
ApoB-Specific CD4+ T Cells in Mouse and Human Atherosclerosis


1
Department of Cardiology and Angiology I, University Heart Center Freiburg, Hugstetterstraße 55, 79106 Freiburg, Germany
2
Faculty of Medicine, University of Freiburg, Breisacherstraße 153, 79110 Freiburg, Germany
3
Facultad de Farmacia y Bioquímica, Universidad de Buenos Aires, CONICET, Instituto de Bioquímica y Medicina Molecular (IBIMOL), Junín 954, C1113 AAD Buenos Aires, Argentina
*
Author to whom correspondence should be addressed.
Cells 2021, 10(2), 446; https://doi.org/10.3390/cells10020446
Submission received: 1 February 2021
/
Revised: 15 February 2021
/
Accepted: 17 February 2021
/
Published: 19 February 2021
(This article belongs to the Special Issue Inflammation and Atherosclerosis)
Abstract
:Atherosclerosis is a chronic inflammatory condition of the arterial wall that leads to the formation of vessel-occluding plaques within the subintimal space of middle-sized and larger arteries. While traditionally understood as a myeloid-driven lipid-storage disease, growing evidence suggests that the accumulation of low-density lipoprotein cholesterol (LDL-C) ignites an autoimmune response with CD4+ T-helper (TH) cells that recognize self-peptides from Apolipoprotein B (ApoB), the core protein of LDL-C. These autoreactive CD4+ T cells home to the atherosclerotic plaque, clonally expand, instruct other cells in the plaque, and induce clinical plaque instability. Recent developments in detecting antigen-specific cells at the single cell level have demonstrated that ApoB-reactive CD4+ T cells exist in humans and mice. Their phenotypes and functions deviate from classical immunological concepts of distinct and terminally differentiated TH immunity. Instead, ApoB-specific CD4+ T cells have a highly plastic phenotype, can acquire several, partially opposing and mixed transcriptional programs simultaneously, and transit from one TH subset into another over time. In this review, we highlight adaptive immune mechanisms in atherosclerosis with a focus on CD4+ T cells, introduce novel technologies to detect ApoB-specific CD4+ T cells at the single cell level, and discuss the potential impact of ApoB-driven autoimmunity in atherosclerosis.
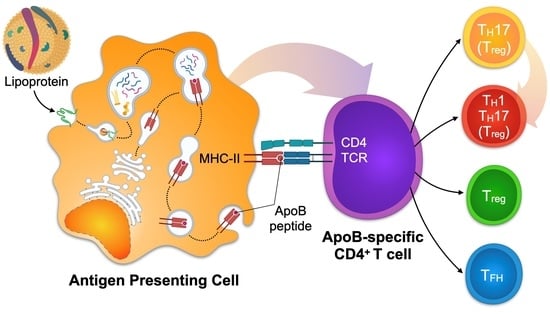
1. Atherosclerosis Is an Immune-Driven, Chronic Inflammatory Disease
Atherosclerosis is a chronic inflammatory disease that is characterized by the build-up of vessel-occluding plaques in the intimal layer of middle- to large-sized arteries [1]. Atherosclerosis precipitates myocardial infarction (MI) and stroke and represents the leading cause of death worldwide [2]. Epidemiologic studies have demonstrated that besides the traditional risk factors of smoking, hypertension, obesity, diabetes, and other environmental factors [3,4], low-density lipoprotein cholesterol (LDL-C) [5,6] is one of the driving factors of developing and progressing atherosclerotic disease. While atherosclerosis was originally perceived as a lipid-storage disease of the arterial wall [7], it is now established that the continuous deposition of LDL-C in the subintimal space is accompanied by a local and systemic low-grade inflammatory and immune response [8]. In the plaque, LDL-C is modified by oxidative processes (oxLDL) and taken up by tissue-resident macrophages. The continuous intracellular accumulation of lipids eventually exceeds the macrophage’s cholesterol storage capacity and intracellular lipid droplets form “foam cells” [9]. Foam cell formation and the per se pro-inflammatory properties of oxLDL [10] initiate a myeloid-dominated immune response with an increasing recruitment of monocytes from the blood circulation [11], a partially self-expanding population of plaque macrophages, and the secretion of proinflammatory cytokines, such as interleukin (IL)-1β by myeloid cells [12]. In addition, a variety of cell types of lymphocytic origin, including B and T cells, accumulate within the plaque and in the surrounding adventitia rendering the cellular architecture of atherosclerotic plaques almost as diverse as that of lymphatic tissue [13]. The continuous secretion of inflammatory mediators from myeloid and lymphoid cells is understood as a self-amplifying inflammatory cascade that ultimately promotes an unstable plaque phenotype, plaque erosion and rupture, and the formation of occlusive arterial thrombi that restrict blood flow and cause critical tissue ischemia in MI and stroke [14]. Of all risk factors, LDL-C provides the strongest causal link between clinical risk and cellular pathology: LDL-C lowering strategies attenuate plaque inflammation, promote plaque regression [15], and have been proven effective in reducing cardiovascular mortality in humans [16]. Likewise, anti-inflammatory treatments targeting the pro-inflammatory cytokine IL-1β [17] and by colchicine [18,19] prevent the progression of cardiovascular disease. A growing body of evidence suggests that the inflammatory milieu in the plaque is accompanied by a powerful autoimmune response [20,21] involving auto-reactive CD4+ T cells [22] and autoantibodies secreted by B cells [23]. While additional autoantigens cannot be excluded, overwhelming evidence shows that LDL-C represents the main culprit of this autoimmune response: While most auto-antibodies are directed against oxidation-specific epitopes in the lipid surface of LDL [23,24], autoreactive T cells recognize peptides from Apolipoprotein B (ApoB) [20], the core protein of LDL-C. Atherosclerosis can therefore be understood as chronic inflammatory disease of the cardiovascular system with a significant autoimmune component [21]. Here, we focus on the role of autoreactive CD4+ T-helper cells and comment on necessary developments to successfully translate novel immunomodulatory strategies into clinical practice.
2. Frequencies, Immune Phenotypes, and Roles of T-Helper Cells in Atherosclerosis
T cells represent the largest and most heterogeneous leukocyte population in human atherosclerotic plaques [25]. In immunohistochemistry (IHC), CD4+ T-helper and CD8+ cytotoxic T cells are detected in the shoulder region, the fibrous cap, and the intima of the plaque as well as in adventitial tissue [26]. In human plaques, T cells account for more than 50% of all lesional leukocytes with the highest T cell density in the shoulder region, while T cells account for less than 15% in the macrophage-dominated necrotic core [13,25,26,27]. In single cell RNA-sequencing (scRNAseq) and mass cytometry by time of flight (CyTOF) of human atherosclerotic plaques, T cells outnumber other hematopoietic lineages and reach a frequency of up to 65% of all leukocytes [28,29]. In atherosclerotic plaques from mice, T cell are less frequent and—depending on the underlying genetic model—range between 6% and 25% of all leukocytes [13,30,31]. In contrast to IHC, protocols employing tissue digestion and cell isolation are at the risk of overestimating non-myeloid cells due to a potential loss of macrophages during tissue digestion [13,25]. Notably, healthy arterial tissue contains CD8+ T cells in small frequencies [13]. Among T cells, CD4+ and CD8+ T cells are found in similar frequencies in atherosclerotic mouse aortas and human atherosclerotic plaques [25,28].
T cells develop from T cell precursors in the thymus. They transit through different developmental stages, including CD4+CD8+ double-positive (DP) T cells, and turn into either cytotoxic CD8+CD4− or helper CD4+CD8− T cells [32]. All T cells express CD3 and a unique T cell receptor (TCR) that binds antigenic peptides loaded on major histocompatibility complex (MHC): CD8+ T cells recognize peptides from intracellular proteins that are degraded by cytosolic and nuclear proteasomes and loaded on MHC-I, which is expressed in all nucleated cells [33]; CD4+ T cells recognize peptides from extracellular proteins that are taken-up via several pathways, degraded in the endosome, and loaded on MHC-II, which is primarily expressed in antigen presenting cells (APCs), such as dendritic cells (DCs) or plaque macrophages [33]. After the MHC-peptide complex is bound by their TCR, T cells are activated and develop into effector T cells (Teff). Antigen-recognition and co-stimulation are central processes for the differentiation and activation of CD4+ T-helper cells (TH) [34]. Teff develop into functionally and phenotypically distinct types of TH immunity that are characterized by the expression of canonical intracellular transcription factors (TF) and cytokines. Solid evidence, mostly from preclinical mouse studies, has established a distinct, but often controversial, role for many known TH-lineages in atherosclerosis.
2.1. TH1 Cells
Studies of CD4+ T cells in human plaques have suggested that—depending on cytokine secretion patterns—30–70% of all CD4+ T cells share features with TH1 cells [35,36]. TH1 cells express the TF T-bet [37], and secrete Interferon (INF)-γ, IL-2, IL-3, Tumor necrosis factor (TNF), and lymphotoxin [20,35,37]. In general immunity, TH1 CD4+ T cells are critical for developing an immune response against pathogens by enhancing the microbicidal activity of macrophages through enhanced IFN-γ production [38]. T cells from atherosclerosis-prone Apolipoprotein E deficient (Apoe−/−) mice secrete IFN-γ [39,40]. A genetic deficiency of IFN-γ [41,42], its receptor [43], and of T-bet [44] protects from atherosclerosis. Consistently, IFN-γ administration aggravates atherosclerosis in mice [45]. C-C chemokine receptor (CCR)-5 serves as homing receptor for TH1 cells [37] and is upregulated in plaque T-helper cells [28]. A genetic deficiency of CCR5 protects from T cell homing into the plaque in mice [40]. Therefore, TH1 cells are generally regarded as pro-atherogenic cells [20].
2.2. T Regulatory Cells
T-regulatory cells (Treg) are defined by the expression of the transcription factor FoxP3 and IL-2 receptor (CD25). Tregs are required to maintain self-tolerance and dampen immunity by secreting the immunosuppressive cytokine IL-10, Transforming growth factor (TGF)-β, and by direct contact-inhibition of Teff cells [46,47]. Tregs are found in human atherosclerotic lesions at a frequency of 1.2–3.9% of all CD3+ T cells [48]. In the mouse, 5% to 10% of CD4+ T cells express the Treg-lineage defining TF FoxP3 suggestive of Tregs [49,50]. Tregs express high levels of CTLA4, GITR, and lack expression of CD127 in humans, which enables the detection and cell sorting of viable Tregs [47,51,52,53]. In mouse atherosclerosis, Tregs are generally regarded as protective [54,55] and expand in regressing plaques [56]. Accordingly, IL-10 deficiency promotes atherosclerosis in mice [46]. Clinically, blood Treg numbers and IL-10 plasma levels are lower in patients with MI compared with healthy individuals [57]. A low fraction of Treg among all CD4+ T cells predicts MI [58]. However, the regulation and function of FoxP3+ Tregs in atherosclerosis remains controversial: In humans, one report has suggested higher frequencies of circulating Tregs in patients with stable atherosclerosis compared with healthy controls [49]. In mice, the population of Tregs in the spleen [59,60] and the liver [61] of Apoe−/− and Ldlr−/− mice increases in the context of hypercholesterolemia. These associative findings argue against a solely protective role of Tregs and may be partially explained by the appearance of Treg-like CD4+ T cells that express FoxP3 and pro-inflammatory cytokines in advanced atherosclerosis [37,40,49]. The function of these abnormal Tregs will be discussed below.
2.3. TH17 Cells
TH17 cells express the TF RORγT and secrete IL-17 [62]. TH17 CD4+ T cells are gatekeepers of mucosal immunity and have been associated with several autoimmune diseases. They are activated by IL-23 and secrete the cytokines IL-17A and -F [63]. Numerous studies have revealed a highly controversial role of TH17 cells in mouse and human atherosclerosis: Some studies showed proatherogenic effects [64,65,66,67] in mice and higher plasma IL-17 levels in humans with unstable angina or MI [68,69]. These findings are consistent with an overall proatherogenic role of TH17 immunity [27]. Other studies demonstrated atheroprotective and plaque-stabilizing properties in mice [70,71,72,73,74] and lower plasma levels of IL-17 in patients with acute MI [75] suggestive of an overall atheroprotective role [41]. In addition, some studies have found no role for TH17 cells in mice [76], which is in accord with unchanged IL-17 plasma levels in humans with or without coronary artery disease (CAD) [77].
2.4. TH2 Cells
TH2 cells are involved in the immune response against parasites, in asthma, and other allergic diseases [78]. They express the TF Gata3 and secrete IL-4, IL-5, IL-10, and IL-13 [13]. Their role in atherosclerosis is unclear. Pro-atherogenic and atheroprotective functions have been proposed in mice [79,80,81,82]. In humans, low TH2 cell numbers and weak IL-4 secretion from CD4+ T cells predict myocardial infarction [83]. In addition, low plasma concentrations of IL-5 associate with subclinical carotid atherosclerosis [84]. Both studies argue for a protective role of TH2 immunity in progressing and de novo atherosclerosis. Likewise, IL-33 administration reduces murine atherosclerosis by increasing levels of IL-4, IL-5, IL-13, and INF-γ in the plasma [85].
2.5. Follicular-Helper T Cells (TFH)
T-follicular helper cells (TFH) provide help for B cells and are required for the antibody isotype switch in germinal center B cells [86]. They express the TF Bcl-6 and the chemokine receptor CXCR5 [86]. Dyslipidemia in Apoe−/− and Ldlr−/− mice induces TFH cells and IgG2c production [87]. Depletion of TFH cells protects from atherosclerosis [50]. TFH cells have been suggested to orchestrate a pro-atherogenic B cell response in mouse atherosclerosis [88]. Ageing increases the number of TFH cells in Apoe−/− mice [89]. TFH cells are found more frequently in advanced atherosclerosis [90]. It has been suggested that TFH cells are at least partially derived from Treg cells [50] and from ApoB-specific CD4+ T cells [91].
2.6. CD4+ Cytotoxic Lymphocytes (CTL)
Lymphocytes with a cytotoxic potential include natural killer (NK) cells, CD8+ T cells, NK T cells, γ/δ T cells, and a subset of human CD4+ T cells that is characterized by a down-regulation of the co-stimulatory molecule CD28 (CD4+CD28null T cells) [92]. CD4+ CTLs represent a highly differentiated subset of memory T cells [93], secrete perforin, granzyme A and B, and TNF-α and IFN-γ [94] and express high levels of the exhaustion marker OX-40 [95]. They have not been detected in mice [96] and are found in human vulnerable atherosclerotic lesions [97,98,99]. Their distinct function in atherosclerosis has not been tested so far [94]. It may be that CD4+ CTLs do not represent a distinct T-helper cell lineage, but instead the fraction of antigen-specific [100,101], terminally differentiated, and exhausted CD4+ T cells that acquire cytotoxic functions [28,102].
2.7. Other Types of T Cell Immunity
TH9 are characterized by the expression of IL-9 in response to TGF-β and IL-4. Their generation is inhibited by INF-γ [103] and depends on several TF, including FoxO1, BATF, and IRF4 [104]. Clinically, IL-9 plasma levels are higher in patients with atherosclerosis [105] and with an acute coronary syndrome, while the count of TH9 cells was unchanged in another study [106]. IL-9 administration seems to promote atherosclerosis in Apoe−/− mice [107], but the overall role of TH9 cells remains unknown. TH22 cells express IL-22 and the TF aryl hydrocarbon receptor (AHR) [108]. Based on one report, TH22 immunity may be pro-atherogenic [109]. TH22 cells in the blood [106,110] and plasma levels of IL-22 are increased in patients with an acute coronary syndrome [106,110]. Other T cell subsets described in atherosclerosis include innate-like NK T cells that express restricted pairs of TCR α- and β- chains and recognize self and foreign lipid antigens presented on the MHC-I like molecules CD1d [111]. NK T cells secrete TH1, TH2, and TH17 cytokines as well as perforin and granzyme B. NKT cells are found in rupture-prone human atherosclerotic plaques [112], but their role in atherosclerosis remains controversial [94,113]. CD8+ T cells represent the main cytotoxic T cell subset that participates in the immune defense against intracellular pathogens and in tumor surveillance [114]. In atherosclerosis, CD8+ T cells have been attributed to a multitude of both pro- and anti-inflammatory roles. CD8+ T cells may suppress inflammation, control macrophage accumulation, and partially by direct cytotoxic effects on lesional macrophages, contribute to endothelial cell surveillance and damage, and exhibit direct effects on myelopoiesis. Whether CD8+ T cells are antigen-specific is a matter of debate. The roles of CD8+ T cells has been extensively reviewed [115] and is beyond the scope of the present review.
2.8. Multi-TH Committed CD4+ T Cells in the Atherosclerotic Plaque
Traditionally, TH-types in CD4+ T cells represent a unidirectional and terminal path of differentiation that is inflexible and irreversible. This concept has been challenged by growing evidence that TH-cells can reprogram towards mixed phenotypes of TH cells or re-differentiate into alternative TH types of cells [116]. Tregs and TH17 cells seem to be particularly prone to such TH cell plasticity [117]: Tregs can acquire features of TH1 (TH1-Tregs) or TH17 cells (TH17-Tregs), or switch into TFH cells [37,40,118,119,120,121,122,123]. Mechanistically, it has been demonstrated that initially immunosuppressive FoxP3+ Tregs downregulate FoxP3 protein expression in direct lineage tracing experiments and give rise to exTregs that express alternative TFs [49,50]. In the atherosclerotic plaque, a considerable fraction of CD4+ T cells express low levels of FoxP3 as well as of IFN-γ, and T-bet [37,40]. These cells promote atherosclerosis after an adoptive transfer, have lost their immunosuppressive properties, and act as effector T cells [40]. Consistently, recent scRNAseq of T cells from mouse atherosclerotic plaques demonstrated CD4+ T cell clusters with mixed TH1/TH2/Treg and TH1/TH17 transcriptomes that account for approximately 50% of all lesional T cells [49]. The co-expression of genes suggestive of TH1- and TH17-phenotypes has also been demonstrated in CD4+ T cells isolated from human carotid plaques [28]. In single cell gene module enrichment analysis, core genes of these mixed phenotypes were enriched in tetramer-selected ApoB-specific CD4+ T cells [49], proposing that Treg lineage instability occurs frequently in antigen-specific T cells in the plaque. In addition to multi-TH committed CD4+ T cells in atherosclerotic plaques, the existence of IL-17 or RORγT expressing Tregs has also been suggested in the blood of patients with cardiovascular disease (CVD) [49,50]. These findings indicate that the generation of CD4+ T cells with features reminiscent of several TH-types is a systemic, rather than a local event in atherosclerosis. Together, these findings question whether traditional TH lineages reflect the actual functional heterogeneity of T-helper cells in the atherosclerotic plaque (Table 1). Whether such high plasticity is driven by antigen-specificity as suggested by other disease models beyond atherosclerosis [124,125,126] will be discussed below.
