
Role of FGF15 in Hepatic Surgery in the Presence of Tumorigenesis: Dr. Jekyll or Mr. Hyde?


 , , and
, , and
Abstract
:1. Introduction
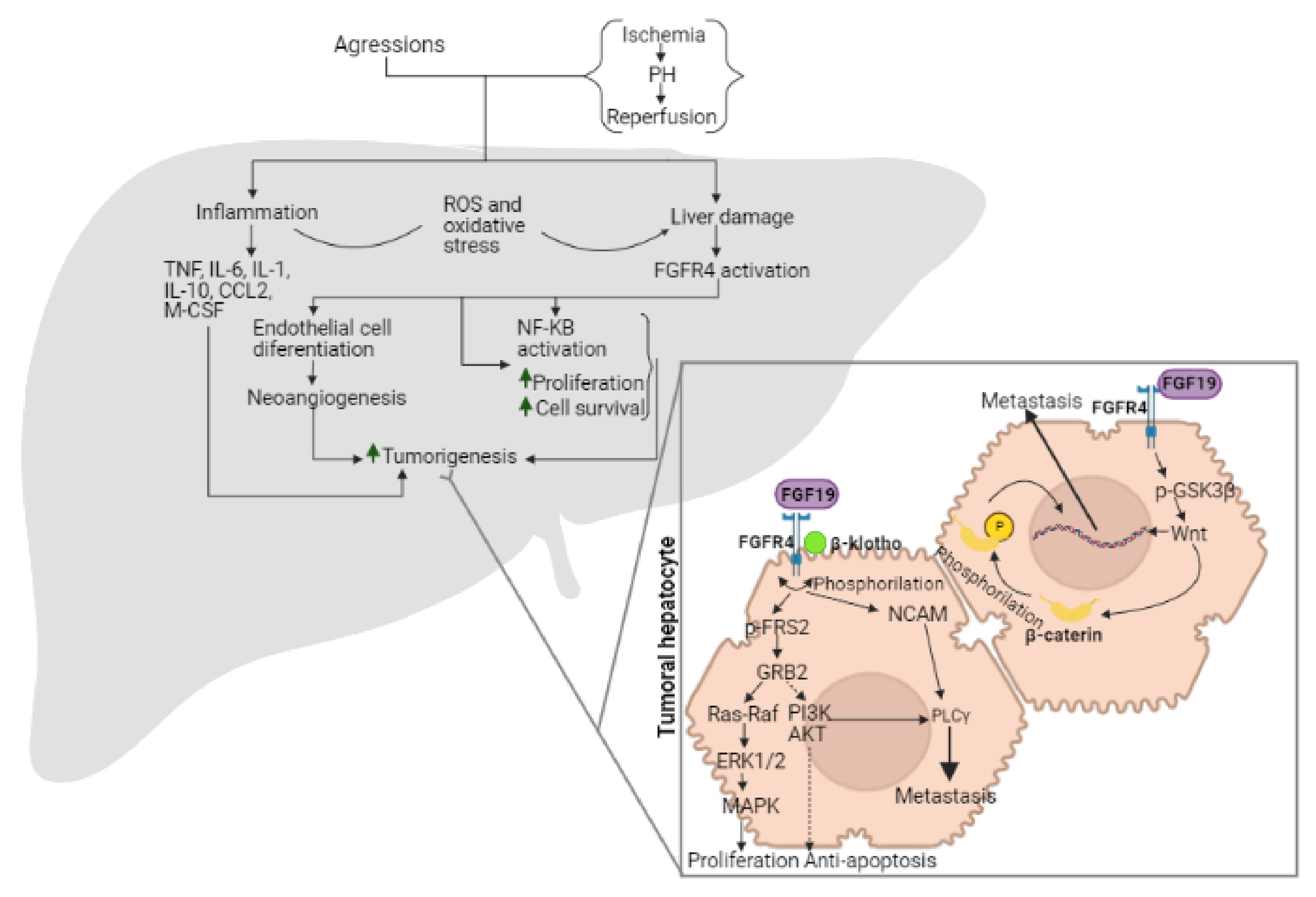
2. Role of FGF15/19 in Liver Tumorigenesis
2.1. Involvement of BA in the Effects of FGF15/19 on Tumorigenesis
2.2. Involvement of FGF15/19 and Inflammation in the Hepatic Microenvironment
2.3. FGF15/19: Prognostic Factor of HCC?
2.4. Mechanisms of Action of FGF15/19-FGFR4
2.5. Involvement of Genetic and Epigenetic FGF15/19 and FGFR4 Alterations in Tumorigenesis
2.6. Role of FGFR4 on Metastasis
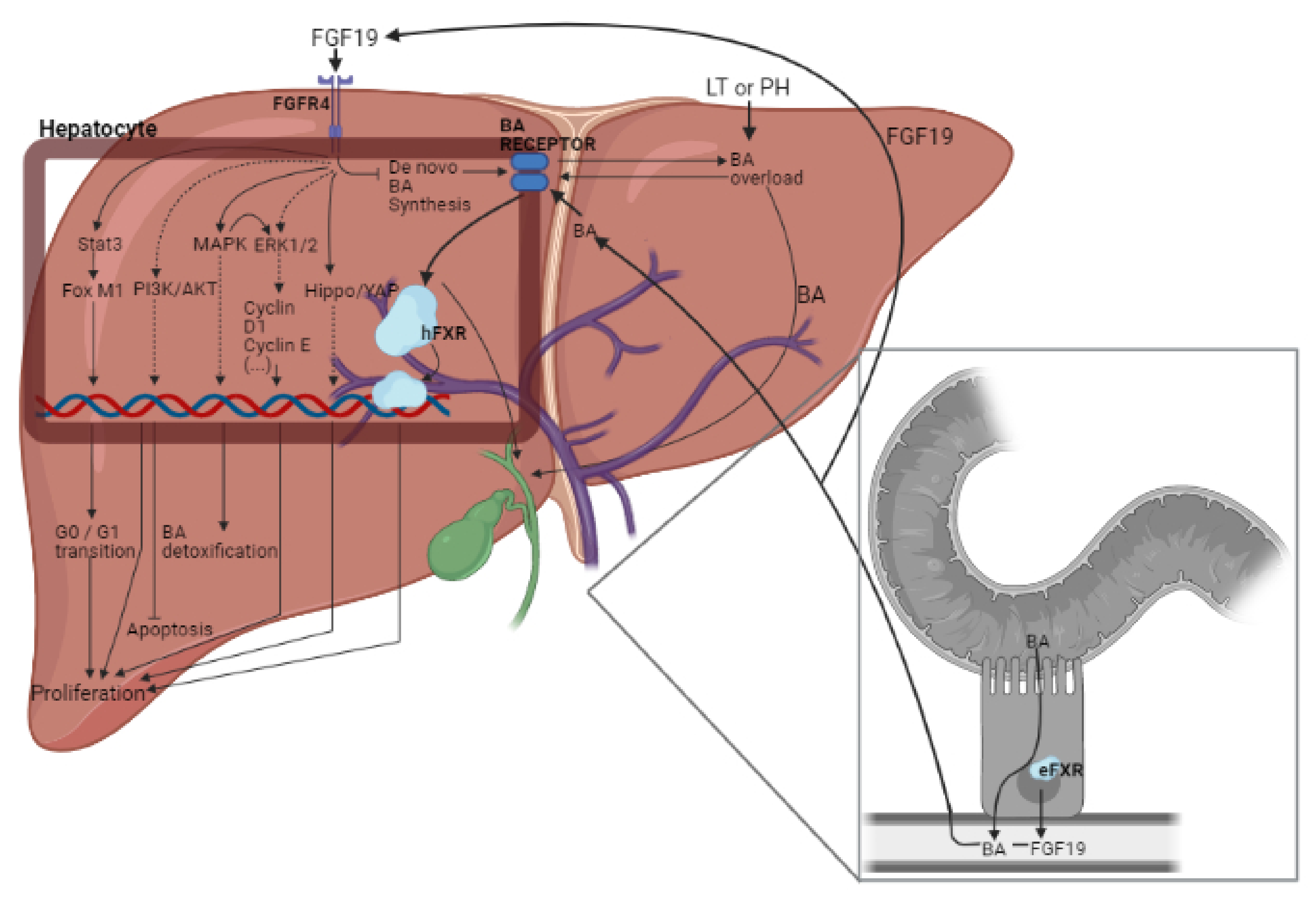
3. Role of FGF15/19 in Liver Regeneration
3.1. Involvement of BAs and YAP in the Effects of FGF15/19
3.2. Signaling Pathways (Different to BAs and YAP) Regulated by FGF15/19
4. Modulation of FGF15/19 Actions
4.1. Non-Protumorigenic Variants of FGF15/19
4.2. Other Drugs That Affect FGF15/19 Signaling Pathways
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Álvarez-Mercado, A.I.; Gulfo, J.; Gómez, M.R.; Jiménez-Castro, M.B.; Gracia-Sancho, J.; Peralta, C. Use of steatotic grafts in liver transplantation: Current status. Liver Transplant. 2019, 25, 771–786. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Castro, M.B.; Gracia-Sancho, J.; Peralta, C. Brain death and marginal grafts in liver transplantation. Cell Death Dis. 2015, 6, e1777. [Google Scholar] [CrossRef] [Green Version]
- Danion, J.; Thuillier, R.; Allain, G.; Bruneval, P.; Tomasi, J.; Pinsard, M.; Hauet, T.; Kerforne, T. Evaluation of liver quality after circulatory death versus brain death: A comparative preclinical pig model study. Int. J. Mol. Sci. 2020, 21, 9040. [Google Scholar] [CrossRef]
- Xu, J.; Sayed, B.A.; Casas-Ferreira, A.M.; Srinivasan, P.; Heaton, N.; Rela, M.; Ma, Y.; Fuggle, S.; Legido-Quigley, C.; Jassem, W. The impact of ischemia/reperfusion injury on liver allografts from deceased after cardiac death versus deceased after brain death donors. PLoS ONE 2016, 11, e0148815. [Google Scholar] [CrossRef]
- Pandya, K.; Sastry, V.; Panlilio, M.T.; Yip, T.C.F.; Salimi, S.; West, C.; Virtue, S.; Wells, M.; Crawford, M.; Pulitano, C.; et al. Differential impact of extended criteria donors after brain death or circulatory death in adult liver transplantation. Liver Transplant. 2020, 26, 1603–1617. [Google Scholar] [CrossRef]
- Micó-Carnero, M.; Rojano-Alfonso, C.; Álvarez-Mercado, A.I.; Gracia-Sancho, J.; Casillas-Ramírez, A.; Peralta, C. Effects of gut metabolites and microbiota in healthy and marginal livers submitted to surgery. Int. J. Mol. Sci. 2021, 22, 44. [Google Scholar] [CrossRef]
