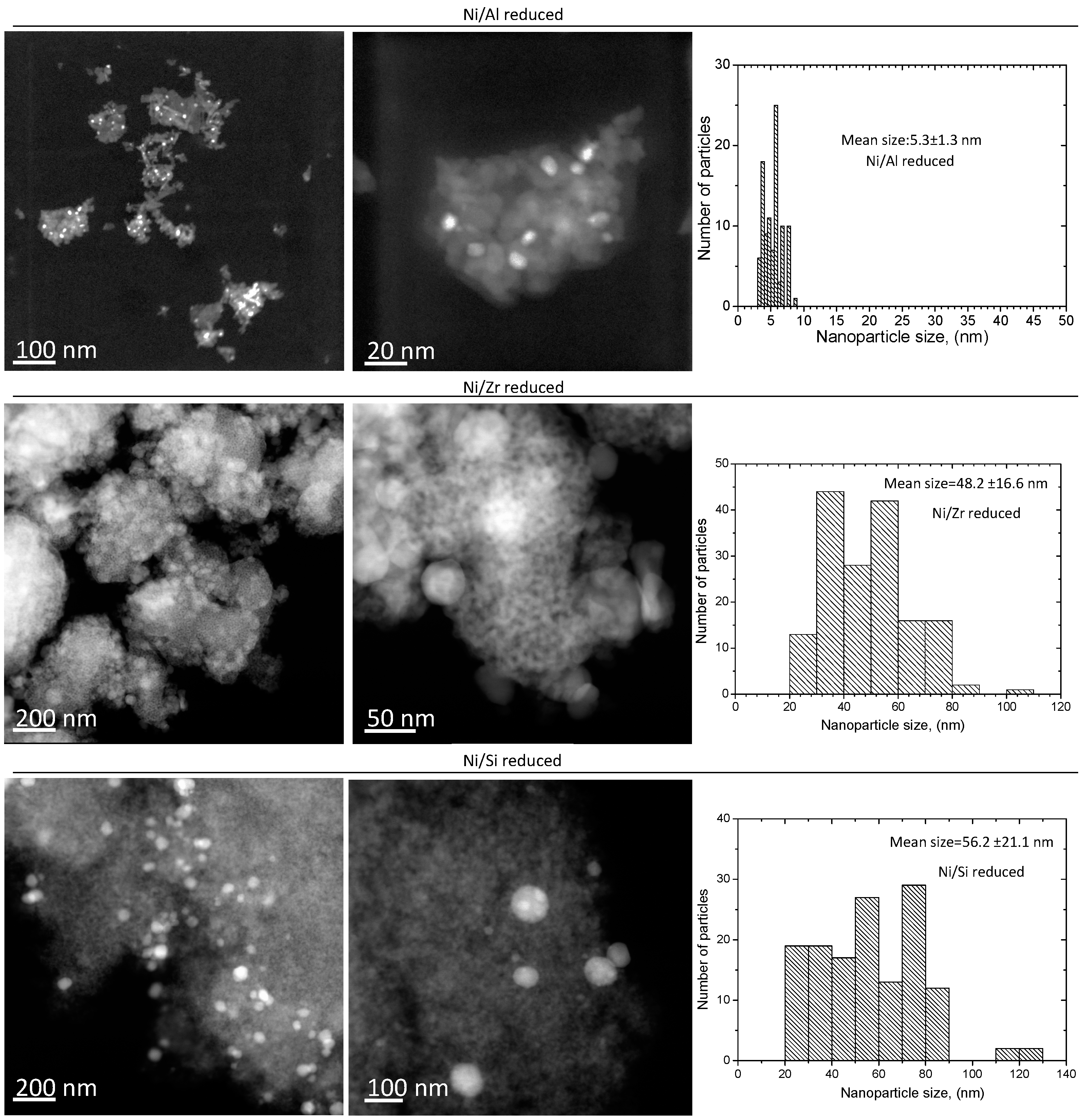
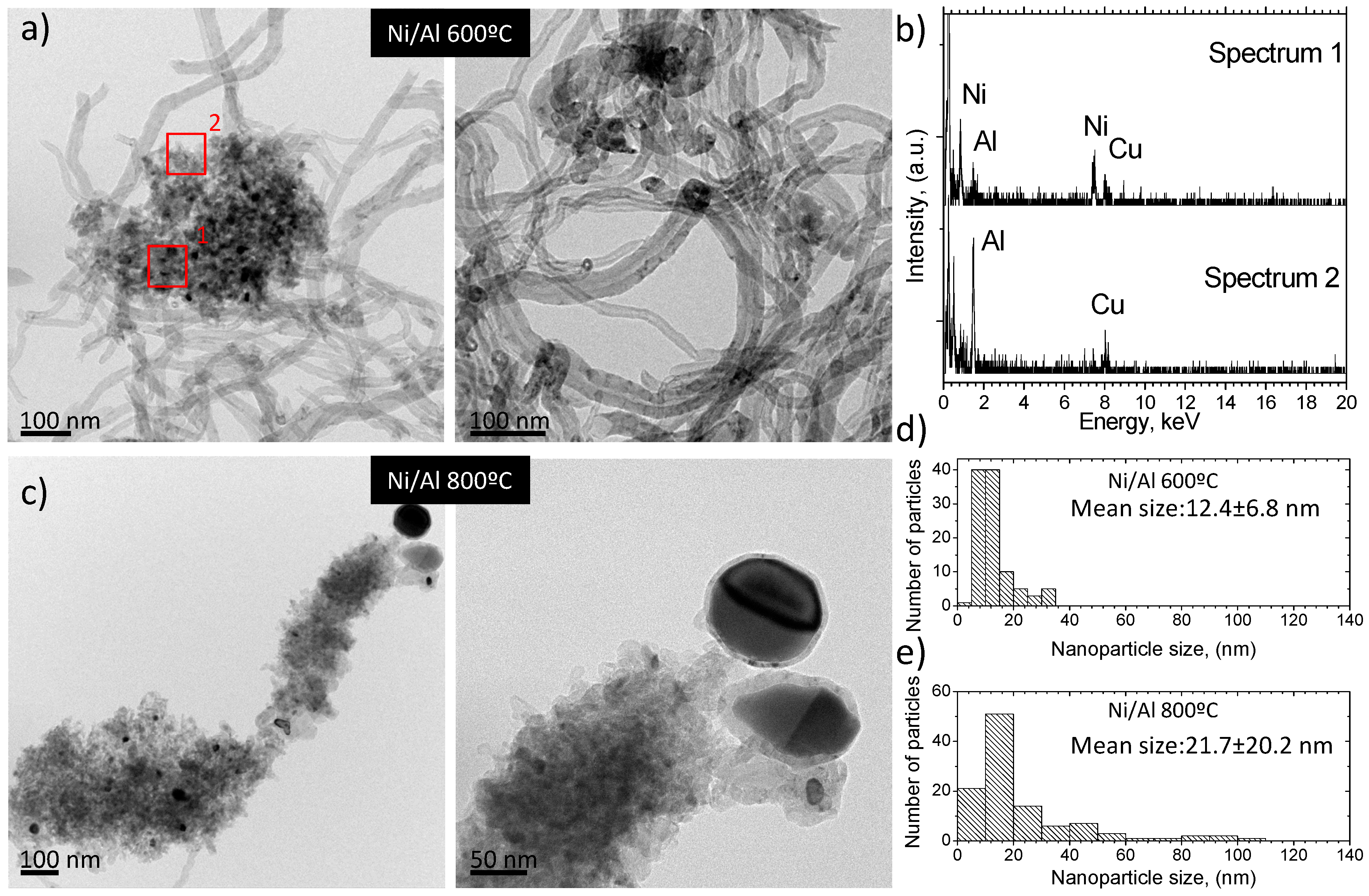
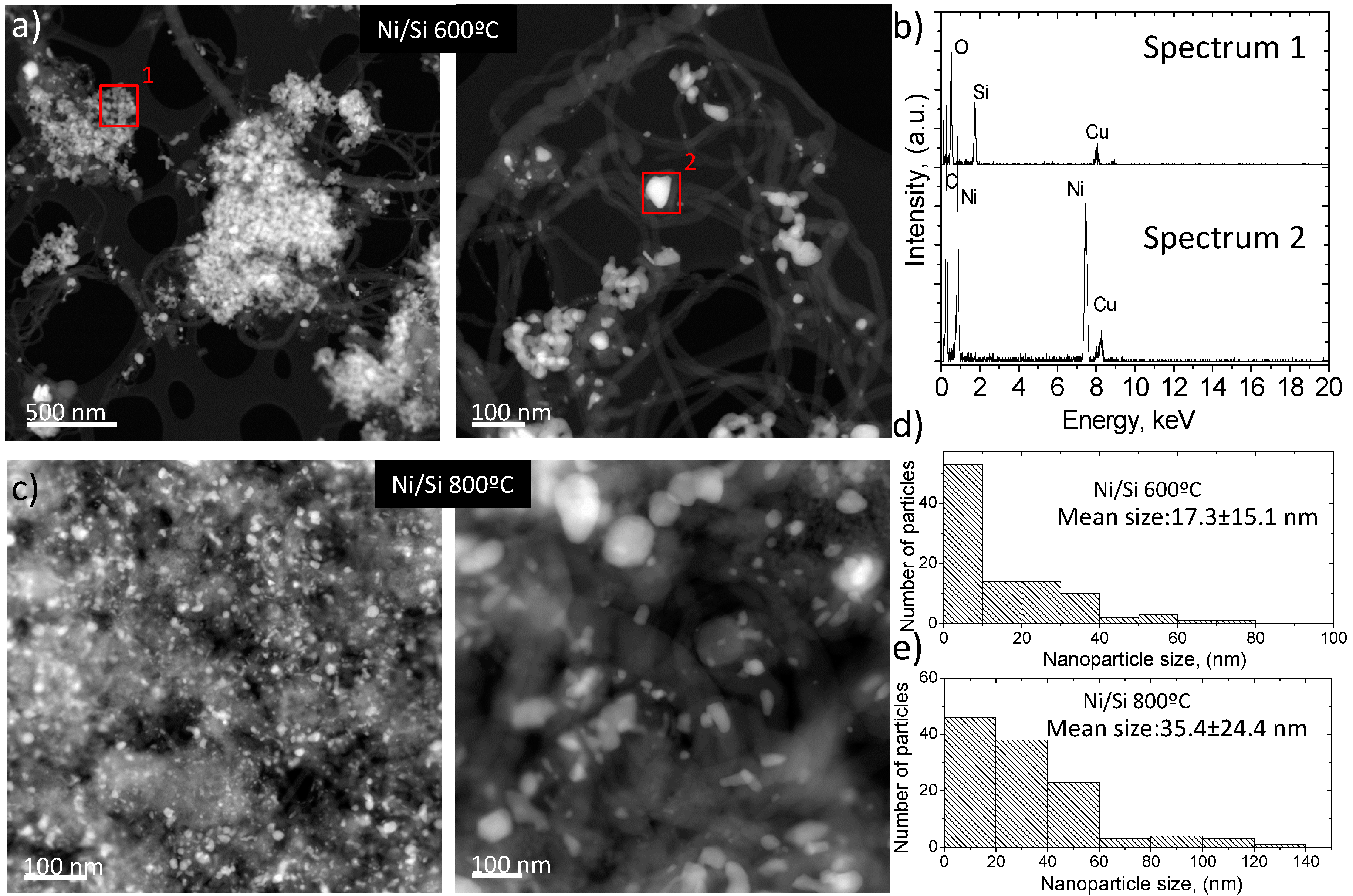
2.2.3. Electron Microscopy Analysis
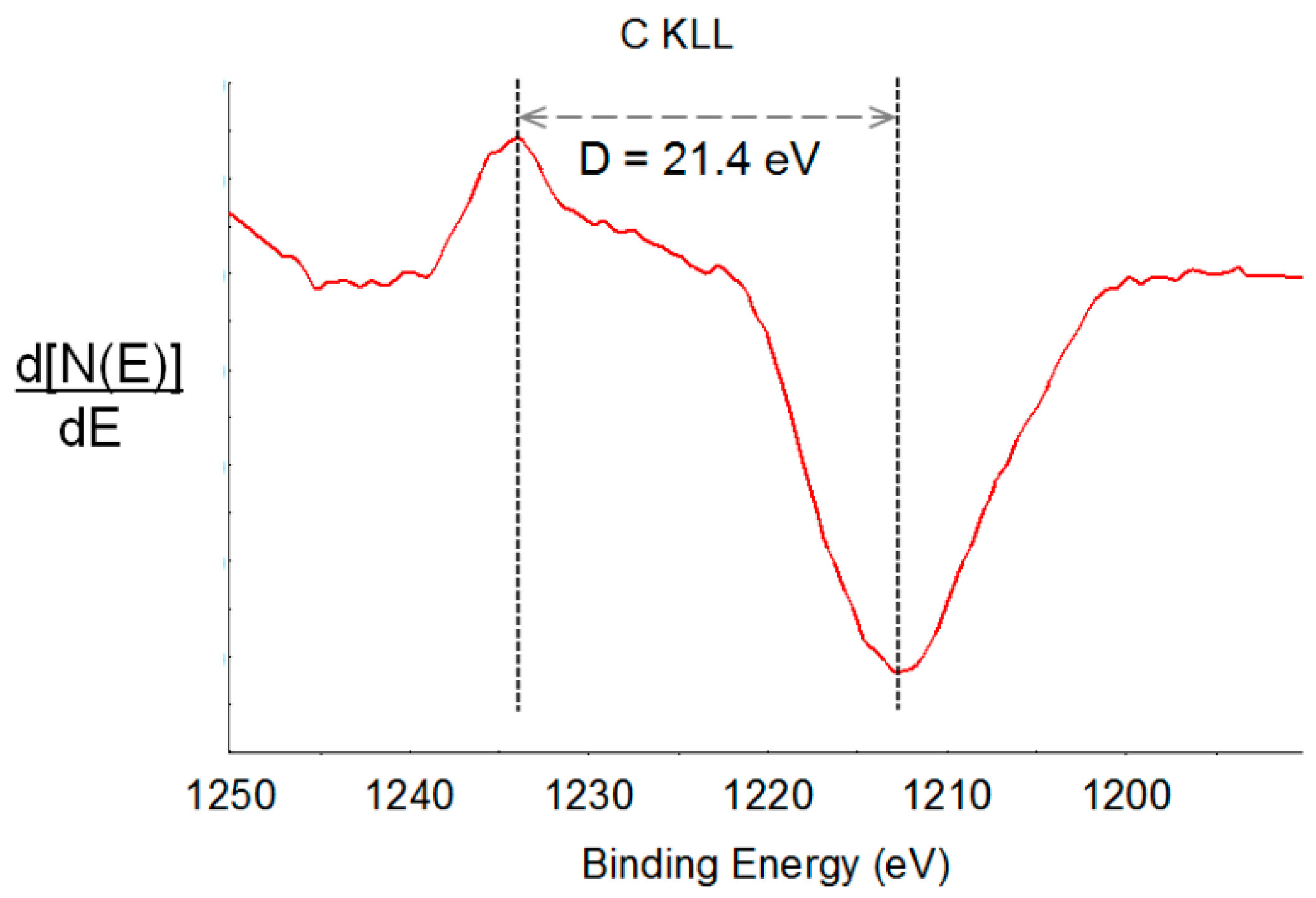
2.2.4. XPS Analysis
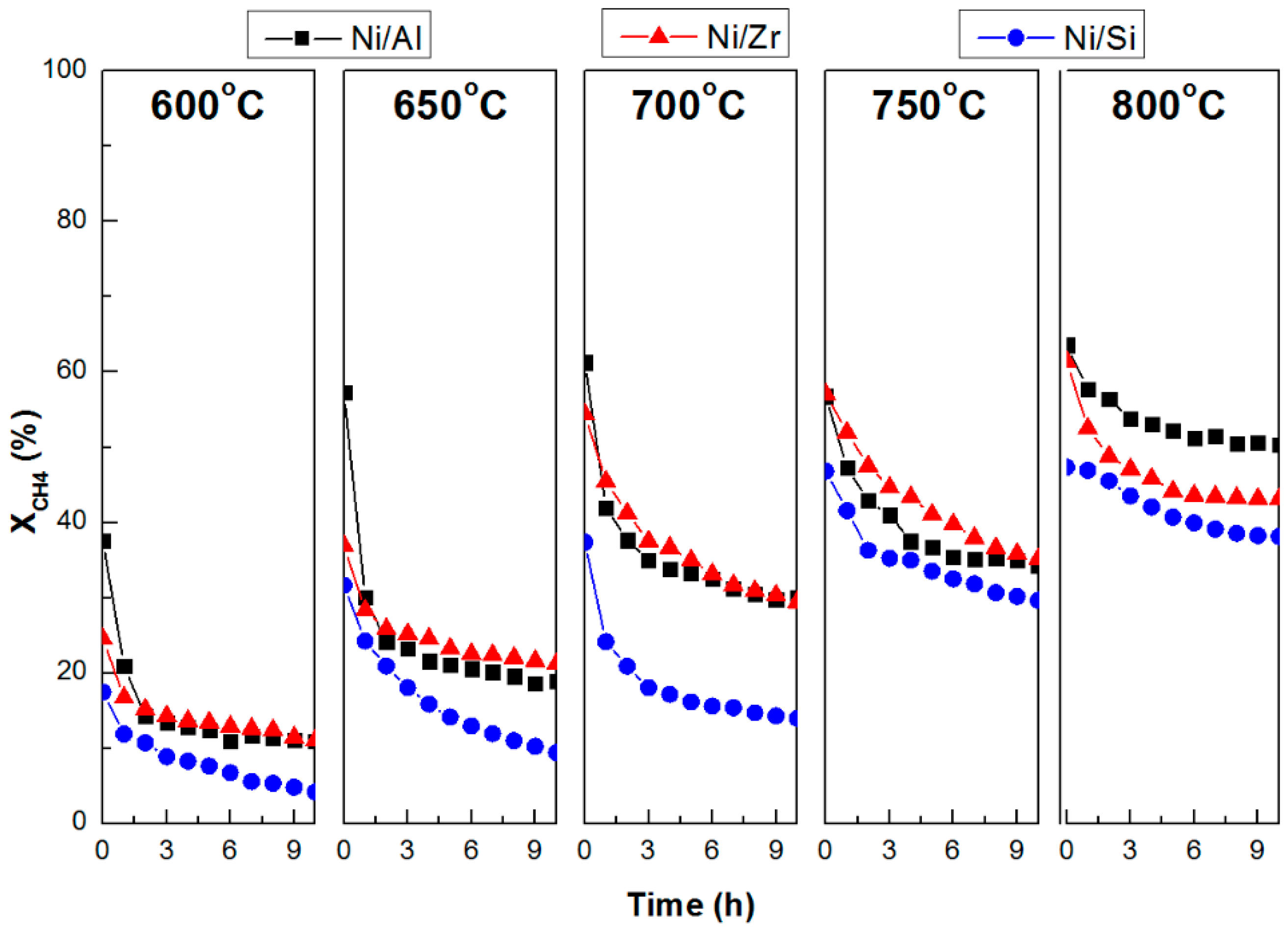
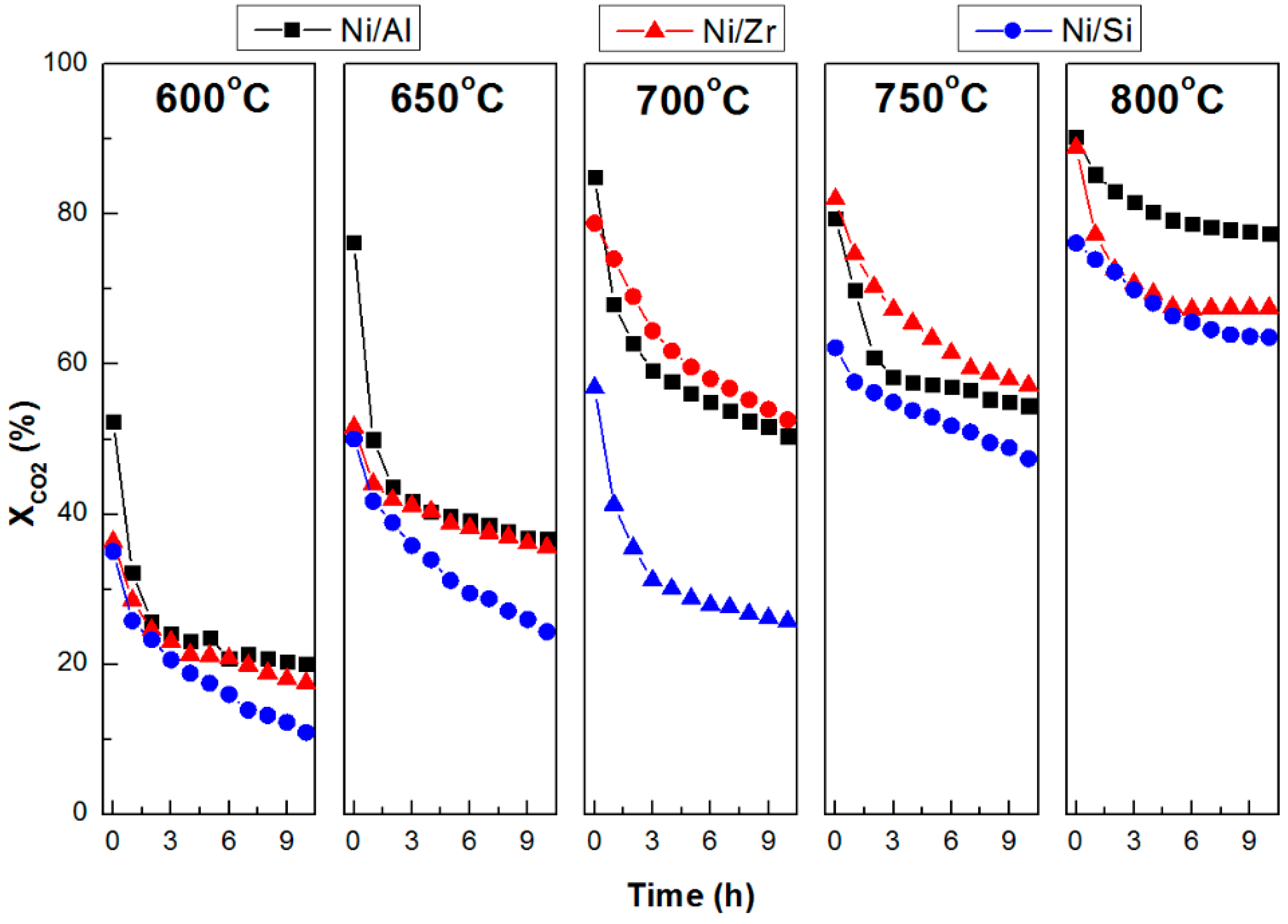
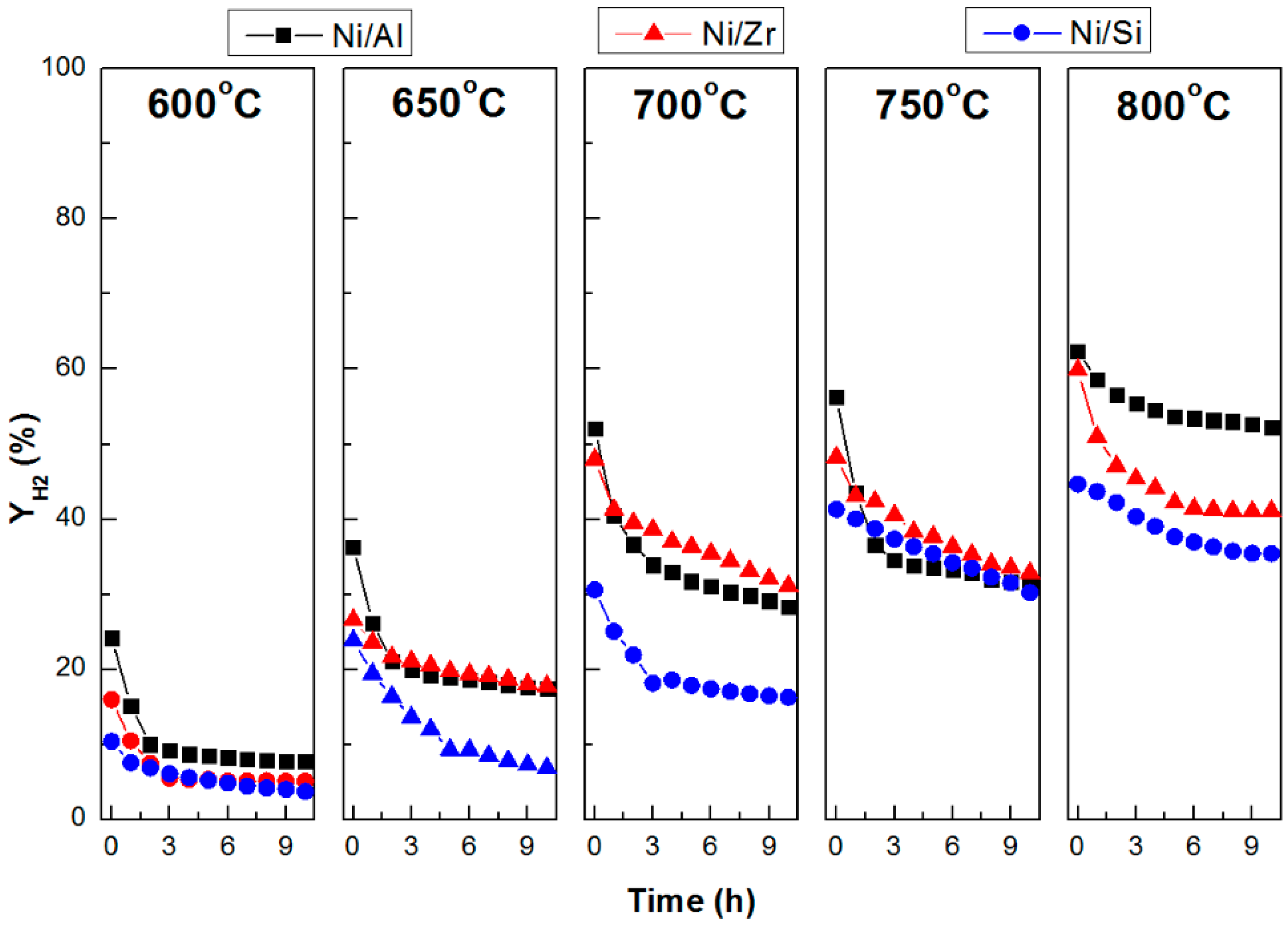
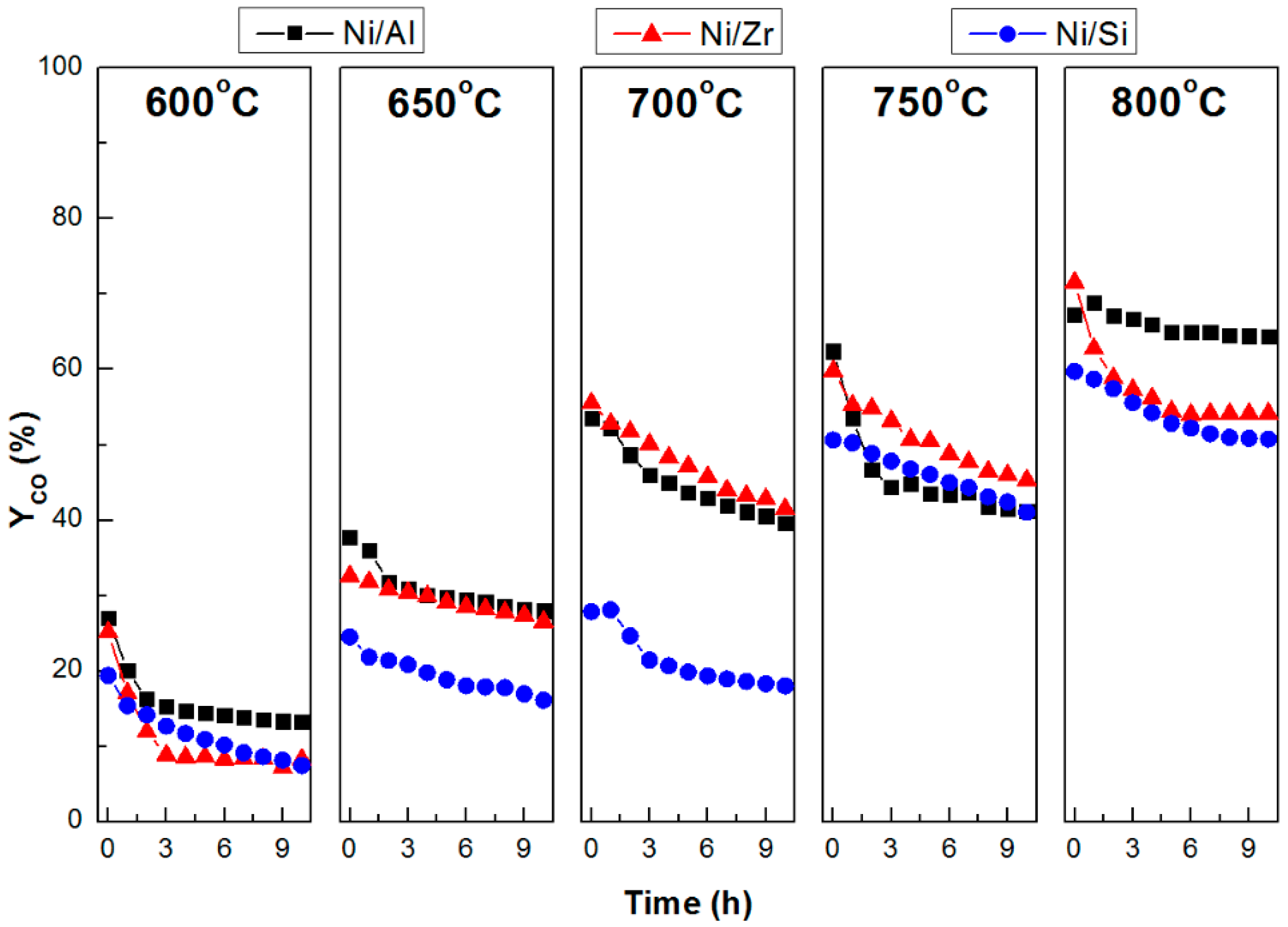
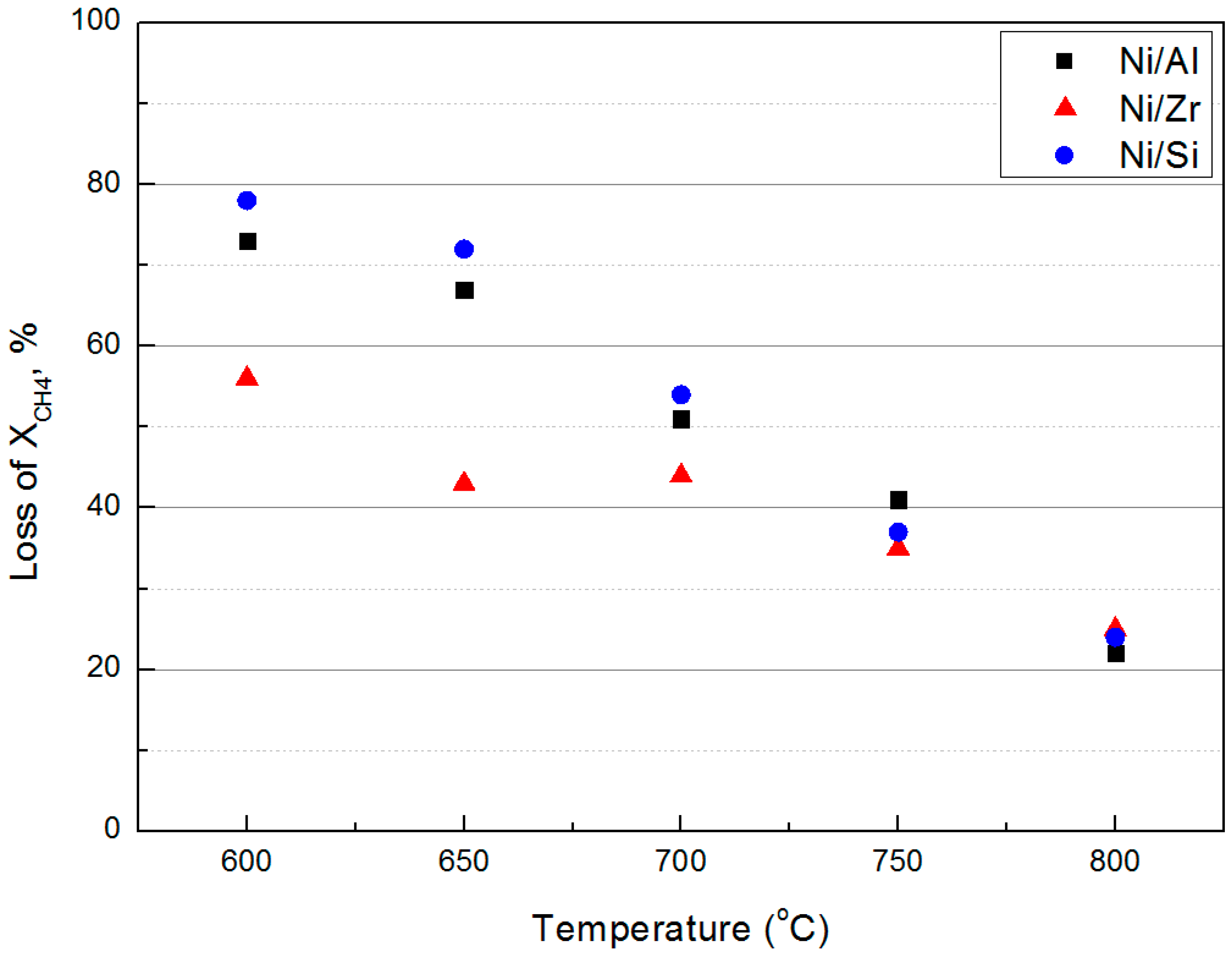
2.3. Catalytic Stability
3. Materials and Methods
3.1. Catalyst Preparation
3.2. Catalyst Characterization
3.3. Catalytic Tests
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dama, S.; Ghodke, S.R.; Bobade, R.; Gurav, H.R.; Chilukuri, S. Active and durable alkaline earth metal substituted perovskite catalysts for dry reforming of methane. Appl. Catal. B Environ. 2018, 224, 146–158. [Google Scholar] [CrossRef]
- Avraam, D.G.; Halkides, T.I.; Liguras, D.K.; Bereketidou, O.A.; Goula, M.A. An experimental and theoretical approach for the biogas steam reforming reaction. Int. J. Hydrog. Energy 2010, 35, 9818–9827. [Google Scholar] [CrossRef]
- Italiano, C.; Luchters, N.T.J.; Pino, L.; Fletcher, J.V.; Specchia, S.; Fletcher, J.C.Q.; Vita, A. High specific surface area supports for highly active Rh catalysts: Syngas production from methane at high space velocity. Int. J. Hydrog. Energy 2018, 43, 11755–11765. [Google Scholar] [CrossRef]
- Charisiou, N.D.; Baklavaridis, A.; Papadakis, V.G.; Goula, M.A. Synthesis gas production via the biogas reforming reaction over Ni/MgO-Al2O3 and Ni/CaO-Al2O3 catalysts. Waste Biomass Valoriz. 2016, 7, 725–736. [Google Scholar] [CrossRef]
- Zhou, W.J.; Zhou, B.; Li, W.Z.; Zhou, Z.H.; Song, S.Q.; Sun, G.Q.; Xin, Q.; Douvartzides, S.; Goula, M.; Tsiakaras, P. Performance comparison of low-temperature direct alcohol fuel cells with different anode catalysts. J. Power Sources 2004, 126, 16–22. [Google Scholar] [CrossRef]
- Weger, L.; Abanades, A.; Butler, T. Methane cracking as a bridge technology to the hydrogen economy. Int. J. Hydrog. Energy 2017, 42, 720–731. [Google Scholar] [CrossRef] [Green Version]