3. Evidence for Autoimmunity in Atherosclerosis
Autoimmune disease is defined as an abnormal response of the immune system against endogenous proteins and other components of the body (autoreactivity). This immune response can lead to the damage, destruction, or functional loss of involved tissues [130]. Naturally occurring Tregs prevent autoimmunity against self-peptides and -antigens [47]. As outlined above, the accumulation of T cells in the plaque and the appearance of circulating autoantibodies in patients with atherosclerosis has inspired the idea of an autoimmune component in addition to antigen-independent inflammation [131]. Several observations support this hypothesis: First, T cells in the plaque of mice and humans exhibit an unexpected strong memory phenotype with hallmarks of chronic stimulation and T cell exhaustion as evidenced by a high proportion of CD4+CD45RAlowCCR7low TEM and of CD4+ T cells expressing high levels of the activation markers CD69 and CD38 [28] and of cytotoxic factors [29]. These findings suggest the presence of antigen-specific T cells that have built up a T cell memory against antigens that are likely present in the plaque or the draining lymphatics [28,60]. The expression of programmed cell death protein 1 (PD-1), a known exhaustion marker, and of several genes associated with exhaustive T cell signaling, such as EOMES and LAG3, in human plaques further argues for chronic antigen recognition by CD4+ T cells in the plaque [28]. T cell exhaustion is understood as a negative regulator and checkpoint of chronic T cell stimulation and -antigen recognition [132]. Inhibition of PD-1, which dampens subsequent cellular activation, results in aggravated atherosclerotic disease in mice [133]. This finding is consistent with the hypothesis that antigen-specific and pathogenic CD4+ T cells in the advanced plaque become resistant to the ongoing recognition of their cognate antigens by exhaustive transcriptional programs. A therapeutic checkpoint inhibition, as performed in several malignancies, could therefore re-activate plaque T cells, and provoke complicated atherosclerosis [28,134]. Second, T cells frequently interact with plaque-resident APCs in live cell imaging in mice, in particular when APCs and T cells originate from atherosclerotic and hypercholesterolemic Apoe−/− mice. As result of this physical interaction, T cells secrete pro-atherogenic cytokines such as IFN-γ [39]. Notably, pro-inflammatory cytokine secretion in this model requires the presence of atherosclerosis-related antigens and is not observed when unrelated model antigens are used as control. Likewise, human and mouse T cells from atherosclerotic plaques secrete cytokines and proliferate when restimulated with LDL or peptides from ApoB [22,49,135]. Third, T cell activation is a result of antigen-recognition and promotes the proliferation of antigen-specific T cells with the same TCR to build oligoclonal populations. In mouse plaques, T cell proliferation is evident in histological analysis [39,136] and in scRNAseq, where clusters of proliferating cells contained T cell signatures [13,137]. Lesional T cells seem to be clonally enriched in TCR-sequencing [138] in mice. In vitro cloned T cell lines reactive against ApoB show a preferential usage of the V-chain segment 31 (TCRBV-31) [135]. Consistently, MHC-II tetramer selected T cells expressed an oligoclonal TCR-repertoire [49]. In humans, T cell clonality in the plaque [97,139] and in coronary thrombi [140] are restricted in TCR-usage. Only one report has suggested a non-restricted repertoire in atherosclerotic aortas [141]. Whether CD8+ or CD4+ T cells or both are the cause of such TCR-restriction is not known, but a recent report has demonstrated a correlation between T cell exhaustion in lesional CD8+ T cells and TCR-clonality [28]. Fourth, several autoantigens have been derived from direct vaccination experiments: LDL-C/ApoB (discussed below), heat shock proteins (HSPs) [142,143], and β2-Glycoprotein I (β2GPI). HSPs are intracellular, highly species-conserved chaperones that protect against stress and physical irritation such as temperature, UV light, and changes in the pH [144]. In humans, antibodies directed against HSP60 correlate with cardiovascular disease [145,146]. In addition, immunization using HSP60/65 and peptides thereof as antigens modulates atherosclerosis [147,148,149,150,151,152,153,154,155,156]. Interestingly, it was proposed that bacteria derived HSP65 induces an autoreactive response against human HSP60. Both molecules express similar immunodominant B cell epitopes [157], which may explain a cross-reactivity between infection-derived epitopes and self-epitopes as recently shown for the cross-reactivity between Streptoccocus pneuomniae and oxidation-specific epitopes in mice [158]. β2GPI is the target of anti-cardiolipin antibodies [159] that cause the anti-phospholipid syndrome, a state of hyper-coagulation in systemic lupus erythematosus patients [160]. β2GPI has been found in human atherosclerotic lesions [161] but direct vaccination experiments using β2GPI have yielded inconsistent results [162,163,164,165,166]. In addition to the aforementioned autoantigens, several targets of IgM- and IgG-autoantibodies, mostly oxidation-specific epitopes of LDL, have been suggested [23,24,167]. It has also been discussed that a fraction of antigen-specific T cells in the atherosclerotic plaque recognizes infectious peptides from bacteria or viruses. This hypothesis is based on numerous observations, foremost the clinical association of infectious disease and atherosclerosis: For instance, observational studies have established that an infection with Varicella Zoster Virus (VZV) and Influenza Virus increases the risk for MI and stroke [168,169]. Vaccination against Influenza is now recommended for secondary prevention of patients with heart disease [170] and improves the cardiovascular outcomes [171,172,173]. Human Cytomegalovirus (HCMV), Herpes Simplex (HSV), Epstein Barr Virus (EBV), VZV, and Influenza Virus were suspected of causing an infection of the arterial vessel wall [174,175,176]. However, only in rare cases, viral particles have been detected within atherosclerotic lesions [176,177,178] and a potential interference of infection and atherosclerosis may be explained by increased inflammatory signaling cascades, local tissue injury, and enhanced thrombotic pathways [176,179] rather than a direct pathogenicity of virus-specific T cells in the atherosclerotic plaque. Such indirect effects also seem to trigger some of the cardiovascular complications of SARS-CoV2 [180]. In addition, it cannot be excluded that some autoreactive T cells in the plaque are cross-reactive to exogenous infection with a structural similarity (molecular mimicry) to autoantigens as shown for S. pneumoniae which induces antibodies that bind oxLDL [158,181,182]. Whether pneumococcal vaccination is beneficial in targeting auto-antigens in the plaque remains controversial [183,184,185]. Of all proposed atherosclerosis-related (auto-) antigens, LDL-C and ApoB provide the strongest causal link between autoimmunity and the pathogenesis of atherosclerosis. We will therefore focus on the role of ApoB-specific CD4+ T-helper cells in the following sections.
4. ApoB-Specific CD4+ T Cells in Mice and Humans
4.1. Mechanisms of CD4+ T Cell Activation
The activation and transition from naïve to effector/memory T cells (TEM) is a process that encompasses two signals: The presentation of the antigen by an APC (Signal 1) and additional co-stimulatory signals (Signal 2). DCs and macrophages are found in healthy arteries and atherosclerotic plaques [8,131,186] and serve as APCs at different stages of the disease [187]. Cellular, sub-cellular, or molecular antigens are taken up by the APC by phagocytosis or endocytosis and processed in the endosome. Antigen-derived peptides may then bind to an MHC in the Golgi apparatus depending on the binding affinity between the MHC and the peptide. The complex of peptide and MHC is next translocated to the cell surface [188]. In the presence of a suited antigen-specific T cell, the MHC-peptide complex is bound by a unique TCR. The bond between TCR and MHC-peptide is stable for several hours [189]. TCR signal-transduction in the T cell is mediated by the complex of CD3, CD4, and the TCR [190], and by down-stream signaling molecules like zeta-chain-associated protein kinase 70 (ZAP-70) and SH2 Domain-containing Leukocyte Protein of 76 KDa (SLP-76) [191]. These signaling events result in the transcription and translation of proteins necessary for the differentiation and proliferation of the activated T cell [192,193]. “Signal 2” describes the additional signaling by a total of 38 possible combinations of co-stimulatory and co-inhibitory ligands and receptors [194]: Co-stimulatory pairs of ligands and receptors, such as CD28/CD80, promote activation whereas others, such as CTLA4 and CD80, prevent subsequent TCR-signaling and cell activation [195,196,197]. TFs and signaling pathways in APCs that induce a tolerogenic response in T cells include IL-10, TGF-β, Flt3-, and Myd88-dependent signaling events [198,199,200,201], while cholesterol accumulation and IRF-8-dependent signaling promote an immunogenic response [202,203]. Further differentiation signals are provided by pro- or anti-inflammatory cytokines secreted by APCs [186,204,205,206]: The anti-inflammatory signals IL-10 and TGF-β induce tolerogenic responses and predispose to Treg differentiation, while IL-6 prompts a TH17, and IL-12 a TH1 response. A firm cellular bond between the APC and the T cell requires the interaction of additional cell–cell adhesion molecules, such as LFA-1/ICAM [207]. The physical interaction site of the T cell and the APC that exhibits a high density of cell adhesion molecules, co-stimulatory molecules, and peptide-loaded MHC/TCR complexes is often referred to as the immunological synapse [208]. Several assays have been designed to identify antigen-specific T cells, including single cell detection by MHC-II multimers, functional restimulation, and cloning of T cell lines [209,210,211].
4.2. Detection of ApoB-Specific CD4+ T Cells in Humans by Functional Restimulation
We have recently introduced an in vitro restimulation assay for the detection of ApoB-specific CD4+ T cells in humans [49] (Figure 1A). In this assay, human PBMCs including APCs and T cells are co-incubated in vitro with a mix of antigenic peptides from ApoB-100. To limit all possible ApoB-100 peptides to the ones that could potentially be loaded on MHC-II, an in-silico screening of human ApoB-100 and direct MHC-II-peptide affinity measurements was performed. This screening strategy generated a pool of ApoB-100 candidate peptides with a high affinity for several human MHC-II alleles, thereby covering 80% of a Caucasian population with unknown MHC-II variants [49]. The upregulation and detection of T cell activation markers as a result of peptide-recognition by T cells serves as marker for peptide-specific CD4+ T cells. In vitro culturing itself is known to decrease cell viability and interferes with T cell differentiation pathways, TF expression, and cytokine secretion. Therefore, the time of restimulation has to be kept to a minimum. Accordingly, the kinetics of cell surface marker expression used for the identification of activated CD4+ T cells needs to be carefully considered. CD25, CD69, CD154 (CD40L), and OX40 are established CD4+ T cell activation markers [212] and are highly expressed in human unstable atherosclerotic plaques [213]. CD25 is the receptor for IL-2 (IL-2R) and peaks 72 h after CD4+ T cell stimulation. CD25 is therefore not suited as an immediate activation marker [214]. OX40 shows similar dynamics and peaks between one and five days after stimulation [215]. Contrastingly, CD69 is upregulated already 30 to 60 min after stimulation with a sharp decrease in expression after 4 to 6 h. Comparative studies have shown that frequencies of antigen specific T cells found after short time stimulation using CD69 and CD40L were similar compared with a stimulation for 8 h using CD25 and OX40 as activation markers [216]. Although its expression profile seems most favorable for in vitro activation assays, CD69 is expressed on naïve and memory T cell subsets and responds to unspecific cellular activation, such as by calcium ionophores. Contrastingly, CD40L has been shown to serve as an immediate activation marker with a high specificity for TCR-signaling events [217], making it an ideal candidate for antigen-specific restimulation assays. A downside of using CD40L remains its transient extracellular expression [218]: After translocation to the cell surface, CD40L is quickly degraded, likely by matrix-metalloproteinases [219], and internalized [220]. We have validated an in vitro assay for the identification of human ApoB-100 specific T-cells employing intracellular CD40L as immediate activation marker in a restimulation assay for 6 h [49]. Using this assay, we were able to show that T cells specific for ApoB-100 peptides exist in the blood circulation of patients with CAD, but not in healthy individuals. In this assay, patients with CAD expressed higher levels of TNF-α, IFN-γ, and IL-17. Contrastingly, the expression of IL-10 decreased in patients with CAD compared to healthy individuals. The findings demonstrate that T-helper cells specific for several ApoB self-peptides exist in humans with atherosclerotic disease.
4.3. Detection of ApoB-Specific CD4+ T Cells in Mice and Humans by Tetramers of MHC-II
Kimura et al. recently introduced multimers of MHC-II loaded with ApoB-specific peptides to detect peptide-reactive CD4+ T cells at the single cell level [118]. These reagents take advantage of the binding of recombinant MHC-II molecules with a pre-defined peptide to a TCR solely specific for this MHC-II-peptide complex. Because the MHC-II-peptide–TCR binding of monomeric MHC-II complexes is weak with a short half-life in the range of seconds, the avidity of this interaction can be increased by coupling several MHC-II-peptide complexes that engage more than one TCR, often as tetramers or dextramers [209,210,211]. The labeling of these reagents with classical fluorochromes for flow cytometry allows the subsequent detection of T cells specifically binding this MHC-II-peptide complex, i.e., T cells with a TCR specific for this (peptide) antigen (Figure 1B). Kimura et al. made use of a combined in silico screening for the 28 most common human MHC-II (HLA-DR) alleles and verification of affinities in competitive binding assays. Several peptide sequences of human ApoB were identified and incorporated into corresponding tetramers. In a sub-study of participants of the Women’s Interagency HIV Study (WIHS) expressing the HLA-DRB1*07:01 allele, 0.17% of all CD4+ T cells were reactive against the peptide-epitope p18 (SLFFSAQPFEITAST). More than half of all ApoB/p18-reactive T cells from donors without atherosclerotic disease expressed FoxP3, which indicates the predominance of an immunosuppressive phenotype. The remaining ApoB/p18-reactive T cells expressed RORγt, Gata3, or T-bet. TF expression in ApoB:p18 specific T cells was not exclusive and often occurred in combinations, indicating the existence of TH17-Tregs and TH1-Tregs. Notably, the fraction of these multi-lineage committed cells increased, in particular the co-expression of the TH1 TF T-bet, in donors with subclinical atherosclerosis, while the fraction of single FoxP3 expressors decreased. These results therefore suggest that ApoB-specific CD4+ T cells in humans shift towards a more inflammatory phenotype in the context of atherosclerosis. In another study, we recently introduced a tetramer of mouse MHC-II (I-Ab) to characterize mouse CD4+ T-helper cells recognizing the ApoB peptide p6 (ApoB978–993) [49]. ApoB-reactive T cells isolated with this tetramer were detectable in lymph nodes of healthy and atherosclerotic mice and showed a predominant Treg and partially atheroprotective phenotype in healthy Apoe−/− mice. Notably, these cells had formed a memory phenotype already in healthy mice in about 20% of all ApoB+ T cells. In the setting of hypercholesterolemia, ApoB+ T cells proliferated, expressed pro-inflammatory genes, partially lost the Treg-defining TF FoxP3, and converted into pathogenic TH1 and TH17-like cells with an only residual Treg gene signature. Both studies demonstrate that tetramers represent a feasible method to detect ApoB-specific CD4+ T cells in human blood and in murine lymphoid tissue. It is noteworthy to point out that tetramers with a single peptide specificity may underestimate other T cells clones binding overlapping, adjacent sequences, and that TCR-binding to a given peptide sequence may be less specific, as supposed in [181]. Together with our recent observation that ApoB-specific T cells overlapped with a fraction of 50% of lesional T cells in atherosclerotic aortas [49], it is highly plausible that T cells with more peptide specificities against ApoB or other autoantigens exist in atherosclerotic plaques. Because TCR-clonality in the aorta may directly link to a functional phenotype as recently suggested [221], combined TCR-sequencing and single cell RNA-sequencing workflows may provide a valuable tool to directly infer antigen-specificity from TCR-clonality in scRNAseq in future (Figure 1C). Although in situ tetramer staining in tissue sections is technically possible [222], it has not been tested on atherosclerotic plaques yet.
4.4. Cloning of CD4+ T Cell Lines and TCR-Transgenic Mice
In the naïve organism, only a small fraction of T cells is expected to be specific for a given (auto) antigen. The absolute size of a population of T cells specific for self or foreign antigens differs considerably and ranges between 10 and 10,000 cells per mouse [181]. Around ~1200 CD4+ T cells specific for the self-peptide p6 are found in Apoe−/− mice [49]. These numbers render it experimentally extremely difficult to assess the function of ApoB+ T cells in in vivo. Recently, Gistera et al. have reported the first TCR-transgenic mouse recognizing human LDL/ApoB [91]. In a series of reports (Figure 1D), the authors first isolated CD4+ T cells from transgenic mice expressing human ApoB100 that were immunized with human oxLDL. T cells from these mice were isolated, restimulated with human oxLDL, native human LDL, or purified human ApoB-100, and reactive single cell clones were identified by IL-2 expression in vitro. V-segments of the T α- and β-chains of the TCR were determined by RT-PCR on IL-2 reactive clones. The TCR-β V-segment 31 was the only V-β segment uniformly expressed across all clones. In non-LDL-reactive clones, V-segment usage was not restricted [135]. Antibodies against the predominating V-chain segment TCRBV31 protected from atherosclerosis in vivo, likely by an elimination of atherosclerosis-relevant T cell clones. In a second study [91], the TCR clone containing the enriched β chain V-segment TRBV31 was used for a TCR-transgenic mouse, in which 90% of T cells expressed TRBV-31 and recognized LDL. In vivo, ApoB-specific CD4+ T cells developed into TFH cells, activated B cells, stimulated the formation of germinal centers, and induced the expansion of plasma cells expressing anti-LDL immunoglobulins. Anti-LDL IgGs enhanced LDL clearance in the liver—a mechanism that led to decreasing LDL-C plasma levels and significantly smaller atherosclerotic lesions than in controls. Thus, ApoB-reactive T cells have the ability to serve as potent TFH. Whether the selection procedure in vivo used in this series of reports predisposes for a specific TCR/phenotype that does not predominate in TCR-WT mice is currently not known. Still, it remains an interesting speculation that low and high affinities between a peptide loaded MHC-II and the TCR induce distinct transcriptional programs in T cells, which may induce distinct TH types as recently suggested [221].