- Dixon, E.; Vollmer, C.M.; Bathe, O.F.; Sutherland, F. Vascular occlusion to decrease blood loss during hepatic resection. Am. J. Surg. 2005, 190, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Bujaldon, E.; Cornide-Petronio, M.E.; Gulfo, J.; Rotondo, F.; Ávalos de León, C.; Negrete-Sánchez, E.; Gracia-Sancho, J.; Novials, A.; Jiménez-Castro, M.B.; Peralta Uroz, C. Relevance of VEGFA in rat livers subjected to partial hepatectomy under ischemia-reperfusion. J. Mol. Med. 2019, 97, 1299–1314. [Google Scholar] [CrossRef] [Green Version]
- Choi, M.; Moschetta, A.; Bookout, A.L.; Peng, L.; Umetani, M.; Holmstrom, S.R.; Suino-Powell, K.; Xu, H.E.; Richardson, J.A.; Gerard, R.D.; et al. Identification of a hormonal basis for gallbladder filling. Nat. Med. 2006, 12, 1253–1255. [Google Scholar] [CrossRef] [PubMed]
- Escalera, L.M.D.L.; Kyrou, I.; Vrbikova, J.; Hainer, V.; Sramkova, P.; Fried, M.; Piya, M.K.; Kumar, S.; Tripathi, G.; McTernan, P.G. Impact of gut hormone FGF-19 on type-2 diabetes and mitochondrial recovery in a prospective study of obese diabetic women undergoing bariatric surgery. BMC Med. 2017, 15, 34. [Google Scholar] [CrossRef] [Green Version]
- Lundåsen, T.; Gälman, C.; Angelin, B.; Rudling, M. Circulating intestinal fibroblast growth factor 19 has a pronounced diurnal variation and modulates hepatic bile acid synthesis in man. J. Intern. Med. 2006, 260, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Kir, S.; Beddow, S.A.; Samuel, V.T.; Miller, P.; Previs, S.F.; Suino-Powell, K.; Xu, H.E.; Shulman, G.I.; Kliewer, S.A.; Mangelsdorf, D.J. FGF19 as a postprandial, insulin-independent activator of hepatic protein and glycogen synthesis. Science 2011, 331, 1621–1624. [Google Scholar] [CrossRef] [Green Version]
- Potthoff, M.J.; Boney-Montoya, J.; Choi, M.; He, T.; Sunny, N.E.; Satapati, S.; Suino-Powell, K.; Xu, H.E.; Gerard, R.D.; Finck, B.N.; et al. FGF15/19 regulates hepatic glucose metabolism by inhibiting the CREB-PGC-1α pathway. Cell Metab. 2011, 13, 729–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurosu, H.; Choi, M.; Ogawa, Y.; Dickson, A.S.; Goetz, R.; Eliseenkova, A.V.; Mohammadi, M.; Rosenblatt, K.P.; Kliewer, S.A.; Kuro-o, M. Tissue-specific expression of βklotho and fibroblast growth factor (FGF) receptor isoforms determines metabolic activity of FGF19 and FGF. J. Biol. Chem. 2007, 282, 26687–26695. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; John, L.M.; Adams, S.H.; Yu, X.X.; Tomlinson, E.; Renz, M.; Williams, P.M.; Soriano, R.; Corpuz, R.; Moffat, B.; et al. Fibroblast growth factor 19 increases metabolic rate and reverses dietary and leptin-deficient diabetes. Endocrinology 2004, 145, 2594–2603. [Google Scholar] [CrossRef] [Green Version]
- Lenicek, M.; Duricova, D.; Komarek, V.; Gabrysova, B.; Lukas, M.; Smerhovsky, Z.; Vitek, L. Bile acid malabsorption in inflammatory bowel disease: Assessment by serum markers. Inflamm. Bowel Dis. 2011, 17, 1322–1327. [Google Scholar] [CrossRef]
- Mráz, M.; Lacinová, Z.; Kaválková, P.; Haluzíková, D.; Trachta, P.; Drápalová, J.; Hanušova, V.; Haluzík, M. Serum concentrations of fibroblast growth factor 19 in patients with obesity and type 2 diabetes mellitus: The influence of acute hyperinsulinemia, very-low calorie diet and PPAR-α agonist treatment. Physiol. Res. 2011, 60, 627–636. [Google Scholar] [CrossRef]
- Schreuder, T.C.M.A.; Marsman, H.A.; Lenicek, M.; Van Werven, J.R.; Nederveen, A.J.; Jansen, P.L.M.; Schaap, F.G. The hepatic response to FGF19 is impaired in patients with nonalcoholic fatty liver disease and insulin resistance. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, 440–445. [Google Scholar] [CrossRef]
- Modica, S.; Petruzzelli, M.; Bellafante, E.; Murzilli, S.; Salvatore, L.; Celli, N.; Tullio, G.D.; Palasciano, G.; Moustafa, T.; Halilbasic, E.; et al. Selective activation of nuclear bile acid receptor FXR in the intestine protects mice against cholestasis. Gastroenterology 2012, 142, 355–365.e4. [Google Scholar] [CrossRef]
- Gulfo, J.; Rotondo, F.; León, C.G.Á.D.; Cornide-Petronio, M.E.; Fuster, C.; Gracia-Sancho, J.; Jiménez-Castro, M.B.; Peralta, C. FGF15 improves outcomes after brain dead donor liver transplantation with steatotic and non-steatotic grafts in rats. J. Hepatol. 2020, 73, 1131–1143. [Google Scholar] [CrossRef]
- León, C.G.Á.D.; Jiménez-Castro, M.B.; Cornide-Petronio, M.E.; Gulfo, J.; Rotondo, F.; Gracia-Sancho, J.; Casillas-Ramírez, A.; Peralta, C. The effect of fibroblast growth factor 15 signaling in non-steatotic and steatotic liver transplantation from cardiocirculatory death. Cells 2019, 8, 1640. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Zhao, Q.; Zhang, C.; Zhang, P.; Hu, A.; Zhang, L.; Schroder, P.M.; Ma, Y.; Guo, Z.; Zhu, X.; et al. The ileal FGF15/19 to hepatic FGFR4 axis regulates liver regeneration after partial hepatectomy in mice. J. Physiol. Biochem. 2018, 74, 247–260. [Google Scholar] [CrossRef]