- Nikoo, M.K.; Amin, N.A.S. Thermodynamic analysis of carbon dioxide reforming of methane in view of solid carbon formation. Fuel Process. Technol. 2011, 92, 678–691. [Google Scholar] [CrossRef] [Green Version]
- Abdullah, B.; Ghani, N.A.A.; Vo, D.-V.N. Recent advances in dry reforming of methane over Ni-based catalysts. J. Clean. Prod. 2017, 162, 170–185. [Google Scholar] [CrossRef] [Green Version]
- Aramouni, N.A.K.; Zeaiter, J.; Kwapinski, W.; Ahmad, M.N. Thermodynamic analysis of methane dry reforming: Effect of the catalyst particle size on carbon formation. Energy Convers. Manag. 2017, 150, 614–622. [Google Scholar] [CrossRef]
- Jang, W.J.; Shim, J.O.; Kim, H.M.; Yoo, S.Y.; Roh, H.S. A review on dry reforming of methane in aspect of catalytic properties. Catal. Today 2019, 324, 15–26. [Google Scholar] [CrossRef]
- Verykios, X.E. Catalytic dry reforming of natural gas for the production of chemicals and hydrogen. Int. J. Hydrog. Energy 2003, 28, 1045–1063. [Google Scholar] [CrossRef]
- Yao, Z.; Jiang, J.; Zhao, Y.; Luan, F.; Zhu, J.; Shi, Y. Insights into the deactivation mechanism of metal carbide catalysts for dry reforming of methane via comparison of nickel-modified molybdenum and tungsten carbides. RSC Adv. 2016, 6, 19944–19951. [Google Scholar] [CrossRef]
- Froment, G.F. Production of synthesis gas by steam- and CO2-reforming of natural gas. J. Mol. Catal. A Chem. 2000, 163, 147–156. [Google Scholar] [CrossRef]
- Horlyck, J.; Lawrey, C.; Lovell, E.C.; Amal, R.; Scott, J. Elucidating the impact of Ni and Co loading on the selectivity of bimetallic NiCo catalysts for dry reforming of methane. Chem. Eng. J. 2018, 352, 572–580. [Google Scholar] [CrossRef]
- Goula, M.A.; Charisiou, N.D.; Papageridis, K.N.; Delimitis, A.; Pachatouridou, E.; Iliopoulou, E.F. Nickel on alumina catalysts for the production of hydrogen rich mixtures via the biogas dry reforming reaction: Influence of the synthesis method. Int. J. Hydrog. Energy 2015, 40, 9183–9200. [Google Scholar] [CrossRef]
- Li, M.; van Veen, A.C. Tuning the catalytic performance of Ni-catalysed dry reforming of methane and carbon deposition via Ni-CeO2-x interaction. Appl. Catal. B Environ. 2018, 237, 641–648. [Google Scholar] [CrossRef]