5. Function of ApoB-Specific CD4+ T Cells
As stated above, LDL-C levels correlate with adverse clinical outcomes [1] and the progression of coronary atherosclerosis [5] and represent one of the best established targets for medical prevention of CVD [223]. Besides the plethora of innate-related inflammatory mechanisms, LDL-C likely serves as autoantigen in the atherosclerotic plaque, which is best demonstrated by the ability of human plaque T cells to secrete pro-inflammatory cytokines and proliferate when restimulated with LDL preparations—an effect highly dependent on MHC-II antigen presentation, suggesting specificity of these results [22,135]. In addition, ApoB-specific TH cells have been detected in humans by MHC-II tetramers [118]. The function of ApoB-specific CD4+ T cells in humans can currently only be inferred from their differentiation into classical TH types of immunity. These findings suggest that in the presence of sub-clinical or clinical atherosclerosis, ApoB+ T cells in humans are more polarized towards TH17 and TH1 cells, while only maintaining a residual Treg signature in healthy individuals. It is therefore plausible that the compartment of ApoB-specific autoreactive TH cells may have protective properties in health but switch into pro-inflammatory TH types in disease. Whether this switch is causal for the pro-inflammatory environment in the plaque or a result of the inflammatory response that accompanies atherosclerotic disease is currently unknown.
Since 1959, when Gero et al. performed the first atheroprotective vaccination of rabbits with LDL [224], numerous studies in rodents have demonstrated that vaccination with either native, modified (oxidized) LDL, or (peptides from) ApoB has the potential to elicit a T-cellular immune response that prevents atherosclerosis [225,226,227,228,229,230,231,232,233,234]. ApoB-100 and its truncated version, ApoB-48, which is present in chylomicrons [235], contain several immunogenic T cell peptide epitopes. In contrast, B cell epitopes are mostly located on lipid moieties of native and modified apolipoproteins [23]. Direct immunization with ApoB-100 and ApoB-100 peptides protects from atherosclerosis, likely by the induction of IL-10 secreting protective Tregs [199,236,237,238]. The immunogenic peptide epitopes from human or mouse ApoB that have been validated by vaccination of mice expressing the wildtype, mouse ApoB-100, or human transgenic ApoB-100 include the peptides p3, p6, p18, p101, p102, p103, p210, p265, and p295 [49,239,240,241]. p18 is the only so far identified peptide that is sequence-identical in mouse and human ApoB. The ApoB-peptide p6 (ApoB978–993, sequence TGAYSNASSTESASY) has been most extensively characterized. p6 is located in the surface region of ApoB-48 and ApoB-100 at the interface of the amphipathic core region [240]. Vaccination with p6 induces an antigen-specific T cell response with cellular proliferation and cytokine secretion [240]. Interestingly, ApoB p6-reactive T helper cells isolated from immunized mice and transferred to donor mice promoted atherosclerosis in abdominal aortas [242], while vaccination with p6 prevented atherosclerosis in Apoe−/− mice in another report [240]. This finding is striking because most reports employing vaccination with ApoB/LDL have suggested a primarily protective phenotype encompassing Tregs in the atherosclerotic aorta and spleen as well as IL-10 secretion [240,243,244] or a decrease of TH1 immunity [245]. Notably, some of these favorable effects were abolished by a depletion of Tregs [245]. The functional dichotomy raised by vaccination studies using p6 was partially clarified in later studies that took use of a tetramer of MHC-II loaded with the peptide p6. It was suggested that ApoB-specific CD4+ T cells stem from a Treg-like TH17 cell with a partially protective phenotype that was lost during progressing natural disease and replaced by a TH1-like phenotype with several pro-inflammatory transcriptional programs including TNF-α, IL-6, and IFN-γ (Figure 2). These findings are consistent with the phenotypes observed in human ApoB restimulation assays and suggest that ApoB-specific CD4+ T cells per se are neither atheroprotective nor pro-atherogenic. Instead, phenotypes of ApoB-reactive CD4+ T cells may be dictated by the microenvironment in the plaque or systemic inflammation [49]. A protective role of antigen-specific CD4+ T cells can also be derived from the observation that a genetic knock-out for MHC-II, which abrogates antigen-recognition and -presentation, promotes de novo atherosclerosis [246,247]. Because hypercholesterolemia (with elevated LDL-C levels) in mice favors the differentiation of Tregs in the early stages of atherosclerosis [60,61] and enhances TCR-signaling events in Tregs [59], it must be hypothesized that in healthy mice, a cellular Treg-driven protective autoimmune response against LDL-C/ApoB exists [247]. It is therefore plausible, but remains experimentally unproven, that early ApoB-reactive Tregs have the ability to prevent atherosclerosis. It is also tempting to speculate how these cells are generated and why they appear even in healthy individuals and mice. The general concept of autoimmunity states that the immune system distinguishes between self and non-self [248]. Some CD4+ T cells expressing a T cell receptor (TCR) that recognizes self-peptides loaded on MHC-II with a high affinity are eliminated by negative selection [249,250]. However, negative selection is not very efficient [251,252] as demonstrated by the existence of self-antigen specific CD4+ T cells in a naïve organism [181]. A part of these surviving T cells develops into pathogenic Teff, while these with a low to intermediate affinity turn into protective Tregs [253,254]. Thus, autoimmunity is understood as a competition of protective and pathogenic CD4+ T cells that both recognize (different) self-peptides from the same autoantigen. Indeed, several reports have demonstrated the existence of autoreactive T-helper cells with a protective Treg and a pathogenic Teff phenotype in autoimmune disease of the central nervous system, graft-versus-host-disease, and in type 1 diabetes [124,126,255,256,257,258]. During development of disease, this fine-tuned balance is thought to shift towards a relative overrepresentation of pathogenic, often TH1 T cell phenotypes. In atherosclerosis, autoreactive CD4+ T cells with a predominant or partial Treg phenotype in the absence of atherosclerosis and a pathogenic phenotype in established disease have been found in two studies employing MHC-II tetramers in humans and mice [49,118]. Whether pathogenic Teff exclusively develop from switching Tregs or independently and how the composition of autoreactive CD4+ T cells with Treg and Teff cells changes over time, is currently unknown. It also remains unclear if the phenotypic switch of ApoB+ T cells to a more pathogenic phenotype is a cause or a consequence of exaggerated inflammation in advanced atherosclerosis. Whether the resulting TH1-like ApoB-reactive T cells are in fact pro-atherogenic has not been directly tested. TF expression from human ApoB-reactive T cells suggests that FoxP3 protein-expressing Tregs dominate the phenotypic repertoire of ApoB-specific TH cells in healthy humans. In healthy mice, only a minor fraction of ApoB-specific CD4+ T cells expresses FoxP3, while the majority shows a transcriptional similarity to Tregs but does not express FoxP3 protein [49]. Lineage-tracing experiments have demonstrated that not all FoxP3neg ApoB-specific T cells stem from initial FoxP3+ Tregs [49]. Therefore, Treg plasticity does not explain the generation of all pathogenic ApoB-specific Teff cells alone [20]. Notably, mouse ApoB+ T cells demonstrate at least a partial TFH signature with Bcl6 and Cxcr5 transcripts [49], indicating that the TFH phenotype observed in the ApoB-TCR-transgenic mouse may represent a natural occurring phenotype, albeit likely overrepresented in the experimental settings in the transgenic mouse model [91]. Potential factors that cause ApoB-reactive T cells to transform from protective (Treg-like) to pathogenic TH-types have not been explicitly investigated and are currently unknown. However, it is plausible that factors that influence the stability of Tregs—hypercholesterolemia, inflammatory cytokines, local hypoxia, and changes in cellular metabolism [20,25,117]—partially overlap with those favoring the pathogenic conversion of functionally protective FoxP3neg ApoB-specific CD4+ T cells.
6. Clinical Translation and Outlook
The fundamental role of inflammation in atherosclerosis has been increasingly appreciated in the last decades [20,259]. Clinical landmark trials, such as the CANTOS and COLCOT trials [17,18,19], have highlighted the potency of anti-inflammatory treatment strategies in cardiovascular disease prevention. However, the limitations of unspecific anti-inflammatory treatments remain considerable as evidenced by increased rates of infection and missing experience on long-term treatments. By contrast, the development of antigen-specific immunomodulation holds the promise of specific antigen-directed therapies with only minimal side-effects [234]. The recent development of technologies to detect ApoB-specific T cells at the single cell level, including MHC-class II tetramers, has greatly widened our understanding of adaptive immune mechanisms in atherosclerosis [25]. It is now clear that ApoB-specific TH cells exist in mice and humans. These undergo dramatic transcriptional, numeric, and phenotypic changes throughout the natural course of atherosclerosis. While the predominant pro-atherogenic TH1 phenotype of ApoB-reactive TH cells in the blood of patients with advanced atherosclerosis and in atherosclerotic plaques is consistent with older findings, it is striking that ApoB-reactive T cells in early disease are transcriptionally closer to immunosuppressive Tregs [49,118]. This observation provides a reasonable explanation for enhanced numbers of ApoB-specific Tregs in numerous mouse vaccination studies and suggests that autoimmunity per se is not detrimental but required to restrain a pathogenic immune response in most healthy individuals [247]. Vaccination with tolerogenic adjuvants and immunogenic ApoB-peptides may therefore have the potential to reinforce the protective limb of ApoB-specific immunity even in patients with established atherosclerosis. The successful implementation of a human atherosclerosis vaccine in clinical practice will depend on several developments that have yet to be made: First, exact doses, routes of delivery, and adjuvants in a vaccine need to be clarified. In addition, additional autoantigens beyond ApoB are likely to exist and may be found in a screening of peptides that are naturally presented on MHC-II in atherosclerotic plaques. Second, patients with an immune-mediated type of atherosclerosis independent of an enhanced diabetes-, lipid-, inflammation-, or thrombosis-associated risk [260] will have to be identified. Third, biomarkers will have to be developed that are suitable for assessing the efficacy of vaccination. While antibody-titers are usually employed to screen vaccination efficiency, it is not clear if ApoB-peptides located within the inner core of LDL-C and other lipoprotein particles will elicit both cellular and humoral immune responses. It is now evident that different types of atherosclerosis-associated risk exist in humans. For instance, certain risk factors associate with different manifestations of atherosclerotic disease, such as evidenced by smoking and the enhanced prevalence for peripheral arterial disease (PAD) [261,262]. Under optimal lipid-lowering therapies, a residual inflammatory risk remains [263] and even with lipid levels in the desired or below target range and in the absence of residual inflammation, event-rates remain high. It may therefore be speculated that a proportion of this excessive, currently not addressable risk is related to autoimmune mechanisms. Notably, even with LDL-C levels at or below the recommend target range of 40 to 55mg/dL [223], LDL-C is not entirely depleted, and this low, but chronic abundance of an autoantigen may suffice to induce pathogenic anti-LDL/ApoB immunity. The possible existence of an independent atherosclerotic immune risk is also justified by the clinical association of chronic immune and atherosclerotic disease [264] in otherwise healthy individuals. Assays that allow the quantification of ApoB-specific T cells, such as by restimulation or tetramers, will be of great use to quantify such immune-risk in future clinical practice.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication. All authors have read and agreed to the published version of the manuscript.
Funding
This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement No 853425).
Acknowledgments
Figures were generated with schematics from Biorender.com (accessed on 31 January 2021).
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef]
- Kruk, M.E.; Gage, A.D.; Joseph, N.T.; Danaei, G.; Garcia-Saiso, S.; Salomon, J.A. Mortality due to low-quality health systems in the universal health coverage era: A systematic analysis of amenable deaths in 137 countries. Lancet 2018. [Google Scholar] [CrossRef] [Green Version]
- Arnett, D.K.; Blumenthal, R.S.; Albert, M.A.; Buroker, A.B.; Goldberger, Z.D.; Hahn, E.J.; Himmelfarb, C.D.; Khera, A.; Lloyd-Jones, D.; McEvoy, J.W.; et al. 2019 ACC/AHA Guideline on the Primary Prevention of Cardiovascular Disease: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2019, 74, 1376–1414. [Google Scholar] [CrossRef]
- Marchini, T.; Zirlik, A.; Wolf, D. Pathogenic Role of Air Pollution Particulate Matter in Cardiometabolic Disease: Evidence from Mice and Humans. Antioxid Redox Signal 2020, 33, 263–279. [Google Scholar] [CrossRef] [PubMed]
- Colantonio, L.D.; Bittner, V.; Reynolds, K.; Levitan, E.B.; Rosenson, R.S.; Banach, M.; Kent, S.T.; Derose, S.F.; Zhou, H.; Safford, M.M.; et al. Association of Serum Lipids and Coronary Heart Disease in Contemporary Observational Studies. Circulation 2016, 133, 256–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libby, P.; Hansson, G.K. From Focal Lipid Storage to Systemic Inflammation: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 74, 1594–1607. [Google Scholar] [CrossRef]
- Ross, R. The pathogenesis of atherosclerosis--an update. N. Eng. J. Med. 1986, 314, 488–500. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.; Zirlik, A.; Ley, K. Beyond vascular inflammation--recent advances in understanding atherosclerosis. Cell Mol. Life Sci. 2015, 72, 3853–3869. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in atherosclerosis: A dynamic balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef]
- Que, X.; Hung, M.Y.; Yeang, C.; Gonen, A.; Prohaska, T.A.; Sun, X.; Diehl, C.; Maatta, A.; Gaddis, D.E.; Bowden, K.; et al. Oxidized phospholipids are proinflammatory and proatherogenic in hypercholesterolaemic mice. Nature 2018, 558, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Swirski, F.K.; Libby, P.; Aikawa, E.; Alcaide, P.; Luscinskas, F.W.; Weissleder, R.; Pittet, M.J. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J. Clin. Invest. 2007, 117, 195–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nahrendorf, M. Myeloid cell contributions to cardiovascular health and disease. Nat. Med. 2018, 24, 711–720. [Google Scholar] [CrossRef]
- Winkels, H.; Ehinger, E.; Vassallo, M.; Buscher, K.; Dinh, H.Q.; Kobiyama, K.; Hamers, A.A.J.; Cochain, C.; Vafadarnejad, E.; Saliba, A.E.; et al. Atlas of the Immune Cell Repertoire in Mouse Atherosclerosis Defined by Single-Cell RNA-Sequencing and Mass Cytometry. Circ. Res. 2018, 122, 1675–1688. [Google Scholar] [CrossRef]