- Kan, N.G.; Junghans, D.; Belmonte, J.C.I. Compensatory growth mechanisms regulated by BMP and FGF signaling mediate liver regeneration in zebrafish after partial hepatectomy. FASEB J. 2009, 23, 3516–3525. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Ren, H.; Zhou, Y.; Shang, L.; Zhang, Y.; Yang, F.; Shi, X. The hypoxia conditioned mesenchymal stem cells promote hepatocellular carcinoma progression through YAP mediated lipogenesis reprogramming. J. Exp. Clin. Cancer Res. 2019, 38, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Moon, H.; Cho, K.; Shin, S.; Kim, D.Y.; Han, K.H.; Ro, S.W. High risk of hepatocellular carcinoma development in fibrotic liver: Role of the hippo-YAP/TAZ signaling pathway. Int. J. Mol. Sci. 2019, 20, 581. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Gea, V.; Toffanin, S.; Friedman, S.L.; Llovet, J.M. Role of the microenvironment in the pathogenesis and treatment of hepatocellular carcinoma. Gastroenterology 2013, 144, 512–527. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.D.; Roberts, L.R. Hepatocellular carcinoma: A global view. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 448–458. [Google Scholar] [CrossRef] [Green Version]
- Choi, K.-J.; Baik, I.H.; Ye, S.-K.; Lee, Y.-H. Molecular targeted therapy for hepatocellular carcinoma: Present status and future directions. Biol. Pharm. Bull. 2015, 38, 986–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altekruse, S.F.; McGlynn, K.A.; Reichman, M.E. Hepatocellular carcinoma incidence, mortality, and survival trends in the United States from 1975 to 2005. J. Clin. Oncol. 2009, 27, 1485–1491. [Google Scholar] [CrossRef] [Green Version]
- Lin, B.C.; Desnoyers, L.R. FGF19 and cancer. Adv. Exp. Med. Biol. 2012, 728, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Yang, C.; Lu, W.; Xie, R.; Jin, C.; Huang, P.; Wang, F.; McKeehan, W.L. Metabolic regulator βklotho interacts with fibroblast growth factor receptor 4 (FGFR4) to induce apoptosis and inhibit tumor cell proliferation. J. Biol. Chem. 2010, 285, 30069–30078. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Yang, C.; Jin, C.; Luo, Y.; Wang, F.; McKeehan, W.L. Resident hepatocyte fibroblast growth factor receptor 4 limits hepatocarcinogenesis. Mol. Carcinog. 2009, 48, 553–562. [Google Scholar] [CrossRef] [Green Version]