- Wei, J.; Iglesia, E. Isotopic and kinetic assessment of the mechanism of reactions of CH4 with CO2 or H2O to form synthesis gas and carbon on nickel catalysts. J. Catal. 2004, 224, 370–383. [Google Scholar] [CrossRef]
- Wei, J.; Iglesia, E. Structural and mechanistic requirements for methane activation and chemical conversion on supported iridium clusters. Angew. Chem. Int. Edit. 2004, 43, 3685–3688. [Google Scholar] [CrossRef]
- Kim, W.Y.; Jang, J.S.; Ra, E.C.; Kim, K.Y.; Kim, E.H.; Lee, J.S. Reduced perovskite LaNiO3 catalysts modified with Co and Mn for low coke formation in dry reforming of methane. Appl. Catal. A Gen. 2019, 575, 198–203. [Google Scholar] [CrossRef]
- Charisiou, N.D.; Siakavelas, G.; Papageridis, K.N.; Baklavaridis, A.; Tzounis, L.; Avraam, D.G.; Goula, M.A. Syngas production via the biogas dry reforming reaction over nickel supported on modified with CeO2 and/or La2O3 alumina catalysts. J. Nat. Gas Sci. Eng. 2016, 31, 164–183. [Google Scholar] [CrossRef]
- Charisiou, N.D.; Siakavelas, G.; Papageridis, K.; Baklavaridis, A.; Tzounis, L.; Goula, G.; Yentekakis, I.V.; Goula, M.A. The effect of WO3 modification of ZrO2 support on the Ni-catalyzed dry reforming of biogas reaction for syngas production. Front. Environ. Sci. 2017, 5, 66. [Google Scholar] [CrossRef]
- Carrara, C.; Munera, J.; Lombardo, E.A.; Cornaglia, L.M. Kinetic and stability studies of Ru/La2O3 used in the dry reforming of methane. Top. Catal. 2008, 51, 98–106. [Google Scholar] [CrossRef]
- Nagaoka, K.; Aika, K.; Seshan, K.; Lercher, J.A. Mechanism of carbon deposit/removal in methane dry reforming on supported metal catalysts. Stud. Surf. Sci. Catal. 2001, 136, 129–136. [Google Scholar] [CrossRef]
- Charisiou, N.D.; Iordanidis, A.; Polychronopoulou, K.; Yentekakis, I.V.; Goula, M.A. Studying the stability of Ni supported on modified with CeO2 alumina catalysts for the biogas dry reforming reaction. Mater. Today Proc. 2018, 5, 27607–27616. [Google Scholar] [CrossRef]
- Usman, M.; Wan Daud, W.M.A.; Abbas, H.F. Dry reforming of methane: Influence of process parameters—A review. Renew. Sustain. Energy Rev. 2015, 45, 710–744. [Google Scholar] [CrossRef]