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of plaque formation and rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R.; Yvan-Charvet, L. Cholesterol, inflammation and innate immunity. Nat. Rev. Immunol. 2015, 15, 104–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mensah, G.A.; Wei, G.S.; Sorlie, P.D.; Fine, L.J.; Rosenberg, Y.; Kaufmann, P.G.; Mussolino, M.E.; Hsu, L.L.; Addou, E.; Engelgau, M.M.; et al. Decline in Cardiovascular Mortality: Possible Causes and Implications. Circ. Res. 2017, 120, 366–380. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Eng. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Tardif, J.C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Eng. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef]
- Nidorf, S.M.; Fiolet, A.T.L.; Mosterd, A.; Eikelboom, J.W.; Schut, A.; Opstal, T.S.J.; The, S.H.K.; Xu, X.F.; Ireland, M.A.; Lenderink, T.; et al. Colchicine in Patients with Chronic Coronary Disease. N. Eng. J. Med. 2020. [Google Scholar] [CrossRef]
- Wolf, D.; Ley, K. Immunity and Inflammation in Atherosclerosis. Circ. Res. 2019, 124, 315–327. [Google Scholar] [CrossRef]
- Kobiyama, K.; Ley, K. Atherosclerosis. Circ. Res. 2018, 123, 1118–1120. [Google Scholar] [CrossRef]
- Stemme, S.; Faber, B.; Holm, J.; Wiklund, O.; Witztum, J.L.; Hansson, G.K. T lymphocytes from human atherosclerotic plaques recognize oxidized low density lipoprotein. Proc. Natl. Acad. Sci. USA 1995, 92, 3893–3897. [Google Scholar] [CrossRef] [Green Version]
- Tsiantoulas, D.; Diehl, C.J.; Witztum, J.L.; Binder, C.J. B cells and humoral immunity in atherosclerosis. Circ. Res. 2014, 114, 1743–1756. [Google Scholar] [CrossRef]
- Chou, M.Y.; Fogelstrand, L.; Hartvigsen, K.; Hansen, L.F.; Woelkers, D.; Shaw, P.X.; Choi, J.; Perkmann, T.; Backhed, F.; Miller, Y.I.; et al. Oxidation-specific epitopes are dominant targets of innate natural antibodies in mice and humans. J. Clin. Invest. 2009, 119, 1335–1349. [Google Scholar] [CrossRef] [Green Version]
- Winkels, H.; Wolf, D. Heterogeneity of T Cells in Atherosclerosis Defined by Single-Cell RNA-Sequencing and Cytometry by Time of Flight. Arter. Thromb. Vasc. Biol. 2020. [Google Scholar] [CrossRef]
- Jonasson, L.; Holm, J.; Skalli, O.; Bondjers, G.; Hansson, G.K. Regional accumulations of T cells, macrophages, and smooth muscle cells in the human atherosclerotic plaque. Arteriosclerosis 1986, 6, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Jonasson, L. The discovery of cellular immunity in the atherosclerotic plaque. Arter. Thromb. Vasc. Biol. 2009, 29, 1714–1717. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, D.M.; Rahman, A.H.; Fernandez, N.F.; Chudnovskiy, A.; Amir, E.D.; Amadori, L.; Khan, N.S.; Wong, C.K.; Shamailova, R.; Hill, C.A.; et al. Single-cell immune landscape of human atherosclerotic plaques. Nat. Med. 2019, 25, 1576–1588. [Google Scholar] [CrossRef] [PubMed]
- Depuydt, M.A.; Prange, K.H.; Slenders, L.; Ord, T.; Elbersen, D.; Boltjes, A.; de Jager, S.C.; Asselbergs, F.W.; de Borst, G.J.; Aavik, E.; et al. Microanatomy of the Human Atherosclerotic Plaque by Single-Cell Transcriptomics. Circ. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Cochain, C.; Vafadarnejad, E.; Arampatzi, P.; Jaroslav, P.; Winkels, H.; Ley, K.; Wolf, D.; Saliba, A.E.; Zernecke, A. Single-Cell RNA-Seq Reveals the Transcriptional Landscape and Heterogeneity of Aortic Macrophages in Murine Atherosclerosis. Circ. Res. 2018. [Google Scholar] [CrossRef]
- Cole, J.E.; Park, I.; Ahern, D.J.; Kassiteridi, C.; Danso Abeam, D.; Goddard, M.E.; Green, P.; Maffia, P.; Monaco, C. Immune cell census in murine atherosclerosis: Cytometry by time of flight illuminates vascular myeloid cell diversity. Cardiovasc Res. 2018, 114, 1360–1371. [Google Scholar] [CrossRef] [Green Version]
- Taniuchi, I. CD4 Helper and CD8 Cytotoxic T Cell Differentiation. Annu. Rev. Immunol. 2018, 36, 579–601. [Google Scholar] [CrossRef]
- Peaper, D.R.; Cresswell, P. Regulation of MHC class I assembly and peptide binding. Annu. Rev. Cell Dev. Biol. 2008, 24, 343–368. [Google Scholar] [CrossRef]
- Neefjes, J.; Jongsma, M.L.; Paul, P.; Bakke, O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 2011, 11, 823–836. [Google Scholar] [CrossRef] [PubMed]
- Frostegard, J.; Ulfgren, A.K.; Nyberg, P.; Hedin, U.; Swedenborg, J.; Andersson, U.; Hansson, G.K. Cytokine expression in advanced human atherosclerotic plaques: Dominance of pro-inflammatory (Th1) and macrophage-stimulating cytokines. Atherosclerosis 1999, 145, 33–43. [Google Scholar] [CrossRef]
- Van Dijk, R.A.; Duinisveld, A.J.; Schaapherder, A.F.; Mulder-Stapel, A.; Hamming, J.F.; Kuiper, J.; de Boer, O.J.; van der Wal, A.C.; Kolodgie, F.D.; Virmani, R.; et al. A change in inflammatory footprint precedes plaque instability: A systematic evaluation of cellular aspects of the adaptive immune response in human atherosclerosis. J. Am. Heart Assoc. 2015, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butcher, M.J.; Filipowicz, A.R.; Waseem, T.C.; McGary, C.M.; Crow, K.J.; Magilnick, N.; Boldin, M.; Lundberg, P.S.; Galkina, E.V. Atherosclerosis-Driven Treg Plasticity Results in Formation of a Dysfunctional Subset of Plastic IFNgamma+ Th1/Tregs. Circ. Res. 2016, 119, 1190–1203. [Google Scholar] [CrossRef] [Green Version]
- Mosmann, T.R.; Coffman, R.L. TH1 and TH2 cells: Different patterns of lymphokine secretion lead to different functional properties. Annu. Rev. Immunol. 1989, 7, 145–173. [Google Scholar] [CrossRef]
- Koltsova, E.K.; Garcia, Z.; Chodaczek, G.; Landau, M.; McArdle, S.; Scott, S.R.; von Vietinghoff, S.; Galkina, E.; Miller, Y.I.; Acton, S.T.; et al. Dynamic T cell-APC interactions sustain chronic inflammation in atherosclerosis. J. Clin. Invest. 2012, 122, 3114–3126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; McArdle, S.; Gholami, A.; Kimura, T.; Wolf, D.; Gerhardt, T.; Miller, J.; Weber, C.; Ley, K. CCR5+T-bet+FoxP3+ Effector CD4 T Cells Drive Atherosclerosis. Circ. Res. 2016, 118, 1540–1552. [Google Scholar] [CrossRef] [Green Version]
- Niwa, T.; Wada, H.; Ohashi, H.; Iwamoto, N.; Ohta, H.; Kirii, H.; Fujii, H.; Saito, K.; Seishima, M. Interferon-gamma produced by bone marrow-derived cells attenuates atherosclerotic lesion formation in LDLR-deficient mice. J. Atheroscler. Thromb. 2004, 11, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Buono, C.; Come, C.E.; Stavrakis, G.; Maguire, G.F.; Connelly, P.W.; Lichtman, A.H. Influence of interferon-gamma on the extent and phenotype of diet-induced atherosclerosis in the LDLR-deficient mouse. Arter. Thromb. Vasc. Biol. 2003, 23, 454–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; Pablo, A.M.; Jiang, X.; Wang, N.; Tall, A.R.; Schindler, C. IFN-gamma potentiates atherosclerosis in ApoE knock-out mice. J. Clin. Invest. 1997, 99, 2752–2761. [Google Scholar] [CrossRef] [PubMed]
- Buono, C.; Binder, C.J.; Stavrakis, G.; Witztum, J.L.; Glimcher, L.H.; Lichtman, A.H. T-bet deficiency reduces atherosclerosis and alters plaque antigen-specific immune responses. Proc. Natl. Acad. Sci. USA 2005, 102, 1596–1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitman, S.C.; Ravisankar, P.; Elam, H.; Daugherty, A. Exogenous interferon-gamma enhances atherosclerosis in apolipoprotein E−/− mice. Am. J. Pathol. 2000, 157, 1819–1824. [Google Scholar] [CrossRef]
- Foks, A.C.; Lichtman, A.H.; Kuiper, J. Treating atherosclerosis with regulatory T cells. Arter. Thromb. Vasc. Biol. 2015, 35, 280–287. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, S. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu. Rev. Immunol. 2004, 22, 531–562. [Google Scholar] [CrossRef]
- De Boer, O.J.; van der Meer, J.J.; Teeling, P.; van der Loos, C.M.; van der Wal, A.C. Low numbers of FOXP3 positive regulatory T cells are present in all developmental stages of human atherosclerotic lesions. PLoS ONE 2007, 2, e779. [Google Scholar] [CrossRef] [Green Version]
- Wolf, D.; Gerhardt, T.; Winkels, H.; Michel, N.A.; Pramod, A.B.; Ghosheh, Y.; Brunel, S.; Buscher, K.; Miller, J.; McArdle, S.; et al. Pathogenic Autoimmunity in Atherosclerosis Evolves From Initially Protective Apolipoprotein B100-Reactive CD4(+) T-Regulatory Cells. Circulation 2020, 142, 1279–1293. [Google Scholar] [CrossRef]
- Gaddis, D.E.; Padgett, L.E.; Wu, R.; McSkimming, C.; Romines, V.; Taylor, A.M.; McNamara, C.A.; Kronenberg, M.; Crotty, S.; Thomas, M.J.; et al. Apolipoprotein AI prevents regulatory to follicular helper T cell switching during atherosclerosis. Nat. Commun 2018, 9, 1095. [Google Scholar] [CrossRef] [PubMed]
- Pitoiset, F.; Barbie, M.; Monneret, G.; Braudeau, C.; Pochard, P.; Pellegrin, I.; Trauet, J.; Labalette, M.; Klatzmann, D.; Rosenzwajg, M. A standardized flow cytometry procedure for the monitoring of regulatory T cells in clinical trials. Cytom. Part B Clin. Cytom. 2018, 94, 621–626. [Google Scholar] [CrossRef]
- Shami, A.; Atzler, D.; Bosmans, L.A.; Winkels, H.; Meiler, S.; Lacy, M.; van Tiel, C.; Ta Megens, R.; Nitz, K.; Baardman, J.; et al. Glucocorticoid-induced tumour necrosis factor receptor family-related protein (GITR) drives atherosclerosis in mice and is associated with an unstable plaque phenotype and cerebrovascular events in humans. Eur. Heart J. 2020, 41, 2938–2948. [Google Scholar] [CrossRef] [PubMed]
- Meiler, S.; Smeets, E.; Winkels, H.; Shami, A.; Pascutti, M.F.; Nolte, M.A.; Beckers, L.; Weber, C.; Gerdes, N.; Lutgens, E. Constitutive GITR Activation Reduces Atherosclerosis by Promoting Regulatory CD4+ T-Cell Responses-Brief Report. Arter. Thromb. Vasc. Biol. 2016, 36, 1748–1752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ait-Oufella, H.; Salomon, B.L.; Potteaux, S.; Robertson, A.K.; Gourdy, P.; Zoll, J.; Merval, R.; Esposito, B.; Cohen, J.L.; Fisson, S.; et al. Natural regulatory T cells control the development of atherosclerosis in mice. Nat. Med. 2006, 12, 178–180. [Google Scholar] [CrossRef]
- Klingenberg, R.; Gerdes, N.; Badeau, R.M.; Gistera, A.; Strodthoff, D.; Ketelhuth, D.F.; Lundberg, A.M.; Rudling, M.; Nilsson, S.K.; Olivecrona, G.; et al. Depletion of FOXP3+ regulatory T cells promotes hypercholesterolemia and atherosclerosis. J. Clin. Invest. 2013, 123, 1323–1334. [Google Scholar] [CrossRef]
- Sharma, M.; Schlegel, M.P.; Afonso, M.S.; Brown, E.J.; Rahman, K.; Weinstock, A.; Sansbury, B.E.; Corr, E.M.; van Solingen, C.; Koelwyn, G.J.; et al. Regulatory T Cells License Macrophage Pro-Resolving Functions During Atherosclerosis Regression. Circ. Res. 2020, 127, 335–353. [Google Scholar] [CrossRef]
- Mor, A.; Luboshits, G.; Planer, D.; Keren, G.; George, J. Altered status of CD4(+)CD25(+) regulatory T cells in patients with acute coronary syndromes. Eur. Heart J. 2006, 27, 2530–2537. [Google Scholar] [CrossRef] [Green Version]
- Wigren, M.; Bjorkbacka, H.; Andersson, L.; Ljungcrantz, I.; Fredrikson, G.N.; Persson, M.; Bryngelsson, C.; Hedblad, B.; Nilsson, J. Low levels of circulating CD4+FoxP3+ T cells are associated with an increased risk for development of myocardial infarction but not for stroke. Arter. Thromb. Vasc. Biol. 2012, 32, 2000–2004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mailer, R.K.W.; Gistera, A.; Polyzos, K.A.; Ketelhuth, D.F.J.; Hansson, G.K. Hypercholesterolemia Enhances T Cell Receptor Signaling and Increases the Regulatory T Cell Population. Sci. Rep. 2017, 7, 15655. [Google Scholar] [CrossRef] [Green Version]
- Maganto-Garcia, E.; Tarrio, M.L.; Grabie, N.; Bu, D.X.; Lichtman, A.H. Dynamic changes in regulatory T cells are linked to levels of diet-induced hypercholesterolemia. Circulation 2011, 124, 185–195. [Google Scholar] [CrossRef] [Green Version]
- Mailer, R.K.W.; Gistera, A.; Polyzos, K.A.; Ketelhuth, D.F.J.; Hansson, G.K. Hypercholesterolemia Induces Differentiation of Regulatory T Cells in the Liver. Circ. Res. 2017, 120, 1740–1753. [Google Scholar] [CrossRef]
- McGeachy, M.J.; Cua, D.J.; Gaffen, S.L. The IL-17 Family of Cytokines in Health and Disease. Immunity 2019, 50, 892–906. [Google Scholar] [CrossRef]
- Miossec, P.; Kolls, J.K. Targeting IL-17 and TH17 cells in chronic inflammation. Nat. Rev. Drug Discov. 2012, 11, 763–776. [Google Scholar] [CrossRef]
- Erbel, C.; Akhavanpoor, M.; Okuyucu, D.; Wangler, S.; Dietz, A.; Zhao, L.; Stellos, K.; Little, K.M.; Lasitschka, F.; Doesch, A.; et al. IL-17A influences essential functions of the monocyte/macrophage lineage and is involved in advanced murine and human atherosclerosis. J. Immunol. 2014, 193, 4344–4355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erbel, C.; Chen, L.; Bea, F.; Wangler, S.; Celik, S.; Lasitschka, F.; Wang, Y.; Bockler, D.; Katus, H.A.; Dengler, T.J. Inhibition of IL-17A attenuates atherosclerotic lesion development in apoE-deficient mice. J. Immunol. 2009, 183, 8167–8175. [Google Scholar] [CrossRef] [Green Version]
- Butcher, M.J.; Gjurich, B.N.; Phillips, T.; Galkina, E.V. The IL-17A/IL-17RA axis plays a proatherogenic role via the regulation of aortic myeloid cell recruitment. Circ. Res. 2012, 110, 675–687. [Google Scholar] [CrossRef] [Green Version]
- Usui, F.; Kimura, H.; Ohshiro, T.; Tatsumi, K.; Kawashima, A.; Nishiyama, A.; Iwakura, Y.; Ishibashi, S.; Takahashi, M. Interleukin-17 deficiency reduced vascular inflammation and development of atherosclerosis in Western diet-induced apoE-deficient mice. Biochem Biophys Res. Commun. 2012, 420, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yuan, X.; Deng, L.; Xu, W.; Zheng, Y.; Yue, C.; Zhang, G.; Xie, F.; Yang, Y.H.; Gantier, M.P.; et al. Imbalanced frequencies of Th17 and Treg cells in acute coronary syndromes are mediated by IL-6-STAT3 signaling. PLoS ONE 2013, 8, e72804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, X.; Yu, X.; Ding, Y.J.; Fu, Q.Q.; Xie, J.J.; Tang, T.T.; Yao, R.; Chen, Y.; Liao, Y.H. The Th17/Treg imbalance in patients with acute coronary syndrome. Clin. Immunol. 2008, 127, 89–97. [Google Scholar] [CrossRef]
- Danzaki, K.; Matsui, Y.; Ikesue, M.; Ohta, D.; Ito, K.; Kanayama, M.; Kurotaki, D.; Morimoto, J.; Iwakura, Y.; Yagita, H.; et al. Interleukin-17A deficiency accelerates unstable atherosclerotic plaque formation in apolipoprotein E-deficient mice. Arter. Thromb. Vasc. Biol. 2012, 32, 273–280. [Google Scholar] [CrossRef] [Green Version]
- Brauner, S.; Jiang, X.; Thorlacius, G.E.; Lundberg, A.M.; Ostberg, T.; Yan, Z.Q.; Kuchroo, V.K.; Hansson, G.K.; Wahren-Herlenius, M. Augmented Th17 differentiation in Trim21 deficiency promotes a stable phenotype of atherosclerotic plaques with high collagen content. Cardiovasc Res. 2018, 114, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Gistera, A.; Robertson, A.K.; Andersson, J.; Ketelhuth, D.F.; Ovchinnikova, O.; Nilsson, S.K.; Lundberg, A.M.; Li, M.O.; Flavell, R.A.; Hansson, G.K. Transforming growth factor-beta signaling in T cells promotes stabilization of atherosclerotic plaques through an interleukin-17-dependent pathway. Sci. Transl. Med. 2013, 5, 196ra100. [Google Scholar] [CrossRef]
- Taleb, S.; Romain, M.; Ramkhelawon, B.; Uyttenhove, C.; Pasterkamp, G.; Herbin, O.; Esposito, B.; Perez, N.; Yasukawa, H.; Van Snick, J.; et al. Loss of SOCS3 expression in T cells reveals a regulatory role for interleukin-17 in atherosclerosis. J. Exp. Med. 2009, 206, 2067–2077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, E.; Prasad, K.M.; Butcher, M.; Dobrian, A.; Kolls, J.K.; Ley, K.; Galkina, E. Blockade of interleukin-17A results in reduced atherosclerosis in apolipoprotein E-deficient mice. Circulation 2010, 121, 1746–1755. [Google Scholar] [CrossRef]
- Simon, T.; Taleb, S.; Danchin, N.; Laurans, L.; Rousseau, B.; Cattan, S.; Montely, J.M.; Dubourg, O.; Tedgui, A.; Kotti, S.; et al. Circulating levels of interleukin-17 and cardiovascular outcomes in patients with acute myocardial infarction. Eur. Heart J. 2013, 34, 570–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madhur, M.S.; Funt, S.A.; Li, L.; Vinh, A.; Chen, W.; Lob, H.E.; Iwakura, Y.; Blinder, Y.; Rahman, A.; Quyyumi, A.A.; et al. Role of interleukin 17 in inflammation, atherosclerosis, and vascular function in apolipoprotein e-deficient mice. Arter. Thromb. Vasc. Biol. 2011, 31, 1565–1572. [Google Scholar] [CrossRef] [Green Version]