- Poh, W.; Wong, W.; Ong, H.; Aung, M.O.; Lim, S.G.; Chua, B.T.; Ho, H.K. Klotho-beta overexpression as a novel target for suppressing proliferation and fibroblast growth factor receptor-4 signaling in hepatocellular carcinoma. Mol. Cancer 2012, 11, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.Y.; Xie, D.M.; Zhu, G.Q.; Huang, G.Q.; Lin, Y.Q.; Wang, L.R.; Shi, K.Q.; Hu, B.; Braddock, M.; Chen, Y.P.; et al. Targeting fibroblast growth factor 19 in liver disease: A potential biomarker and therapeutic target. Expert Opin. Ther. Targets 2015, 19, 675–685. [Google Scholar] [CrossRef]
- Jaeschke, H.; Li, T.; Heger, M. Post-hepatectomy liver regeneration in the context of bile acid homeostasis and the gut-liver signaling axis. J. Clin. Transl. Res. 2018, 4, 1–46. [Google Scholar] [CrossRef] [Green Version]
- Raja, A.; Park, I.; Haq, F.; Ahn, S.-M. FGF19–FGFR4 signaling in hepatocellular carcinoma. Cells 2019, 8, 536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kir, S.; Kliewer, S.A.; Mangelsdorf, D.J. Roles of FGF19 in liver metabolism. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 139–144. [Google Scholar] [CrossRef] [Green Version]
- Payne, C. Hydrophobic bile acids, genomic instability, Darwinian selection, and colon carcinogenesis. Clin. Exp. Gastroenterol. 2008, 1, 19. [Google Scholar] [CrossRef] [Green Version]
- Barrasa, J.I.; Olmo, N.; Lizarbe, M.A.; Turnay, J. Bile acids in the colon, from healthy to cytotoxic molecules. Toxicol. Vitr. 2013, 27, 964–977. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, X.; So, C.K.; Wang, S.; Wang, P.; Yan, L.; Myers, R.; Chen, Z.; Patterson, A.P.; Yang, C.S.; et al. Regulation of Cdx2 expression by promoter methylation, and effects of Cdx2 transfection on morphology and gene expression of human esophageal epithelial cells. Carcinogenesis 2007, 28, 488–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, H.; Xu, M.; Dong, W.; Deng, B.; Wang, S.; Zhang, Y.; Wang, S.; Luo, S.; Wang, W.; Qi, Y.; et al. Secondary bile acid-induced dysbiosis promotes intestinal carcinogenesis. Int. J. Cancer 2017, 140, 2545–2556. [Google Scholar] [CrossRef] [Green Version]
- Takano, S.; Akagi, M.; Bryan, G.T. Stimulation of ornithine decarboxylase activity and DNA synthesis by phorbol esters or bile acids in rat colon. Gan 1984, 75, 29–35. [Google Scholar]
- Garewal, H.; Bernstein, H.; Bernstein, C.; Sampliner, R.; Payne, C. Reduced bile acid-induced apoptosis in “normal” colorectal mucosa: A potential biological marker for cancer risk. Cancer Res. 1996, 56, 1480–1483. [Google Scholar] [PubMed]