- Byckkov, V.Y.; Tyulenin, Y.P.; Firsova, A.A.; Shafranovsky, E.A.; Gorenberg, A.Y.; Korchak, V.N. Carbonization of nickel catalysts and its effect on methane dry reforming. Appl. Catal. A Gen. 2013, 453, 71–79. [Google Scholar] [CrossRef]
- Charisiou, N.D.; Papageridis, K.N.; Siakavelas, G.; Tzounis, L.; Goula, M.A. Effect of active metal supported on SiO2 for selective hydrogen production from the glycerol steam reforming reaction. Bioresources 2016, 11, 10173–10189. [Google Scholar] [CrossRef]
- Xu, Y.; Du, X.-H.; Li, J.; Wang, P.; Zhu, J.; GE, F.-J.; Zhou, J.; Song, M.; Zhu, W.-Y. A comparison of Al2O3 and SiO2 supported Ni-based catalysts in their performance for the dry reforming of methane. J. Fuel Chem. Technol. 2019, 47, 199–208. [Google Scholar] [CrossRef]
- Cai, W.; Ye, L.; Zhang, L.; Ren, Y.; Yue, B.; Chen, X.; He, H. Highly dispersed nickel-containing mesoporous silica with superior stability in carbon dioxide reforming of methane: The effect of anchoring. Materials 2014, 7, 2340–2355. [Google Scholar] [CrossRef]
- Anchieta, C.G.; Assaf, E.M.; Assaf, J.M. Effect of ionic liquid in Ni/ZrO2 catalysts applied to syngas production by methane tri-reforming. Int. J. Hydrog. Energy 2019, 44, 9316–9327. [Google Scholar] [CrossRef]
- Charisiou, N.D.; Papageridis, K.N.; Siakavelas, G.; Sebastian, V.; Hinder, S.J.; Baker, M.A.; Polychronopoulou, K.; Goula, M.A. The influence of SiO2 doping on the Ni/ZrO2 supported catalyst for hydrogen production through the glycerol steam reforming reaction. Catal. Today 2019, 319, 206–219. [Google Scholar] [CrossRef]
- Sokolov, S.; Kondratenk, E.V.; Pohl, M.M.; Barkschat, A.; Rodemerck, U. Stable low-temperature dry reforming of methane over mesoporous La2O3-ZrO2 supported Ni catalysts. Appl. Catal. B Environ. 2012, 113–114, 19–30. [Google Scholar] [CrossRef]
- Hou, Z.; Chen, P.; Fang, H.; Zheng, X.; Yashima, T. Production of synthesis gas via methane reforming with CO2 on noble metals and small amount of noble-(Rh-) promoted Ni catalysts. Int. J. Hydrog. Energy 2006, 31, 555–561. [Google Scholar] [CrossRef]
- Arandiyan, H.; Peng, Y.; Liu, C.; Chang, H.; Li, J. Effects of noble metals doped on mesoporous LaAlNi mixed oxide catalyst and identification of carbon deposit for reforming CH4 with CO2. J. Chem. Technol. Biot. 2013, 89, 372–381. [Google Scholar] [CrossRef]