- Eid, R.E.; Rao, D.A.; Zhou, J.; Lo, S.F.; Ranjbaran, H.; Gallo, A.; Sokol, S.I.; Pfau, S.; Pober, J.S.; Tellides, G. Interleukin-17 and interferon-gamma are produced concomitantly by human coronary artery-infiltrating T cells and act synergistically on vascular smooth muscle cells. Circulation 2009, 119, 1424–1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, J.A.; McKenzie, A.N.J. TH2 cell development and function. Nat. Rev. Immunol. 2018, 18, 121–133. [Google Scholar] [CrossRef]
- King, V.L.; Szilvassy, S.J.; Daugherty, A. Interleukin-4 deficiency decreases atherosclerotic lesion formation in a site-specific manner in female LDL receptor−/− mice. Arter. Thromb. Vasc. Biol. 2002, 22, 456–461. [Google Scholar] [CrossRef] [Green Version]
- Cardilo-Reis, L.; Gruber, S.; Schreier, S.M.; Drechsler, M.; Papac-Milicevic, N.; Weber, C.; Wagner, O.; Stangl, H.; Soehnlein, O.; Binder, C.J. Interleukin-13 protects from atherosclerosis and modulates plaque composition by skewing the macrophage phenotype. EMBO Mol. Med. 2012, 4, 1072–1086. [Google Scholar] [CrossRef]
- King, V.L.; Cassis, L.A.; Daugherty, A. Interleukin-4 does not influence development of hypercholesterolemia or angiotensin II-induced atherosclerotic lesions in mice. Am. J. Pathol. 2007, 171, 2040–2047. [Google Scholar] [CrossRef] [Green Version]
- Davenport, P.; Tipping, P.G. The role of interleukin-4 and interleukin-12 in the progression of atherosclerosis in apolipoprotein E-deficient mice. Am. J. Pathol. 2003, 163, 1117–1125. [Google Scholar] [CrossRef] [Green Version]
- Engelbertsen, D.; Andersson, L.; Ljungcrantz, I.; Wigren, M.; Hedblad, B.; Nilsson, J.; Bjorkbacka, H. T-helper 2 immunity is associated with reduced risk of myocardial infarction and stroke. Arter. Thromb. Vasc. Biol. 2013, 33, 637–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silveira, A.; McLeod, O.; Strawbridge, R.J.; Gertow, K.; Sennblad, B.; Baldassarre, D.; Veglia, F.; Deleskog, A.; Persson, J.; Leander, K.; et al. Plasma IL-5 concentration and subclinical carotid atherosclerosis. Atherosclerosis 2015, 239, 125–130. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.M.; Xu, D.; Asquith, D.L.; Denby, L.; Li, Y.; Sattar, N.; Baker, A.H.; McInnes, I.B.; Liew, F.Y. IL-33 reduces the development of atherosclerosis. J. Exp. Med. 2008, 205, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Crotty, S. T Follicular Helper Cell Biology: A Decade of Discovery and Diseases. Immunity 2019, 50, 1132–1148. [Google Scholar] [CrossRef]
- Ryu, H.; Lim, H.; Choi, G.; Park, Y.J.; Cho, M.; Na, H.; Ahn, C.W.; Kim, Y.C.; Kim, W.U.; Lee, S.H.; et al. Atherogenic dyslipidemia promotes autoimmune follicular helper T cell responses via IL-27. Nat. Immunol. 2018, 19, 583–593. [Google Scholar] [CrossRef]
- Nus, M.; Sage, A.P.; Lu, Y.; Masters, L.; Lam, B.Y.H.; Newland, S.; Weller, S.; Tsiantoulas, D.; Raffort, J.; Marcus, D.; et al. Marginal zone B cells control the response of follicular helper T cells to a high-cholesterol diet. Nat. Med. 2017, 23, 601–610. [Google Scholar] [CrossRef] [Green Version]
- Clement, M.; Guedj, K.; Andreata, F.; Morvan, M.; Bey, L.; Khallou-Laschet, J.; Gaston, A.T.; Delbosc, S.; Alsac, J.M.; Bruneval, P.; et al. Control of the T follicular helper-germinal center B-cell axis by CD8(+) regulatory T cells limits atherosclerosis and tertiary lymphoid organ development. Circulation 2015, 131, 560–570. [Google Scholar] [CrossRef] [Green Version]
- Ghamar Talepoor, A.; Khosropanah, S.; Doroudchi, M. Functional subsets of circulating follicular helper T cells in patients with atherosclerosis. Physiol Rep. 2020, 8, e14637. [Google Scholar] [CrossRef] [PubMed]
- Gistera, A.; Klement, M.L.; Polyzos, K.A.; Mailer, R.K.W.; Duhlin, A.; Karlsson, M.C.I.; Ketelhuth, D.F.J.; Hansson, G.K. Low-Density Lipoprotein-Reactive T Cells Regulate Plasma Cholesterol Levels and Development of Atherosclerosis in Humanized Hypercholesterolemic Mice. Circulation 2018, 138, 2513–2526. [Google Scholar] [CrossRef]
- Dumitriu, I.E. The life (and death) of CD4+ CD28(null) T cells in inflammatory diseases. Immunology 2015, 146, 185–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acuto, O.; Michel, F. CD28-mediated co-stimulation: A quantitative support for TCR signalling. Nat. Rev. Immunol. 2003, 3, 939–951. [Google Scholar] [CrossRef]
- Kyaw, T.; Peter, K.; Li, Y.; Tipping, P.; Toh, B.H.; Bobik, A. Cytotoxic lymphocytes and atherosclerosis: Significance, mechanisms and therapeutic challenges. Br. J. Pharmacol. 2017, 174, 3956–3972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumitriu, I.E.; Baruah, P.; Finlayson, C.J.; Loftus, I.M.; Antunes, R.F.; Lim, P.; Bunce, N.; Kaski, J.C. High levels of costimulatory receptors OX40 and 4-1BB characterize CD4+CD28null T cells in patients with acute coronary syndrome. Circ. Res. 2012, 110, 857–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saigusa, R.; Winkels, H.; Ley, K. T cell subsets and functions in atherosclerosis. Nat. Rev. Cardiol. 2020. [Google Scholar] [CrossRef]
- Liuzzo, G.; Goronzy, J.J.; Yang, H.; Kopecky, S.L.; Holmes, D.R.; Frye, R.L.; Weyand, C.M. Monoclonal T-cell proliferation and plaque instability in acute coronary syndromes. Circulation 2000, 101, 2883–2888. [Google Scholar] [CrossRef] [Green Version]
- Liuzzo, G.; Kopecky, S.L.; Frye, R.L.; O’Fallon, W.M.; Maseri, A.; Goronzy, J.J.; Weyand, C.M. Perturbation of the T-cell repertoire in patients with unstable angina. Circulation 1999, 100, 2135–2139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakajima, T.; Schulte, S.; Warrington, K.J.; Kopecky, S.L.; Frye, R.L.; Goronzy, J.J.; Weyand, C.M. T-cell-mediated lysis of endothelial cells in acute coronary syndromes. Circulation 2002, 105, 570–575. [Google Scholar] [CrossRef] [Green Version]
- Zal, B.; Kaski, J.C.; Arno, G.; Akiyu, J.P.; Xu, Q.; Cole, D.; Whelan, M.; Russell, N.; Madrigal, J.A.; Dodi, I.A.; et al. Heat-shock protein 60-reactive CD4+CD28null T cells in patients with acute coronary syndromes. Circulation 2004, 109, 1230–1235. [Google Scholar] [CrossRef] [Green Version]
- Zal, B.; Kaski, J.C.; Akiyu, J.P.; Cole, D.; Arno, G.; Poloniecki, J.; Madrigal, A.; Dodi, A.; Baboonian, C. Differential pathways govern CD4+ CD28- T cell proinflammatory and effector responses in patients with coronary artery disease. J. Immunol. 2008, 181, 5233–5241. [Google Scholar] [CrossRef]
- Li, H.; van der Leun, A.M.; Yofe, I.; Lubling, Y.; Gelbard-Solodkin, D.; van Akkooi, A.C.J.; van den Braber, M.; Rozeman, E.A.; Haanen, J.; Blank, C.U.; et al. Dysfunctional CD8 T Cells Form a Proliferative, Dynamically Regulated Compartment within Human Melanoma. Cell 2019, 176, 775–789 e718. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, M.H. Th9 cells: Differentiation and disease. Immunol. Rev. 2013, 252, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Loo, T.T.; Gao, Y.; Lazarevic, V. Transcriptional regulation of CD4(+) TH cells that mediate tissue inflammation. J. Leukoc. Biol. 2018, 104, 1069–1085. [Google Scholar] [CrossRef]
- Gregersen, I.; Skjelland, M.; Holm, S.; Holven, K.B.; Krogh-Sorensen, K.; Russell, D.; Askevold, E.T.; Dahl, C.P.; Orn, S.; Gullestad, L.; et al. Increased systemic and local interleukin 9 levels in patients with carotid and coronary atherosclerosis. PLoS ONE 2013, 8, e72769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.Z.; Wu, B.W.; Lu, Z.D.; Huang, Y.; Shi, Y.; Liu, H.; Liu, L.; Zeng, Q.T.; Wang, X.; Ji, Q.W. Circulating Th22 and Th9 levels in patients with acute coronary syndrome. Mediat. Inflamm. 2013, 2013, 635672. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Tang, T.; Nie, D.; Wen, S.; Jia, C.; Zhu, Z.; Xia, N.; Nie, S.; Zhou, S.; Jiao, J.; et al. IL-9 aggravates the development of atherosclerosis in ApoE−/− mice. Cardiovasc Res. 2015, 106, 453–464. [Google Scholar] [CrossRef]
- Azizi, G.; Yazdani, R.; Mirshafiey, A. Th22 cells in autoimmunity: A review of current knowledge. Eur. Ann. Allergy Clin. Immunol. 2015, 47, 108–117. [Google Scholar]
- Shi, L.; Ji, Q.; Liu, L.; Shi, Y.; Lu, Z.; Ye, J.; Zeng, T.; Xue, Y.; Yang, Z.; Liu, Y.; et al. IL-22 produced by Th22 cells aggravates atherosclerosis development in ApoE(−/−) mice by enhancing DC-induced Th17 cell proliferation. J. Cell Mol. Med. 2020, 24, 3064–3078. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wang, T.; Wang, X.Q.; Du, R.Z.; Zhang, K.N.; Liu, X.G.; Ma, D.X.; Yu, S.; Su, G.H.; Li, Z.H.; et al. Elevated frequencies of circulating Th22 cell in addition to Th17 cell and Th17/Th1 cell in patients with acute coronary syndrome. PLoS ONE 2013, 8, e71466. [Google Scholar] [CrossRef] [PubMed]
- Bendelac, A.; Savage, P.B.; Teyton, L. The biology of NKT cells. Annu. Rev. Immunol. 2007, 25, 297–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobryshev, Y.V.; Lord, R.S. Co-accumulation of dendritic cells and natural killer T cells within rupture-prone regions in human atherosclerotic plaques. J. Histochem. Cytochem. 2005, 53, 781–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Getz, G.S.; Reardon, C.A. Natural killer T cells in atherosclerosis. Nat. Rev. Cardiol. 2017, 14, 304–314. [Google Scholar] [CrossRef]
- Harty, J.T.; Tvinnereim, A.R.; White, D.W. CD8+ T cell effector mechanisms in resistance to infection. Annu. Rev. Immunol. 2000, 18, 275–308. [Google Scholar] [CrossRef]
- Schafer, S.; Zernecke, A. CD8(+) T Cells in Atherosclerosis. Cells 2020, 10, 37. [Google Scholar] [CrossRef] [PubMed]
- DuPage, M.; Bluestone, J.A. Harnessing the plasticity of CD4(+) T cells to treat immune-mediated disease. Nat. Rev. Immunol. 2016, 16, 149–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, A.J.; Makings, J.; Ley, K. Regulatory T Cell Stability and Plasticity in Atherosclerosis. Cells 2020, 9, 2665. [Google Scholar] [CrossRef]
- Kimura, T.; Kobiyama, K.; Winkels, H.; Tse, K.; Miller, J.; Vassallo, M.; Wolf, D.; Ryden, C.; Orecchioni, M.; Dileepan, T.; et al. Regulatory CD4(+) T Cells Recognize MHC-II-Restricted Peptide Epitopes of Apolipoprotein B. Circulation 2018, 138, 1130–1143. [Google Scholar] [CrossRef]
- Tan, T.G.; Mathis, D.; Benoist, C. Singular role for T-BET+CXCR3+ regulatory T cells in protection from autoimmune diabetes. Proc. Natl. Acad. Sci. USA 2016, 113, 14103–14108. [Google Scholar] [CrossRef] [Green Version]
- Tartar, D.M.; VanMorlan, A.M.; Wan, X.; Guloglu, F.B.; Jain, R.; Haymaker, C.L.; Ellis, J.S.; Hoeman, C.M.; Cascio, J.A.; Dhakal, M.; et al. FoxP3+RORgammat+ T helper intermediates display suppressive function against autoimmune diabetes. J. Immunol. 2010, 184, 3377–3385. [Google Scholar] [CrossRef] [Green Version]
- Rizzo, A.; Di Giovangiulio, M.; Stolfi, C.; Franze, E.; Fehling, H.J.; Carsetti, R.; Giorda, E.; Colantoni, A.; Ortenzi, A.; Rugge, M.; et al. RORgammat-Expressing Tregs Drive the Growth of Colitis-Associated Colorectal Cancer by Controlling IL6 in Dendritic Cells. Cancer Immunol. Res. 2018, 6, 1082–1092. [Google Scholar] [CrossRef] [Green Version]
- Ohnmacht, C.; Park, J.H.; Cording, S.; Wing, J.B.; Atarashi, K.; Obata, Y.; Gaboriau-Routhiau, V.; Marques, R.; Dulauroy, S.; Fedoseeva, M.; et al. MUCOSAL IMMUNOLOGY. The microbiota regulates type 2 immunity through RORgammat(+) T cells. Science 2015, 349, 989–993. [Google Scholar] [CrossRef] [PubMed]
- Di Giovangiulio, M.; Rizzo, A.; Franze, E.; Caprioli, F.; Facciotti, F.; Onali, S.; Favale, A.; Stolfi, C.; Fehling, H.J.; Monteleone, G.; et al. Tbet Expression in Regulatory T Cells Is Required to Initiate Th1-Mediated Colitis. Front. Immunol. 2019, 10, 2158. [Google Scholar] [CrossRef] [PubMed]
- Bailey-Bucktrout, S.L.; Martinez-Llordella, M.; Zhou, X.; Anthony, B.; Rosenthal, W.; Luche, H.; Fehling, H.J.; Bluestone, J.A. Self-antigen-driven activation induces instability of regulatory T cells during an inflammatory autoimmune response. Immunity 2013, 39, 949–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsu, N.; Okamoto, K.; Sawa, S.; Nakashima, T.; Oh-hora, M.; Kodama, T.; Tanaka, S.; Bluestone, J.A.; Takayanagi, H. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat. Med. 2014, 20, 62–68. [Google Scholar] [CrossRef]
- Korn, T.; Reddy, J.; Gao, W.; Bettelli, E.; Awasthi, A.; Petersen, T.R.; Backstrom, B.T.; Sobel, R.A.; Wucherpfennig, K.W.; Strom, T.B.; et al. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat. Med. 2007, 13, 423–431. [Google Scholar] [CrossRef]
- Ranjbaran, H.; Sokol, S.I.; Gallo, A.; Eid, R.E.; Iakimov, A.O.; D’Alessio, A.; Kapoor, J.R.; Akhtar, S.; Howes, C.J.; Aslan, M.; et al. An inflammatory pathway of IFN-gamma production in coronary atherosclerosis. J. Immunol. 2007, 178, 592–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, F.J.; Ott, I.; Gawaz, M.; Richardt, G.; Holzapfel, H.; Jochum, M.; Schomig, A. Cardiac release of cytokines and inflammatory responses in acute myocardial infarction. Circulation 1995, 92, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Okba, A.M.; Abd El Raouf Raafat, M.; Nazmy Farres, M.; Abd El Nour Melek, N.; Amin, M.M.; Gendy, N.N. Expanded peripheral CD4(+)CD28(null) T cells and its association with atherosclerotic changes in patients with end stage renal disease on hemodialysis. Hum. Immunol. 2019, 80, 748–754. [Google Scholar] [CrossRef]
- Rosenblum, M.D.; Remedios, K.A.; Abbas, A.K. Mechanisms of human autoimmunity. J. Clin. Invest. 2015, 125, 2228–2233. [Google Scholar] [CrossRef] [Green Version]
- Adler, R. Janeway’s immunobiology. Choice Curr. Rev. Acad. Libr. 2008, 45, 1793–1794. [Google Scholar]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining ’T cell exhaustion’. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef]
- Bu, D.X.; Tarrio, M.; Maganto-Garcia, E.; Stavrakis, G.; Tajima, G.; Lederer, J.; Jarolim, P.; Freeman, G.J.; Sharpe, A.H.; Lichtman, A.H. Impairment of the programmed cell death-1 pathway increases atherosclerotic lesion development and inflammation. Arter. Thromb. Vasc. Biol. 2011, 31, 1100–1107. [Google Scholar] [CrossRef] [Green Version]
- Kusters, P.J.H.; Lutgens, E.; Seijkens, T.T.P. Exploring immune checkpoints as potential therapeutic targets in atherosclerosis. Cardiovasc Res. 2018, 114, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Hermansson, A.; Ketelhuth, D.F.; Strodthoff, D.; Wurm, M.; Hansson, E.M.; Nicoletti, A.; Paulsson-Berne, G.; Hansson, G.K. Inhibition of T cell response to native low-density lipoprotein reduces atherosclerosis. J. Exp. Med. 2010, 207, 1081–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutgens, E.; de Muinck, E.D.; Kitslaar, P.J.; Tordoir, J.H.; Wellens, H.J.; Daemen, M.J. Biphasic pattern of cell turnover characterizes the progression from fatty streaks to ruptured human atherosclerotic plaques. Cardiovasc Res. 1999, 41, 473–479. [Google Scholar] [CrossRef] [Green Version]
- Zernecke, A.; Winkels, H.; Cochain, C.; Williams, J.W.; Wolf, D.; Soehnlein, O.; Robbins, C.S.; Monaco, C.; Park, I.; McNamara, C.A.; et al. Meta-Analysis of Leukocyte Diversity in Atherosclerotic Mouse Aortas. Circ. Res. 2020, 127, 402–426. [Google Scholar] [CrossRef]
- Paulsson, G.; Zhou, X.; Tornquist, E.; Hansson, G.K. Oligoclonal T cell expansions in atherosclerotic lesions of apolipoprotein E-deficient mice. Arter. Thromb. Vasc. Biol. 2000, 20, 10–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Z.; Qian, S.; Gong, Y.; Ren, J.; Zhao, L.; Wang, D.; Wang, X.; Zhang, Y.; Wang, Z.; Zhang, Q. Deep sequencing of the T cell receptor beta repertoire reveals signature patterns and clonal drift in atherosclerotic plaques and patients. Oncotarget 2017, 8, 99312–99322. [Google Scholar] [CrossRef] [PubMed]
- Klingenberg, R.; Brokopp, C.E.; Grives, A.; Courtier, A.; Jaguszewski, M.; Pasqual, N.; Vlaskou Badra, E.; Lewandowski, A.; Gaemperli, O.; Hoerstrup, S.P.; et al. Clonal restriction and predominance of regulatory T cells in coronary thrombi of patients with acute coronary syndromes. Eur. Heart J. 2015, 36, 1041–1048. [Google Scholar] [CrossRef] [Green Version]