- Magnuson, B.A.; Shirtliff, N.; Bird, R.P. Resistance of aberrant crypt foci to apoptosis induced by azoxymethane in rats chronically fed cholic acid. Carcinogenesis 1994, 15, 1459–1462. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Xie, G.; Jia, W. Bile acid–microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 111–128. [Google Scholar] [CrossRef] [Green Version]
- Amaral, J.D.; Viana, R.J.S.; Ramalho, R.M.; Steer, C.J.; Rodrigues, C.M.P. Bile acids: Regulation of apoptosis by ursodeoxycholic acid. J. Lipid Res. 2009, 50, 1721–1734. [Google Scholar] [CrossRef] [Green Version]
- Péan, N.; Doignon, I.; Tordjmann, T. Bile acids and liver carcinogenesis: TGR5 as a novel piece in the puzzle? Clin. Res. Hepatol. Gastroenterol. 2013, 37, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.A.; Lan, T.; Rao, A. Thematic review series: Bile acids. Bile acid transporters. J. Lipid Res. 2009, 50, 2340–2357. [Google Scholar] [CrossRef] [Green Version]
- Wosczyna, M.N.; Biswas, A.A.; Cogswell, C.A.; Goldhamer, D.J. Multipotent progenitors resident in the skeletal muscle interstitium exhibit robust BMP-dependent osteogenic activity and mediate heterotopic ossification. J. Bone Miner. Res. 2012, 27, 1004–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Wei, L.; Fan, F.; Ji, S.; Zhang, S.; Geng, J.; Hong, L.; Fan, X.; Chen, Q.; Tian, J.; et al. Integration of Hippo signalling and the unfolded protein response to restrain liver overgrowth and tumorigenesis. Nat. Commun. 2015, 6, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Chen, Q.; Liu, Q.; Li, Y.; Sun, X.; Hong, L.; Ji, S.; Liu, C.; Geng, J.; Zhang, W.; et al. Hippo signaling suppresses cell ploidy and tumorigenesis through Skp2. Cancer Cell 2017, 31, 669–684.e7. [Google Scholar] [CrossRef] [Green Version]
- Ji, S.; Liu, Q.; Zhang, S.; Chen, Q.; Wang, C.; Zhang, W.; Xiao, C.; Li, Y.; Nian, C.; Li, J.; et al. FGF15 activates hippo signaling to suppress bile acid metabolism and liver tumorigenesis. Dev. Cell 2019, 48, 460–474.e9. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.L.; Shen, Y.C.; Zhu, A.X. Targeting fibroblast growth factor receptor signaling in hepatocellular carcinoma. Oncology 2012, 81, 372–380. [Google Scholar] [CrossRef]
- Feng, S.; Zhou, L.; Nice, E.C.; Huang, C. Fibroblast growth factor receptors: Multifactorialcontributors to tumor initiation and progression. Histol. Histopathol. 2015, 30, 13–31. [Google Scholar] [CrossRef] [PubMed]