- Gao, Y.; Jiang, J.; Meng, Y.; Yan, F.; Aihemaiti, A. A review of recent developments in hydrogen production via biogas dry reforming. Energy Convers. Manag. 2018, 171, 133–155. [Google Scholar] [CrossRef]
- Bereketidou, O.A.; Goula, M.A. Biogas reforming for syngas production over nickel supported on ceria-alumina catalysts. Catal. Today 2012, 195, 93–100. [Google Scholar] [CrossRef]
- Shen, J.; Reule, A.A.C.; Semagina, N. Ni/MgAl2O4 catalyst for low-temperature oxidative dry methane reforming with CO2. Int. J. Hydrog. Energy 2019, 44, 4616–4629. [Google Scholar] [CrossRef]
- Rosha, P.; Mohapatra, S.K.; Mahla, S.K.; Dhir, A. Catalytic reforming of synthetic biogas for hydrogen enrichment over Ni supported on ZnO–CeO2 mixed catalyst. Biomass Bioenerg. 2019, 125, 70–78. [Google Scholar] [CrossRef]
- Goula, M.A.; Charisiou, N.D.; Siakavelas, G.; Tzounis, L.; Tsiaoussis, I.; Panagiotopoulou, P.; Goula, G.; Yentekakis, I.V. Syngas production via the biogas dry reforming reaction over Ni supported on zirconia modified with CeO2 or La2O3 catalysts. Int. J. Hydrog. Energy 2017, 42, 13724–13740. [Google Scholar] [CrossRef]
- Ghelamallah, M.; Granger, P. Supported-induced effect on the catalytic properties of Rh and Pt-Rh particles deposited on La2O3 and mixed α-Al2O3-La2O3 in the dry reforming of methane. Appl. Catal. A Gen. 2014, 485, 172–180. [Google Scholar] [CrossRef]
- Aramouni, N.A.K.; Touma, J.G.; Tarboush, B.A.; Zeaiter, J.; Ahmad, M.N. Catalyst design for dry reforming of methane: Analysis review. Renew. Sustain. Energy Rev. 2018, 82, 2570–2585. [Google Scholar] [CrossRef]
- Charisiou, N.D.; Siakavelas, G.; Tzounis, L.; Sebastian, V.; Monzon, A.; Baker, M.A.; Hinder, S.J.; Polychronopoulou, K.; Yentekakis, I.V.; Goula, M.A. An in depth investigation of deactivation through carbon formation during the biogas dry reforming reaction for Ni supported on modified with CeO2 and La2O3 zirconia catalysts. Int. J. Hydrog. Energy 2018, 43, 18955–18976. [Google Scholar] [CrossRef]
- Sehested, J. Four challenges for nickel steam-reforming catalysts. Catal. Today 2006, 111, 103–110. [Google Scholar] [CrossRef]