- Oksenberg, J.R.; Stavri, G.T.; Jeong, M.C.; Garovoy, N.; Salisbury, J.R.; Erusalimsky, J.D. Analysis of the T-cell receptor repertoire in human atherosclerosis. Cardiovasc Res. 1997, 36, 256–267. [Google Scholar] [CrossRef] [Green Version]
- Kilic, A.; Mandal, K. Heat Shock Proteins: Pathogenic Role in Atherosclerosis and Potential Therapeutic Implications. Autoimmune Dis. 2012, 2012, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wick, C. Tolerization against atherosclerosis using heat shock protein 60. Cell Stress Chaperones 2015. [Google Scholar] [CrossRef] [Green Version]
- Wick, G.; Jakic, B.; Buszko, M.; Wick, M.C.; Grundtman, C. The role of heat shock proteins in atherosclerosis. Nat. Rev. Cardiol. 2014, 11, 516–529. [Google Scholar] [CrossRef]
- Zhu, J.; Quyyumi, A.A.; Rott, D.; Csako, G.; Wu, H.; Halcox, J.; Epstein, S.E. Antibodies to human heat-shock protein 60 are associated with the presence and severity of coronary artery disease: Evidence for an autoimmune component of atherogenesis. Circulation 2001, 103, 1071–1075. [Google Scholar] [CrossRef] [Green Version]
- Kervinen, H.; Huittinen, T.; Vaarala, O.; Leinonen, M.; Saikku, P.; Manninen, V.; Manttari, M. Antibodies to human heat shock protein 60, hypertension and dyslipidemia. A study of joint effects on coronary risk. Atherosclerosis 2003, 169, 339–344. [Google Scholar] [CrossRef]
- Xu, Q.; Dietrich, H.; Steiner, H.J.; Gown, A.M.; Schoel, B.; Mikuz, G.; Kaufmann, S.H.; Wick, G. Induction of arteriosclerosis in normocholesterolemic rabbits by immunization with heat shock protein 65. Arter. Thromb. 1992, 12, 789–799. [Google Scholar] [CrossRef] [Green Version]
- George, J.; Shoenfeld, Y.; Afek, A.; Gilburd, B.; Keren, P.; Shaish, A.; Kopolovic, J.; Wick, G.; Harats, D. Enhanced fatty streak formation in C57BL/6J mice by immunization with heat shock protein-65. Arter. Thromb. Vasc. Biol. 1999, 19, 505–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Xiong, Q.; Hu, X.; Sun, Y.; Tan, X.; Zhang, H.; Lu, Y.; Liu, J. A novel atherogenic epitope from Mycobacterium tuberculosis heat shock protein 65 enhances atherosclerosis in rabbit and LDL receptor-deficient mice. Heart Vessel. 2012, 27, 411–418. [Google Scholar] [CrossRef]
- Afek, A.; George, J.; Gilburd, B.; Rauova, L.; Goldberg, I.; Kopolovic, J.; Harats, D.; Shoenfeld, Y. Immunization of low-density lipoprotein receptor deficient (LDL-RD) mice with heat shock protein 65 (HSP-65) promotes early atherosclerosis. J Autoimmun 2000, 14, 115–121. [Google Scholar] [CrossRef]
- George, J.; Afek, A.; Gilburd, B.; Shoenfeld, Y.; Harats, D. Cellular and humoral immune responses to heat shock protein 65 are both involved in promoting fatty-streak formation in LDL-receptor deficient mice. J. Am. Coll. Cardiol. 2001, 38, 900–905. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Q.; Feng, J.; Zhang, Y.; Sun, Y.; Lu, Y.; Li, T.; Zhang, X.; Cao, R.; Jin, L.; Wu, J. Promotion of atherosclerosis in high cholesterol diet-fed rabbits by immunization with the P277 peptide. Immunol. Lett. 2016, 170, 80–87. [Google Scholar] [CrossRef]
- Klingenberg, R.; Ketelhuth, D.F.; Strodthoff, D.; Gregori, S.; Hansson, G.K. Subcutaneous immunization with heat shock protein-65 reduces atherosclerosis in Apoe(-)/(-) mice. Immunobiology 2012, 217, 540–547. [Google Scholar] [CrossRef]
- Long, J.; Lin, J.; Yang, X.; Yuan, D.; Wu, J.; Li, T.; Cao, R.; Liu, J. Nasal immunization with different forms of heat shock protein-65 reduced high-cholesterol-diet-driven rabbit atherosclerosis. Int. Immunopharmacol. 2012, 13, 82–87. [Google Scholar] [CrossRef]
- Van Puijvelde, G.H.; van Es, T.; van Wanrooij, E.J.; Habets, K.L.; de Vos, P.; van der Zee, R.; van Eden, W.; van Berkel, T.J.; Kuiper, J. Induction of oral tolerance to HSP60 or an HSP60-peptide activates T cell regulation and reduces atherosclerosis. Arter. Thromb. Vasc. Biol. 2007, 27, 2677–2683. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Y.; Tang, H.; Wang, X.; Zeng, Q.; Liu, Y.; Zhao, X.I.; Yu, K.; Shi, H.; Zhu, R.; Mao, X. Intranasal immunization with heat shock protein 60 induces CD4(+) CD25(+) GARP(+) and type 1 regulatory T cells and inhibits early atherosclerosis. Clin. Exp. Immunol. 2016, 183, 452–468. [Google Scholar] [CrossRef] [Green Version]
- Perschinka, H.; Mayr, M.; Millonig, G.; Mayerl, C.; van der Zee, R.; Morrison, S.G.; Morrison, R.P.; Xu, Q.; Wick, G. Cross-reactive B-cell epitopes of microbial and human heat shock protein 60/65 in atherosclerosis. Arter. Thromb. Vasc. Biol. 2003, 23, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Binder, C.J.; Horkko, S.; Dewan, A.; Chang, M.K.; Kieu, E.P.; Goodyear, C.S.; Shaw, P.X.; Palinski, W.; Witztum, J.L.; Silverman, G.J. Pneumococcal vaccination decreases atherosclerotic lesion formation: Molecular mimicry between Streptococcus pneumoniae and oxidized LDL. Nat. Med. 2003, 9, 736–743. [Google Scholar] [CrossRef]
- Tsutsumi, A.; Matsuura, E.; Ichikawa, K.; Fujisaku, A.; Mukai, M.; Kobayashi, S.; Koike, T. Antibodies to beta 2-glycoprotein I and clinical manifestations in patients with systemic lupus erythematosus. Arthritis Rheum. 1996, 39, 1466–1474. [Google Scholar] [CrossRef] [PubMed]
- Kandiah, D.A.; Krilis, S.A. Beta 2-glycoprotein I. Lupus 1994, 3, 207–212. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Harats, D.; Gilburd, B.; Afek, A.; Levy, Y.; Schneiderman, J.; Barshack, I.; Kopolovic, J.; Shoenfeld, Y. Immunolocalization of beta2-glycoprotein I (apolipoprotein H) to human atherosclerotic plaques: Potential implications for lesion progression. Circulation 1999, 99, 2227–2230. [Google Scholar] [CrossRef] [Green Version]
- George, J.; Afek, A.; Gilburd, B.; Blank, M.; Levy, Y.; Aron-Maor, A.; Levkovitz, H.; Shaish, A.; Goldberg, I.; Kopolovic, J.; et al. Induction of early atherosclerosis in LDL-receptor-deficient mice immunized with beta2-glycoprotein I. Circulation 1998, 98, 1108–1115. [Google Scholar] [CrossRef] [Green Version]
- Dunoyer-Geindre, S.; Kwak, B.R.; Pelli, G.; Roth, I.; Satta, N.; Fish, R.J.; Reber, G.; Mach, F.; Kruithof, E.K.; de Moerloose, P. Immunization of LDL receptor-deficient mice with beta2-glycoprotein 1 or human serum albumin induces a more inflammatory phenotype in atherosclerotic plaques. Thromb. Haemost. 2007, 97, 129–138. [Google Scholar] [CrossRef] [Green Version]
- Afek, A.; George, J.; Shoenfeld, Y.; Gilburd, B.; Levy, Y.; Shaish, A.; Keren, P.; Janackovic, Z.; Goldberg, I.; Kopolovic, J.; et al. Enhancement of atherosclerosis in beta-2-glycoprotein I-immunized apolipoprotein E-deficient mice. Pathobiology 1999, 67, 19–25. [Google Scholar] [CrossRef]
- De Haro, J.; Esparza, L.; Bleda, S.; Varela, C.; Sanchez, C.; Acin, F. Attenuation of early atherosclerotic lesions by immunotolerance with beta2 glycoprotein I and the immunomodulatory effectors interleukin 2 and 10 in a murine model. J. Vasc. Surg. 2015, 62, 1625–1631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, J.; Harats, D.; Gilburd, B.; Afek, A.; Shaish, A.; Kopolovic, J.; Shoenfeld, Y. Adoptive transfer of beta(2)-glycoprotein I-reactive lymphocytes enhances early atherosclerosis in LDL receptor-deficient mice. Circulation 2000, 102, 1822–1827. [Google Scholar] [CrossRef] [Green Version]
- Sage, A.P.; Tsiantoulas, D.; Binder, C.J.; Mallat, Z. The role of B cells in atherosclerosis. Nat. Rev. Cardiol. 2019, 16, 180–196. [Google Scholar] [CrossRef]
- Hebsur, S.; Vakil, E.; Oetgen, W.J.; Kumar, P.N.; Lazarous, D.F. Influenza and coronary artery disease: Exploring a clinical association with myocardial infarction and analyzing the utility of vaccination in prevention of myocardial infarction. Rev. Cardiovasc Med. 2014, 15, 168–175. [Google Scholar] [PubMed]
- Lin, H.C.; Chien, C.W.; Ho, J.D. Herpes zoster ophthalmicus and the risk of stroke: A population-based follow-up study. Neurology 2010, 74, 792–797. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.M.; Taubert, K.; Benin, A.L.; Brown, D.W.; Mensah, G.A.; Baddour, L.M.; Dunbar, S.; Krumholz, H.M.; American Heart, A.; American College of, C.; et al. Influenza vaccination as secondary prevention for cardiovascular disease: A science advisory from the American Heart Association/American College of Cardiology. J. Am. Coll. Cardiol. 2006, 48, 1498–1502. [Google Scholar] [CrossRef] [Green Version]
- Udell, J.A.; Zawi, R.; Bhatt, D.L.; Keshtkar-Jahromi, M.; Gaughran, F.; Phrommintikul, A.; Ciszewski, A.; Vakili, H.; Hoffman, E.B.; Farkouh, M.E.; et al. Association between influenza vaccination and cardiovascular outcomes in high-risk patients: A meta-analysis. JAMA 2013, 310, 1711–1720. [Google Scholar] [CrossRef]
- Siriwardena, A.N.; Asghar, Z.; Coupland, C.C. Influenza and pneumococcal vaccination and risk of stroke or transient ischaemic attack-matched case control study. Vaccine 2014, 32, 1354–1361. [Google Scholar] [CrossRef]
- Macintyre, C.R.; Heywood, A.E.; Kovoor, P.; Ridda, I.; Seale, H.; Tan, T.; Gao, Z.; Katelaris, A.L.; Siu, H.W.; Lo, V.; et al. Ischaemic heart disease, influenza and influenza vaccination: A prospective case control study. Heart 2013, 99, 1843–1848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Famularo, G.; Trinchieri, V.; Santini, G.; De Simone, C. Infections, atherosclerosis, and coronary heart disease. Ann. Ital. Med. Int. 2000, 15, 144–155. [Google Scholar] [PubMed]
- Hemmat, N.; Ebadi, A.; Badalzadeh, R.; Memar, M.Y.; Baghi, H.B. Viral infection and atherosclerosis. Eur. J. Clin. Microbiol. Infect. Dis. 2018, 37, 2225–2233. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, M.E.; Campbell, L.A. Pathogens and atherosclerosis: Update on the potential contribution of multiple infectious organisms to the pathogenesis of atherosclerosis. Thromb. Haemost 2011, 106, 858–867. [Google Scholar] [CrossRef] [PubMed]
- Haidari, M.; Wyde, P.R.; Litovsky, S.; Vela, D.; Ali, M.; Casscells, S.W.; Madjid, M. Influenza virus directly infects, inflames, and resides in the arteries of atherosclerotic and normal mice. Atherosclerosis 2010, 208, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Lawson, J.S.; Glenn, W.K.; Tran, D.D.; Ngan, C.C.; Duflou, J.A.; Whitaker, N.J. Identification of Human Papilloma Viruses in Atheromatous Coronary Artery Disease. Front Cardiovasc Med. 2015, 2, 17. [Google Scholar] [CrossRef] [Green Version]
- Naghavi, M.; Wyde, P.; Litovsky, S.; Madjid, M.; Akhtar, A.; Naguib, S.; Siadaty, M.S.; Sanati, S.; Casscells, W. Influenza infection exerts prominent inflammatory and thrombotic effects on the atherosclerotic plaques of apolipoprotein E-deficient mice. Circulation 2003, 107, 762–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendren, N.S.; Drazner, M.H.; Bozkurt, B.; Cooper, L.T., Jr. Description and Proposed Management of the Acute COVID-19 Cardiovascular Syndrome. Circulation 2020, 141, 1903–1914. [Google Scholar] [CrossRef]
- Nelson, R.W.; Beisang, D.; Tubo, N.J.; Dileepan, T.; Wiesner, D.L.; Nielsen, K.; Wuthrich, M.; Klein, B.S.; Kotov, D.I.; Spanier, J.A.; et al. T cell receptor cross-reactivity between similar foreign and self peptides influences naive cell population size and autoimmunity. Immunity 2015, 42, 95–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suthers, B.; Hansbro, P.; Thambar, S.; McEvoy, M.; Peel, R.; Attia, J. Pneumococcal vaccination may induce anti-oxidized low-density lipoprotein antibodies that have potentially protective effects against cardiovascular disease. Vaccine 2012, 30, 3983–3985. [Google Scholar] [CrossRef] [PubMed]
- Worzella, S.L.; Hayney, M.S. Inflammatory chronic diseases: Preventable by vaccines? J. Am. Pharm. Assoc. 2014, 54, 446–448. [Google Scholar] [CrossRef]
- Vila-Corcoles, A.; Ochoa-Gondar, O.; Rodriguez-Blanco, T.; de Diego, C.; Satue, E.; Group, E.S. Ineffectiveness of pneumococcal vaccination in cardiovascular prevention: The CAPAMIS study. JAMA Intern. Med. 2013, 173, 1918–1920. [Google Scholar] [CrossRef]
- Shiri-Sverdlov, R.; Dos Reis, I.M.; Oligschlaeger, Y.; Hendrikx, T.; Meesters, D.M.; Vanclooster, A.; Vanhoutvin, N.; Koek, G.H.; Westerterp, M.; Binder, C.J.; et al. The Influence of a Conjugated Pneumococcal Vaccination on Plasma Antibody Levels against Oxidized Low-Density Lipoprotein in Metabolic Disease Patients: A Single-Arm Pilot Clinical Trial. Antioxidants 2021, 10, 129. [Google Scholar] [CrossRef] [PubMed]
- Zernecke, A. Dendritic Cells in Atherosclerosis: Evidence in Mice and Humans. Arter. Thromb. Vasc. Biol. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacRitchie, N.; Grassia, G.; Noonan, J.; Cole, J.E.; Hughes, C.E.; Schroeder, J.; Benson, R.A.; Cochain, C.; Zernecke, A.; Guzik, T.J.; et al. The aorta can act as a site of naive CD4+ T-cell priming. Cardiovasc Res. 2020, 116, 306–316. [Google Scholar] [CrossRef]
- Roche, P.A.; Furuta, K. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat. Rev. Immunol. 2015, 15, 203–216. [Google Scholar] [CrossRef]
- Hennecke, J.; Wiley, D.C. T cell receptor-MHC interactions up close. Cell 2001, 104, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Dong, D.; Zheng, L.; Lin, J.; Zhang, B.; Zhu, Y.; Li, N.; Xie, S.; Wang, Y.; Gao, N.; Huang, Z. Structural basis of assembly of the human T cell receptor-CD3 complex. Nature 2019, 573, 546–552. [Google Scholar] [CrossRef]
- Saito, T.; Yokosuka, T. Immunological synapse and microclusters: The site for recognition and activation of T cells. Curr. Opin. Immunol. 2006, 18, 305–313. [Google Scholar] [CrossRef]
- Frauwirth, K.A.; Thompson, C.B. Regulation of T lymphocyte metabolism. J. Immunol. 2004, 172, 4661–4665. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Yamane, H.; Paul, W.E. Differentiation of effector CD4 T cell populations (*). Annu. Rev. Immunol. 2010, 28, 445–489. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef]
- Vandenborre, K.; Van Gool, S.W.; Kasran, A.; Ceuppens, J.L.; Boogaerts, M.A.; Vandenberghe, P. Interaction of CTLA-4 (CD152) with CD80 or CD86 inhibits human T-cell activation. Immunology 1999, 98, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Zirlik, A.; Lutgens, E. An inflammatory link in atherosclerosis and obesity. Co-stimulatory molecules. Hamostaseologie 2015, 35, 272–278. [Google Scholar] [CrossRef]
- Ley, K.; Gerdes, N.; Winkels, H. ATVB Distinguished Scientist Award: How Costimulatory and Coinhibitory Pathways Shape Atherosclerosis. Arter. Thromb. Vasc. Biol. 2017, 37, 764–777. [Google Scholar] [CrossRef] [Green Version]
- Lievens, D.; Habets, K.L.; Robertson, A.K.; Laouar, Y.; Winkels, H.; Rademakers, T.; Beckers, L.; Wijnands, E.; Boon, L.; Mosaheb, M.; et al. Abrogated transforming growth factor beta receptor II (TGFbetaRII) signalling in dendritic cells promotes immune reactivity of T cells resulting in enhanced atherosclerosis. Eur. Heart J. 2013, 34, 3717–3727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermansson, A.; Johansson, D.K.; Ketelhuth, D.F.; Andersson, J.; Zhou, X.; Hansson, G.K. Immunotherapy with tolerogenic apolipoprotein B-100-loaded dendritic cells attenuates atherosclerosis in hypercholesterolemic mice. Circulation 2011, 123, 1083–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.H.; Cheong, C.; Dandamudi, D.B.; Park, C.G.; Rodriguez, A.; Mehandru, S.; Velinzon, K.; Jung, I.H.; Yoo, J.Y.; Oh, G.T.; et al. Flt3 signaling-dependent dendritic cells protect against atherosclerosis. Immunity 2011, 35, 819–831. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, M.; Thorp, E.; Hansson, G.K.; Tabas, I. Treg-mediated suppression of atherosclerosis requires MYD88 signaling in DCs. J. Clin. Invest. 2013, 123, 179–188. [Google Scholar] [CrossRef] [Green Version]
- Westerterp, M.; Gautier, E.L.; Ganda, A.; Molusky, M.M.; Wang, W.; Fotakis, P.; Wang, N.; Randolph, G.J.; D’Agati, V.D.; Yvan-Charvet, L.; et al. Cholesterol Accumulation in Dendritic Cells Links the Inflammasome to Acquired Immunity. Cell Metab. 2017, 25, 1294–1304 e1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clement, M.; Haddad, Y.; Raffort, J.; Lareyre, F.; Newland, S.A.; Master, L.; Harrison, J.; Ozsvar-Kozma, M.; Bruneval, P.; Binder, C.J.; et al. Deletion of IRF8 (Interferon Regulatory Factor 8)-Dependent Dendritic Cells Abrogates Proatherogenic Adaptive Immunity. Circ. Res. 2018, 122, 813–820. [Google Scholar] [CrossRef]