- Peláez-García, A.; Barderas, R.; Torres, S.; Hernández-Varas, P.; Teixidó, J.; Bonilla, F.; Herreros, A.G.D.; Casal, J.I. FGFR4 role in epithelial-mesenchymal transition and its therapeutic value in colorectal cancer. PLoS ONE 2013, 8, e63695. [Google Scholar] [CrossRef] [Green Version]
- Bharadwaj, U.; Kasembeli, M.M.; Robinson, P.; Tweardy, D.J. Targeting janus kinases and signal transducer and activator of transcription 3 to treat inflammation, fibrosis, and cancer: Rationale, progress, and caution. Pharmacol. Rev. 2020, 72, 486–526. [Google Scholar] [CrossRef] [Green Version]
- Brenner, C.; Galluzzi, L.; Kepp, O.; Kroemer, G. Decoding cell death signals in liver inflammation. J. Hepatol. 2013, 59, 583–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serafín, A.; Roselló-Catafau, J.; Prats, N.; Xaus, C.; Gelpí, E.; Peralta, C. Ischemic preconditioning increases the tolerance of fatty liver to hepatic ischemia-reperfusion injury in the rat. Am. J. Pathol. 2002, 161, 587–601. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Zhao, F.; Cao, Y.; Zhang, J.; Shi, P.; Sun, X.; Zhang, F.; Tong, L. DHA attenuates hepatic ischemia reperfusion injury by inhibiting pyroptosis and activating PI3K/Akt pathway. Eur. J. Pharmacol. 2018, 835, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Higashitsuji, H.; Arii, S.; Furutani, M.; Mise, M.; Monden, K.; Fujita, S.-I.; Ishiguro, S.; Kitao, T.; Nakamura, T.; Nakayama, H.; et al. Expression of cytokine genes during liver regeneration after partial hepatectomy in rats. J. Surg. Res. 1995, 58, 267–274. [Google Scholar] [CrossRef]
- Gracia-Sancho, J.; Casillas-Ramírez, A.; Peralta, C. Molecular pathways in protecting the liver from ischaemia/reperfusion injury: A 2015 update. Clin. Sci. 2015, 129, 345–362. [Google Scholar] [CrossRef] [PubMed]
- Krause, P.; Flikweert, H.; Monin, M.; Hosseini, A.S.A.; Helms, G.; Cantanhede, G.; Ghadimi, B.M.; Koenig, S. Increased growth of colorectal liver metastasis following partial hepatectomy. Clin. Exp. Metastasis 2013, 30, 681–693. [Google Scholar] [CrossRef] [Green Version]
- Miura, S.; Mitsuhashi, N.; Shimizu, H.; Kimura, F.; Yoshidome, H.; Otsuka, M.; Kato, A.; Shida, T.; Okamura, D.; Miyazaki, M. Fibroblast growth factor 19 expression correlates with tumor progression and poorer prognosis of hepatocellular carcinoma. BMC Cancer 2012, 12, 56. [Google Scholar] [CrossRef] [Green Version]