- Rostrup-Nielsen, J.R.S.; Norskov, J.K. Hydrogen and synthesis gas by steam- and CO2 reforming. Adv. Catal. 2002, 47, 65–139. [Google Scholar] [CrossRef]
- Charisiou, N.D.; Tzounis, L.; Sebastian, V.; Hinder, S.J.; Baker, M.A.; Polychronopoulou, K.; Goula, M.A. Investigating the correlation between deactivation and the carbon deposited on the surface of Ni/Al2O3 and Ni/La2O3 -Al2O3 catalysts during the biogas reforming reaction. Appl. Surf. Sci. 2019, 474, 42–56. [Google Scholar] [CrossRef]
- Charisiou, N.D.; Papageridis, K.N.; Siakavelas, G.; Tzounis, L.; Kousi, K.; Baker, M.A.; Hinder, S.J.; Sebastian, V.; Polychronopoulou, K.; Goula, M.A. Glycerol steam reforming for hydrogen production over Nickel supported on alumina, zirconia and silica catalysts. Top. Catal. 2017, 60, 1226–1250. [Google Scholar] [CrossRef]
- Bartholomew, C.H. Mechanisms of catalyst deactivation. Appl. Catal. A Gen. 2001, 212, 17–60. [Google Scholar] [CrossRef]
- Tzounis, L.; Kirsten, M.; Simon, F.; Mäder, E.; Stamm, M. The interphase microstructure and electrical properties of glass fibers covalently and non-covalently bonded with multiwall carbon nanotubes. Carbon 2014, 73, 310–324. [Google Scholar] [CrossRef]
- Charisiou, N.D.; Papageridis, K.N.; Tzounis, L.; Sebastian, V.; Hinder, S.J.; Baker, M.A.; Polychronopoulou, K.; Goula, M.A. Ni supported on CaO-MgO-Al2O3 as a highly selective and stable catalyst for H2 production via the glycerol steam reforming reaction. Int. J. Hydrog. Energy 2018, 44, 256–273. [Google Scholar] [CrossRef]
- Velasquez, M.; Batiot-Dupeyrat, C.B.; Gallego, J.; Fernandez, J.J.; Santamaria, A. Synthesis of carbon nano-chains from glycerol-ethanol decomposition over Ni-Fe alloy catalyst. Diam. Relat. Mater. 2016, 70, 105–113. [Google Scholar] [CrossRef]
- Velasquez, M.; Batiot-Dupeyrat, C.B.; Gallego, J.; Santamaria, A. Chemical and morphological characterization of multi-walled-carbon nanotubes synthesized by carbon deposition from an ethanol–glycerol blend. Diam. Relat. Mater. 2014, 50, 38–48. [Google Scholar] [CrossRef]