- Saravia, J.; Chapman, N.M.; Chi, H. Helper T cell differentiation. Cell. Mol. Immunol. 2019, 16, 634–643. [Google Scholar] [CrossRef]
- Ley, K. The second touch hypothesis: T cell activation, homing and polarization. F1000Research 2014, 3, 37. [Google Scholar] [CrossRef]
- Koltsova, E.K.; Ley, K. How dendritic cells shape atherosclerosis. Trends Immunol. 2011, 32, 540–547. [Google Scholar] [CrossRef] [Green Version]
- Dustin, M.L. A dynamic view of the immunological synapse. Semin Immunol 2005, 17, 400–410. [Google Scholar] [CrossRef]
- Huppa, J.B.; Davis, M.M. T-cell-antigen recognition and the immunological synapse. Nat. Rev. Immunol. 2003, 3, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Altman, J.D.; Davis, M.M. MHC-peptide tetramers to visualize antigen-specific T cells. Curr. Protoc. Immunol. 2003, 53, 17.3.1–17.3.33. [Google Scholar] [CrossRef] [PubMed]
- Newell, E.W.; Davis, M.M. Beyond model antigens: High-dimensional methods for the analysis of antigen-specific T cells. Nat. Biotechnol. 2014, 32, 149–157. [Google Scholar] [CrossRef]
- Altman, J.D.; Moss, P.A.; Goulder, P.J.; Barouch, D.H.; McHeyzer-Williams, M.G.; Bell, J.I.; McMichael, A.J.; Davis, M.M. Phenotypic analysis of antigen-specific T lymphocytes. Science. 1996. 274: 94-96. J. Immunol. 2011, 187, 7–9. [Google Scholar] [PubMed]
- Reiss, S.; Baxter, A.E.; Cirelli, K.M.; Dan, J.M.; Morou, A.; Daigneault, A.; Brassard, N.; Silvestri, G.; Routy, J.P.; Havenar-Daughton, C.; et al. Comparative analysis of activation induced marker (AIM) assays for sensitive identification of antigen-specific CD4 T cells. PLoS ONE 2017, 12, e0186998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosono, M.; de Boer, O.J.; van der Wal, A.C.; van der Loos, C.M.; Teeling, P.; Piek, J.J.; Ueda, M.; Becker, A.E. Increased expression of T cell activation markers (CD25, CD26, CD40L and CD69) in atherectomy specimens of patients with unstable angina and acute myocardial infarction. Atherosclerosis 2003, 168, 73–80. [Google Scholar] [CrossRef]
- Poulton, T.A.; Gallagher, A.; Potts, R.C.; Beck, J.S. Changes in activation markers and cell membrane receptors on human peripheral blood T lymphocytes during cell cycle progression after PHA stimulation. Immunology 1988, 64, 419–425. [Google Scholar]
- Croft, M.; So, T.; Duan, W.; Soroosh, P. The significance of OX40 and OX40L to T-cell biology and immune disease. Immunol. Rev. 2009, 229, 173–191. [Google Scholar] [CrossRef] [Green Version]
- Cibrian, D.; Sanchez-Madrid, F. CD69: From activation marker to metabolic gatekeeper. Eur. J. Immunol. 2017, 47, 946–953. [Google Scholar] [CrossRef]
- Frentsch, M.; Arbach, O.; Kirchhoff, D.; Moewes, B.; Worm, M.; Rothe, M.; Scheffold, A.; Thiel, A. Direct access to CD4+ T cells specific for defined antigens according to CD154 expression. Nat. Med. 2005, 11, 1118–1124. [Google Scholar] [CrossRef]
- Roy, M.; Waldschmidt, T.; Aruffo, A.; Ledbetter, J.A.; Noelle, R.J. The regulation of the expression of gp39, the CD40 ligand, on normal and cloned CD4+ T cells. J. Immunol. 1993, 151, 2497–2510. [Google Scholar] [PubMed]
- Michel, N.A.; Zirlik, A.; Wolf, D. CD40L and Its Receptors in Atherothrombosis-An Update. Front Cardiovasc Med. 2017, 4, 40. [Google Scholar] [CrossRef] [Green Version]
- Yellin, M.J.; Sippel, K.; Inghirami, G.; Covey, L.R.; Lee, J.J.; Sinning, J.; Clark, E.A.; Chess, L.; Lederman, S. CD40 molecules induce down-modulation and endocytosis of T cell surface T cell-B cell activating molecule/CD40-L. Potential role in regulating helper effector function. J. Immunol. 1994, 152, 598–608. [Google Scholar]
- Han, A.; Glanville, J.; Hansmann, L.; Davis, M.M. Linking T-cell receptor sequence to functional phenotype at the single-cell level. Nat. Biotechnol. 2014, 32, 684–692. [Google Scholar] [CrossRef]
- Abdelaal, H.M.; Cartwright, E.K.; Skinner, P.J. Detection of Antigen-Specific T Cells Using In Situ MHC Tetramer Staining. Int. J. Mol. Sci. 2019, 20, 5165. [Google Scholar] [CrossRef] [Green Version]
- Authors/Task Force, M.; Guidelines, E.S.C.C.f.P.; Societies, E.S.C.N.C. 2019 ESC/EAS guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Atherosclerosis 2019, 290, 140–205. [Google Scholar] [CrossRef] [Green Version]
- Gero, S.; Gergely, J.; Jakab, L.; Szekely, J.; Virag, S.; Farkas, K.; Czuppon, A. Inhibition of cholesterol atherosclerosis by immunisation with beta-lipoprotein. Lancet 1959, 2, 6–7. [Google Scholar] [CrossRef]
- Freigang, S.; Horkko, S.; Miller, E.; Witztum, J.L.; Palinski, W. Immunization of LDL receptor-deficient mice with homologous malondialdehyde-modified and native LDL reduces progression of atherosclerosis by mechanisms other than induction of high titers of antibodies to oxidative neoepitopes. Arter. Thromb. Vasc. Biol. 1998, 18, 1972–1982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palinski, W.; Miller, E.; Witztum, J.L. Immunization of low density lipoprotein (LDL) receptor-deficient rabbits with homologous malondialdehyde-modified LDL reduces atherogenesis. Proc. Natl. Acad. Sci. USA 1995, 92, 821–825. [Google Scholar] [CrossRef] [Green Version]
- Ameli, S.; Hultgardh-Nilsson, A.; Regnstrom, J.; Calara, F.; Yano, J.; Cercek, B.; Shah, P.K.; Nilsson, J. Effect of immunization with homologous LDL and oxidized LDL on early atherosclerosis in hypercholesterolemic rabbits. Arter. Thromb. Vasc. Biol. 1996, 16, 1074–1079. [Google Scholar] [CrossRef]
- George, J.; Afek, A.; Gilburd, B.; Levkovitz, H.; Shaish, A.; Goldberg, I.; Kopolovic, Y.; Wick, G.; Shoenfeld, Y.; Harats, D. Hyperimmunization of apo-E-deficient mice with homologous malondialdehyde low-density lipoprotein suppresses early atherogenesis. Atherosclerosis 1998, 138, 147–152. [Google Scholar] [CrossRef]
- Zhou, X.; Caligiuri, G.; Hamsten, A.; Lefvert, A.K.; Hansson, G.K. LDL Immunization Induces T-Cell-Dependent Antibody Formation and Protection Against Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 108–114. [Google Scholar] [CrossRef] [Green Version]
- Chyu, K.Y.; Reyes, O.S.; Zhao, X.; Yano, J.; Dimayuga, P.; Nilsson, J.; Cercek, B.; Shah, P.K. Timing affects the efficacy of LDL immunization on atherosclerotic lesions in apo E (−/−) mice. Atherosclerosis 2004, 176, 27–35. [Google Scholar] [CrossRef]
- Zhou, X.; Robertson, A.K.; Rudling, M.; Parini, P.; Hansson, G.K. Lesion development and response to immunization reveal a complex role for CD4 in atherosclerosis. Circ. Res. 2005, 96, 427–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Y.; Wang, X.; Ji, Q.; Mao, X.; Tang, H.; Yi, G.; Meng, K.; Yang, X.; Zeng, Q. CD4+LAP + and CD4 +CD25 +Foxp3 + regulatory T cells induced by nasal oxidized low-density lipoprotein suppress effector T cells response and attenuate atherosclerosis in ApoE−/− mice. J. Clin. Immunol. 2012, 32, 1104–1117. [Google Scholar] [CrossRef] [PubMed]
- Kobiyama, K.; Saigusa, R.; Ley, K. Vaccination against atherosclerosis. Curr. Opin. Immunol. 2019, 59, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Nettersheim, F.S.; De Vore, L.; Winkels, H. Vaccination in Atherosclerosis. Cells 2020, 9, 2560. [Google Scholar] [CrossRef]
- Davidson, N.O.; Shelness, G.S. APOLIPOPROTEIN B: mRNA editing, lipoprotein assembly, and presecretory degradation. Annu. Rev. Nutr. 2000, 20, 169–193. [Google Scholar] [CrossRef] [PubMed]
- Klingenberg, R.; Lebens, M.; Hermansson, A.; Fredrikson, G.N.; Strodthoff, D.; Rudling, M.; Ketelhuth, D.F.; Gerdes, N.; Holmgren, J.; Nilsson, J.; et al. Intranasal immunization with an apolipoprotein B-100 fusion protein induces antigen-specific regulatory T cells and reduces atherosclerosis. Arter. Thromb. Vasc. Biol. 2010, 30, 946–952. [Google Scholar] [CrossRef] [Green Version]
- Kimura, T.; Tse, K.; McArdle, S.; Gerhardt, T.; Miller, J.; Mikulski, Z.; Sidney, J.; Sette, A.; Wolf, D.; Ley, K. Atheroprotective vaccination with MHC-II-restricted ApoB peptides induces peritoneal IL-10-producing CD4 T cells. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H781–H790. [Google Scholar] [CrossRef]
- Kobiyama, K.; Vassallo, M.; Mitzi, J.; Winkels, H.; Pei, H.; Kimura, T.; Miller, J.; Wolf, D.; Ley, K. A clinically applicable adjuvant for an atherosclerosis vaccine in mice. Eur. J. Immunol. 2018, 48, 1580–1587. [Google Scholar] [CrossRef]
- Fredrikson, G.N.; Hedblad, B.; Berglund, G.; Alm, R.; Ares, M.; Cercek, B.; Chyu, K.Y.; Shah, P.K.; Nilsson, J. Identification of immune responses against aldehyde-modified peptide sequences in apoB associated with cardiovascular disease. Arter. Thromb. Vasc. Biol. 2003, 23, 872–878. [Google Scholar] [CrossRef]
- Tse, K.Y.-B.; Gonen, A.; Sidney, J.; Ouyang, H.; Witztum, J.L.; Sette, A.; Tse, H.Y.; Ley, K. Atheroprotective vaccination with MHC-II restricted peptides from ApoB-100. Front. Immunol. 2013, 4, 493. [Google Scholar] [CrossRef] [PubMed]
- Gistera, A.; Hermansson, A.; Strodthoff, D.; Klement, M.L.; Hedin, U.; Fredrikson, G.N.; Nilsson, J.; Hansson, G.K.; Ketelhuth, D.F. Vaccination against T-cell epitopes of native ApoB100 reduces vascular inflammation and disease in a humanized mouse model of atherosclerosis. J. Intern. Med. 2017, 281, 383–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, M.K.; Tse, K.Y.; Zhao, X.; Welch, K.; Eitzman, D.T.; Thipparthi, R.R.; Montgomery, P.C.; Thummel, R.; Tse, H.Y. T-Cells Specific for a Self-Peptide of ApoB-100 Exacerbate Aortic Atheroma in Murine Atherosclerosis. Front Immunol. 2017, 8, 95. [Google Scholar] [CrossRef] [PubMed]
- Tse, K.; Tse, H.; Sidney, J.; Sette, A.; Ley, K. T cells in atherosclerosis. Int. Immunol. 2013, 25, 615–622. [Google Scholar] [CrossRef] [Green Version]
- Wigren, M.; Bengtsson, D.; Duner, P.; Olofsson, K.; Bjorkbacka, H.; Bengtsson, E.; Fredrikson, G.N.; Nilsson, J. Atheroprotective effects of Alum are associated with capture of oxidized LDL antigens and activation of regulatory T cells. Circ. Res. 2009, 104, e62–e70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbin, O.; Ait-Oufella, H.; Yu, W.; Fredrikson, G.N.; Aubier, B.; Perez, N.; Barateau, V.; Nilsson, J.; Tedgui, A.; Mallat, Z. Regulatory T-cell response to apolipoprotein B100-derived peptides reduces the development and progression of atherosclerosis in mice. Arter. Thromb. Vasc. Biol. 2012, 32, 605–612. [Google Scholar] [CrossRef] [Green Version]
- Wigren, M.; Rattik, S.; Yao Mattisson, I.; Tomas, L.; Gronberg, C.; Soderberg, I.; Alm, R.; Sundius, L.; Ljungcrantz, I.; Bjorkbacka, H.; et al. Lack of Ability to Present Antigens on Major Histocompatibility Complex Class II Molecules Aggravates Atherosclerosis in ApoE(−/−) Mice. Circulation 2019, 139, 2554–2566. [Google Scholar] [CrossRef] [PubMed]
- Ley, K. 2015 Russell Ross Memorial Lecture in Vascular Biology: Protective Autoimmunity in Atherosclerosis. Arter. Thromb. Vasc. Biol. 2016, 36, 429–438. [Google Scholar] [CrossRef] [Green Version]
- Burnet, F.M. Immunological recognition of self. Science 1961, 133, 307–311. [Google Scholar] [CrossRef]
- Klein, L.; Kyewski, B.; Allen, P.M.; Hogquist, K.A. Positive and negative selection of the T cell repertoire: What thymocytes see (and don’t see). Nat. Rev. Immunol. 2014, 14, 377–391. [Google Scholar] [CrossRef] [Green Version]
- Metzger, T.C.; Anderson, M.S. Control of central and peripheral tolerance by Aire. Immunol. Rev. 2011, 241, 89–103. [Google Scholar] [CrossRef] [Green Version]
- Juang, J.; Ebert, P.J.; Feng, D.; Garcia, K.C.; Krogsgaard, M.; Davis, M.M. Peptide-MHC heterodimers show that thymic positive selection requires a more restricted set of self-peptides than negative selection. J. Exp. Med. 2010, 207, 1223–1234. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Jiang, N.; Ebert, P.J.; Kidd, B.A.; Muller, S.; Lund, P.J.; Juang, J.; Adachi, K.; Tse, T.; Birnbaum, M.E.; et al. Clonal Deletion Prunes but Does Not Eliminate Self-Specific alphabeta CD8(+) T Lymphocytes. Immunity 2015, 42, 929–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudensky, A.Y. Regulatory T cells and Foxp3. Immunol. Rev. 2011, 241, 260–268. [Google Scholar] [CrossRef] [Green Version]
- Dominguez-Villar, M.; Baecher-Allan, C.M.; Hafler, D.A. Identification of T helper type 1-like, Foxp3+ regulatory T cells in human autoimmune disease. Nat. Med. 2011, 17, 673–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurence, A.; Amarnath, S.; Mariotti, J.; Kim, Y.C.; Foley, J.; Eckhaus, M.; O’Shea, J.J.; Fowler, D.H. STAT3 transcription factor promotes instability of nTreg cells and limits generation of iTreg cells during acute murine graft-versus-host disease. Immunity 2012, 37, 209–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClymont, S.A.; Putnam, A.L.; Lee, M.R.; Esensten, J.H.; Liu, W.; Hulme, M.A.; Hoffmuller, U.; Baron, U.; Olek, S.; Bluestone, J.A.; et al. Plasticity of human regulatory T cells in healthy subjects and patients with type 1 diabetes. J. Immunol. 2011, 186, 3918–3926. [Google Scholar] [CrossRef]
- Pugliese, A. Autoreactive T cells in type 1 diabetes. J. Clin. Invest. 2017, 127, 2881–2891. [Google Scholar] [CrossRef]
- Libby, P.; Lichtman, A.H.; Hansson, G.K. Immune effector mechanisms implicated in atherosclerosis: From mice to humans. Immunity 2013, 38, 1092–1104. [Google Scholar] [CrossRef] [Green Version]
- Reiner, Z.; Laufs, U.; Cosentino, F.; Landmesser, U. The year in cardiology 2018: Prevention. Eur. Heart J. 2019, 40, 336–344. [Google Scholar] [CrossRef]
- Aday, A.W.; Ridker, P.M. Targeting Residual Inflammatory Risk: A Shifting Paradigm for Atherosclerotic Disease. Front Cardiovasc Med. 2019, 6, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, P.; Rudan, D.; Zhu, Y.; Fowkes, F.J.I.; Rahimi, K.; Fowkes, F.G.R.; Rudan, I. Global, regional, and national prevalence and risk factors for peripheral artery disease in 2015: An updated systematic review and analysis. Lancet Glob. Health 2019, 7, e1020–e1030. [Google Scholar] [CrossRef] [Green Version]
- Peikert, A.; Kaier, K.; Merz, J.; Manhart, L.; Schafer, I.; Hilgendorf, I.; Hehn, P.; Wolf, D.; Willecke, F.; Sheng, X.; et al. Residual inflammatory risk in coronary heart disease: Incidence of elevated high-sensitive CRP in a real-world cohort. Clin. Res. Cardiol. 2020, 109, 315–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frostegard, J. Atherosclerosis in patients with autoimmune disorders. Arter. Thromb. Vasc. Biol. 2005, 25, 1776–1785. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Approaches to detect Apolipoprotein B (ApoB)-specific CD4+ T cells in mice and humans. Functional restimulation with ApoB-peptides requires a peptide mapping of the entire ApoB-sequence and a selection of peptides with the highest predicted and tested affinity towards the MHC-II complex. Antigen-presenting cells are loaded with selected peptides and co-incubated with CD4+ T cells. Expression of activation markers is subsequently used to identify activated, ApoB-reactive T cells (A) Design of tetramers of MHC-II loaded with the peptide of interest. Tetramers are labelled with fluorochromes for the detection of tetramer-binding T cells in flow cytometry. (B) In simultaneous T cell receptor (TCR) and gene expression RNA-sequencing, T cells with an oligoclonal repertoire of the TCR are detected on a single cell level, which allows to selectively analyze gene expression in these cells. (C) Workflow for the generation of a TCR-transgenic mouse: peptide-specific T cells are expanded in vivo by immunization with LDL or a self-peptide from ApoB, isolated, and further expanded ex vivo by restimulation with LDL or peptide-loaded APC. Resulting clones undergo TCR-sequencing and the most promising TCR-sequences are used as templates for the generation of a TCR-transgenic mouse (D).
Figure 1.
Approaches to detect Apolipoprotein B (ApoB)-specific CD4+ T cells in mice and humans. Functional restimulation with ApoB-peptides requires a peptide mapping of the entire ApoB-sequence and a selection of peptides with the highest predicted and tested affinity towards the MHC-II complex. Antigen-presenting cells are loaded with selected peptides and co-incubated with CD4+ T cells. Expression of activation markers is subsequently used to identify activated, ApoB-reactive T cells (A) Design of tetramers of MHC-II loaded with the peptide of interest. Tetramers are labelled with fluorochromes for the detection of tetramer-binding T cells in flow cytometry. (B) In simultaneous T cell receptor (TCR) and gene expression RNA-sequencing, T cells with an oligoclonal repertoire of the TCR are detected on a single cell level, which allows to selectively analyze gene expression in these cells. (C) Workflow for the generation of a TCR-transgenic mouse: peptide-specific T cells are expanded in vivo by immunization with LDL or a self-peptide from ApoB, isolated, and further expanded ex vivo by restimulation with LDL or peptide-loaded APC. Resulting clones undergo TCR-sequencing and the most promising TCR-sequences are used as templates for the generation of a TCR-transgenic mouse (D).

Figure 2.
Activation and differentiation of ApoB-specific CD4+ T cells in mice. The ApoB-containing apolipoproteins LDL, VLDL, and chylomicrons (CM) are taken up by antigen-presenting cells (APCs) by endocytosis. After their intracellular processing, apolipoprotein-derived peptides are loaded on MHC-II molecules, before the entire MHC-II-peptide complex is transposed to the cell membrane. There, MHC-II-peptide complexes can be recognized and bound by a specific T cell receptor (TCR). In combination with sufficient co-stimulatory signaling events provided by the APC, a naïve CD4+ T cell is activated and may differentiate into distinct, partially overlapping TH-types of immunity: Most ApoB-specific CD4+ T cells (ApoB+) express transcriptomes and markers of TH17 and T-regulatory cells (Treg). They express CCR5 and CXCR6, two known chemokine receptors (CCRs) required for aortic homing. Over time, ApoB+ cells acquire additional pro-inflammatory transcriptional programs and express the TH1 transcription factor T-bet, as well as the CCRs CXCR5 and CCR6. The initially detectable protective Treg signature is lost in this process. After vaccination, IL-10 secreting FoxP3+ApoB+ cells have been described at the site of vaccination. In transgenic mice, only expressing a TCR that recognizes a specific ApoB-peptide, a part of ApoB+ cells differentiates into TFH that promote plasma cells generation and the production of LDL-lowering anti-LDL antibodies.
Figure 2.
Activation and differentiation of ApoB-specific CD4+ T cells in mice. The ApoB-containing apolipoproteins LDL, VLDL, and chylomicrons (CM) are taken up by antigen-presenting cells (APCs) by endocytosis. After their intracellular processing, apolipoprotein-derived peptides are loaded on MHC-II molecules, before the entire MHC-II-peptide complex is transposed to the cell membrane. There, MHC-II-peptide complexes can be recognized and bound by a specific T cell receptor (TCR). In combination with sufficient co-stimulatory signaling events provided by the APC, a naïve CD4+ T cell is activated and may differentiate into distinct, partially overlapping TH-types of immunity: Most ApoB-specific CD4+ T cells (ApoB+) express transcriptomes and markers of TH17 and T-regulatory cells (Treg). They express CCR5 and CXCR6, two known chemokine receptors (CCRs) required for aortic homing. Over time, ApoB+ cells acquire additional pro-inflammatory transcriptional programs and express the TH1 transcription factor T-bet, as well as the CCRs CXCR5 and CCR6. The initially detectable protective Treg signature is lost in this process. After vaccination, IL-10 secreting FoxP3+ApoB+ cells have been described at the site of vaccination. In transgenic mice, only expressing a TCR that recognizes a specific ApoB-peptide, a part of ApoB+ cells differentiates into TFH that promote plasma cells generation and the production of LDL-lowering anti-LDL antibodies.


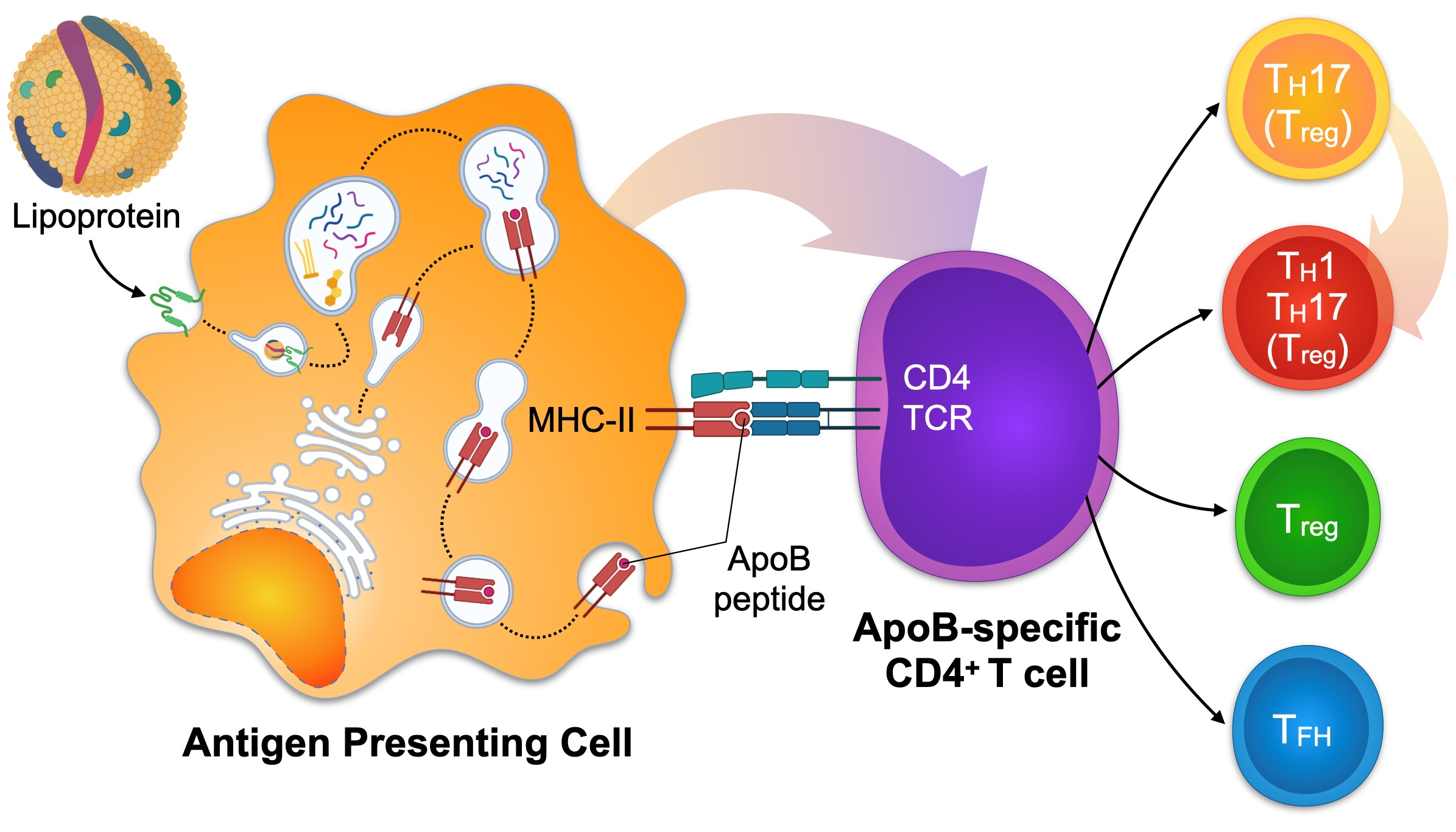{kind=link}
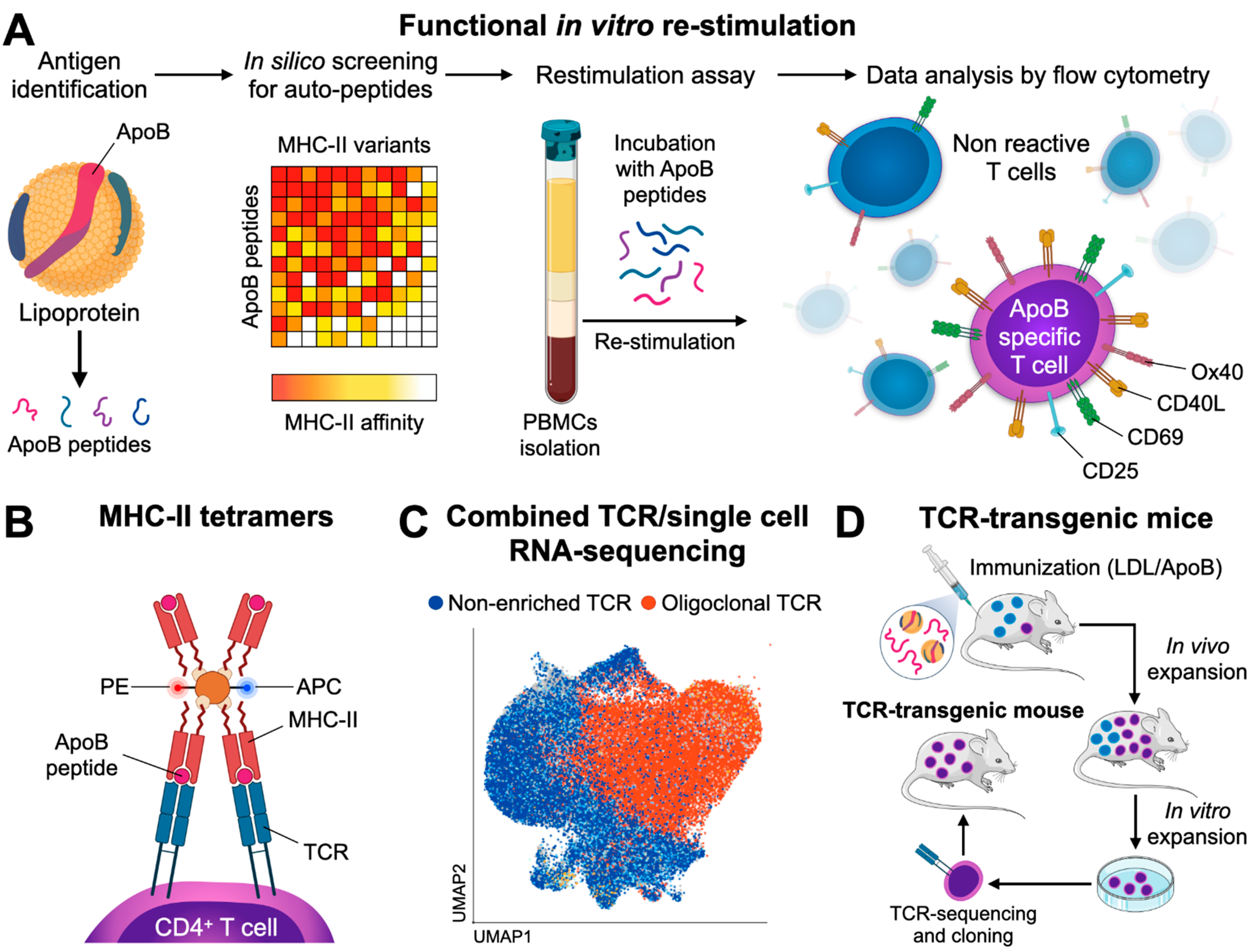{kind=link}
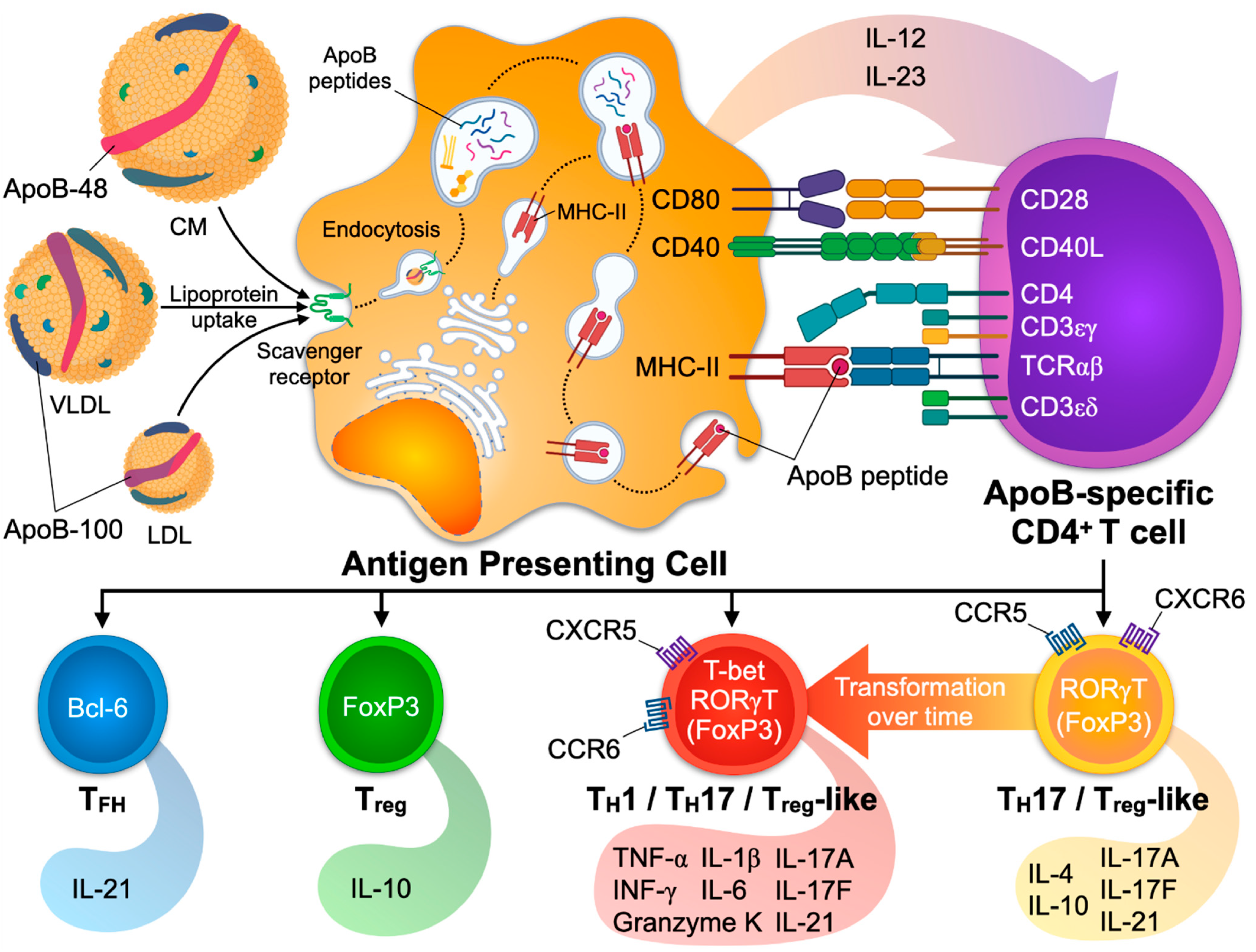{kind=link}
Table 1.
CD4+ T cell subsets and functions in atherosclerosis.
| Lineage | TF | Effector Cytokines | Role in Mouse Atherosclerosis | Regulation in Human Atherosclerosis |
|---|---|---|---|---|
| TH1 | T-bet | INF-γ, IL-2, IL-3, IL-6, TNF-α, lymphotoxin | Pro-atherogenic [37,39,40,41,42,43,44] | TH1 dominance in atherosclerotic lesions [35,36], higher IFN-γ plasma levels in patients with CAD [127], higher IL-6 and TNF-α plasma levels in patients with MI [128]. |
| TH2 | GATA3 | IL-4, IL-5, IL-10, IL-13 | Pro-atherogenic [79,82] Atheroprotective [80] No effect [81] | Lower TH2 cell numbers and decreased IL-4 secretion by CD4+ in patients with MI [83], lower IL-5 plasma levels in patients with subclinical atherosclerosis [84] |
| TH9 | FoxO1, BATF, IRF4 | IL-9 | Pro-atherogenic [107] | Higher IL-9 plasma levels in patients with atherosclerosis and ACS [105] Unchanged TH9 numbers in patients with ACS [106] |
| TH17 | RORγT | IL-17A, IL-17-F, IL-21, IL-22 | Pro-atherogenic [64,65,66,67] Atheroprotective [70,71,72,73,74] No effect [76] | Higher IL-17 plasma levels in patients with unstable angina or MI [68,69], lower IL-17 plasma levels in patients with MI [75], unchanged IL-17 plasma levels patients in patients with CAD [77] |
| TH22 | AHR | IL-22 | Pro-atherogenic [109] | Higher TH22 cell counts and IL-22 plasma level in patient with an ACS [106,110] |
| Treg | FoxP3, CD25 | IL-10, TGF-β | Atheroprotective [54,55,56] | Lower Treg numbers in blood from patients with MI [57] and ACS [68,69], low Treg numbers predict MI [58], higher Treg numbers in blood of patients with CAD [49] |
| TFH | Bcl6 | IL-21 | Pro-atherogenic [50,87,88] | Higher TFH count in patients with advanced atherosclerosis [90] |
| CD4+ CTL | TNF-α, INF-γ, perforin, granzyme A, B | Not present in mice | Higher numbers in blood from patients with ACS [95] and with end-stage renal disease and atherosclerosis [129], enrichment in unstable atherosclerotic lesions [97,98,99] | |
| NK T cells | Multiple, including perforin and granzymes | Controversial [113] | Accumulation of NKT cells in rupture-prone atherosclerotic plaques [112]. |
TF, transcription factor; TH, T-helper; Treg, T regulatory cell; TFH, T follicular helper cell; AHR, aryl hydrocarbon receptor; MI, Myocardial Infarction; ACS, Acute Coronary Syndrome; CAD, Coronary Artery Disease; CTL, cytotoxic lymphocyte; NK, natural killer.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Marchini, T.; Hansen, S.; Wolf, D. ApoB-Specific CD4+ T Cells in Mouse and Human Atherosclerosis. Cells 2021, 10, 446. https://doi.org/10.3390/cells10020446
AMA Style
Marchini T, Hansen S, Wolf D. ApoB-Specific CD4+ T Cells in Mouse and Human Atherosclerosis. Cells. 2021; 10(2):446. https://doi.org/10.3390/cells10020446
Chicago/Turabian StyleMarchini, Timoteo, Sophie Hansen, and Dennis Wolf. 2021. "ApoB-Specific CD4+ T Cells in Mouse and Human Atherosclerosis" Cells 10, no. 2: 446. https://doi.org/10.3390/cells10020446
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.