- Zhang, Z.L.; Verykios, X.E. Carbon dioxide reforming of methane to synthesis gas over supported Ni catalysts. Catal. Today 1994, 21, 581–595. [Google Scholar] [CrossRef]
- Goula, M.A.; Lemonidou, A.A.; Efstathiou, A.M. Characterization of carbonaceous species formed during reforming of CH4 with CO2 over Ni/CaO-Al2O3 catalysts studied by various transient techniques. J. Catal. 1996, 161, 626–640. [Google Scholar] [CrossRef]
- Barros, E.B.; Demir, N.S.; Souza Filho, A.G.; Mendes Filho, J.; Jorio, A.; Dresselhaus, G.; Dresselhaus, M.S. Raman spectroscopy of graphitic foams. Phys. Rev. B 2005, 71, 165422. [Google Scholar] [CrossRef]
- Dresselhaus, M.A.; Dresselhaus, G.; Saito, R.; Jorio, A. Raman spectroscopy of carbon nanotubes. Phys. Rep. 2005, 409, 47–99. [Google Scholar] [CrossRef]
- Helveg, S.; López-Cartes, C.; Sehested, J.; Hansen, P.L.; Clausen, B.S.; Rostrup-Nielsen, J.R.; Abild-Pedersen, F.; Nørskov, J.K. Atomic-scale imaging of carbon nanofibre growth. Nature 2004, 427, 426–429. [Google Scholar] [CrossRef]
- Das, S.; Jangam, A.; Du, Y.; Hidajat, K.; Kawi, S. Highly dispersed nickel catalysts via a facile pyrolysis generated protective carbon layer. Chem. Commun. 2019, 55, 6074–6077. [Google Scholar] [CrossRef] [Green Version]
- Mezzi, A.; Kaciulis, S. Surface investigation of carbon films: From diamond to graphite. Surf. Interface Anal. 2010, 42, 1082–1084. [Google Scholar] [CrossRef]
- Mizokawa, Y.; Miyasato, T.; Nakamura, S.; Geib, K.M.; Wilmsen, C.W. Comparison of the CKLL first-derivative auger spectra from XPS and AES using diamond, graphite, SiC and diamond-like-carbon films. Surf. Sci. 1987, 182, 431–438. [Google Scholar] [CrossRef]
- Cao, A.; Lu, R.; Veser, G. Stabilizing metal nanoparticles for heterogeneous catalysts. Phys. Chem. Chem. Phys. 2010, 12, 13499–13510. [Google Scholar] [CrossRef]
- Meshkani, F.; Rezaei, M. Ni catalysts supported on nanocrystalline magnesium oxide for syngas production by CO2 reforming of CH4. J. Nat. Gas Chem. 2011, 20, 198–203. [Google Scholar] [CrossRef]
- Pakhare, D.; Spivey, J. A review of dry (CO2) reforming of methane over noble metal catalysts. Chem. Soc. Rev. 2014, 43, 7813–7837. [Google Scholar] [CrossRef] [PubMed]
- Naeem, M.A.; Al-Fatesh, A.S.; Khan, W.U.; Abaseeed, A.E.; Fakeeha, A.H. Syngas production from dry reforming of methane over nano Ni polyol catalysts. Int. J. Chem. Eng. Appl. 2013, 4, 315–320. [Google Scholar] [CrossRef]
- Polychronopoulou, Κ.; Costa, C.N.; Efstathiou, A.M. The role of oxygen and hydroxyl support species on the mechanism of H2 production in the steam reforming of phenol over metal oxide-supported-Rh and -Fe catalysts. Catal. Today 2006, 112, 89–93. [Google Scholar] [CrossRef]
- Polychronopoulou, K.; Efstathiou, A.M. Spillover of labile OH, H, and O species in the H2 production by steam reforming of phenol over supported-Rh catalysts. Catal. Today 2006, 116, 341–347. [Google Scholar] [CrossRef]
- Campbell, C.T. Catalyst-support interactions: Electronic perturbations. Nat. Chem. 2012, 4, 597–598. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Temperature (°C) | 600 | 650 | 700 | 750 | 800 |
| D-parameter (eV) | 20.8 | 21.4 | 21.4 | 21.8 | 21.8 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Charisiou, N.D.; Douvartzides, S.L.; Siakavelas, G.I.; Tzounis, L.; Sebastian, V.; Stolojan, V.; Hinder, S.J.; Baker, M.A.; Polychronopoulou, K.; Goula, M.A. The Relationship between Reaction Temperature and Carbon Deposition on Nickel Catalysts Based on Al2O3, ZrO2 or SiO2 Supports during the Biogas Dry Reforming Reaction. Catalysts 2019, 9, 676. https://doi.org/10.3390/catal9080676
Charisiou ND, Douvartzides SL, Siakavelas GI, Tzounis L, Sebastian V, Stolojan V, Hinder SJ, Baker MA, Polychronopoulou K, Goula MA. The Relationship between Reaction Temperature and Carbon Deposition on Nickel Catalysts Based on Al2O3, ZrO2 or SiO2 Supports during the Biogas Dry Reforming Reaction. Catalysts. 2019; 9(8):676. https://doi.org/10.3390/catal9080676
Chicago/Turabian StyleCharisiou, Nikolaos D., Savvas L. Douvartzides, Georgios I. Siakavelas, Lazaros Tzounis, Victor Sebastian, Vlad Stolojan, Steven J. Hinder, Mark A. Baker, Kyriaki Polychronopoulou, and Maria A. Goula. 2019. "The Relationship between Reaction Temperature and Carbon Deposition on Nickel Catalysts Based on Al2O3, ZrO2 or SiO2 Supports during the Biogas Dry Reforming Reaction" Catalysts 9, no. 8: 676. https://doi.org/10.3390/catal9080676