- Desnoyers, L.R.; Pai, R.; Ferrando, R.E.; Hötzel, K.; Le, T.; Ross, J.; Carano, R.; D’Souza, A.; Qing, J.; Mohtashemi, I.; et al. Targeting FGF19 inhibits tumor growth in colon cancer xenograft and FGF19 transgenic hepatocellular carcinoma models. Oncogene 2008, 27, 85–97. [Google Scholar] [CrossRef] [Green Version]
- Pai, R.; Dunlap, D.; Qing, J.; Mohtashemi, I.; Hotzel, K.; French, D.M. Inhibition of fibroblast growth factor 19 reduces tumor growth by modulating β-catenin signaling. Cancer Res. 2008, 68, 5086–5095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, X.; Liu, Z.; Niu, B.; Zhang, J.; Tan, T.K.; Lee, S.R.; Zhao, Y.; Harris, D.C.H.; Zheng, G. E-Cadherin/β-catenin complex and the epithelial barrier. J. Biomed. Biotechnol. 2011, 2011. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Li, J.; Xie, K.; Zhang, T.; Lei, Y.; Chen, Y.; Zhang, L.; Huang, K.; Wang, K.; Wu, H.; et al. FGFR4 promotes stroma-induced epithelial-to-mesenchymal transition in colorectal cancer. Cancer Res. 2013, 73, 5926–5935. [Google Scholar] [CrossRef] [Green Version]
- Heinzle, C.; Erdem, Z.; Paur, J.; Grasl-Kraupp, B.; Holzmann, K.; Grusch, M.; Berger, W.; Marian, B. Is fibroblast growth factor receptor 4 a suitable target of cancer therapy? Curr. Pharm. Des. 2014, 20, 2881–2898. [Google Scholar] [CrossRef] [Green Version]
- Kanda, S.; Tomasini-Johansson, B.; Klint, P.; Dixelius, J.; Rubin, K.; Claesson-Welsh, L. Signaling via fibroblast growth factor receptor-1 is dependent on extracellular matrix in capillary endothelial cell differentiation. Exp. Cell Res. 1999, 248, 203–213. [Google Scholar] [CrossRef]
- Roidl, A.; Berger, H.J.; Kumar, S.; Bange, J.; Knyazev, P.; Ullrich, A. Resistance to chemotherapy is associated with fibroblast growth factor receptor 4 up-regulation. Clin. Cancer Res. 2009, 15, 2058–2066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loguercio, C.; Simone, T.D.; D’Auria, M.V.; Sio, I.D.; Federico, A.; Tuccillo, C.; Abbatecola, A.M.; Blanco, C.D.V. Non-alcoholic fatty liver disease: A multicentre clinical study by the Italian Association for the Study of the Liver. Dig. Liver Dis. 2004, 36, 398–405. [Google Scholar] [CrossRef]
- Rinella, M.E.; Alonso, E.; Rao, S.; Whitington, P.; Fryer, J.; Abecassis, M.; Superina, R.; Flamm, S.L.; Blei, A.T. Body mass index as as a predictor of hepatic steatosis in living liver donors. Liver Transplant. 2001, 7, 409–414. [Google Scholar] [CrossRef]
- Poynard, T.; Ratziu, V.; Naveau, S.; Thabut, D.; Charlotte, F.; Messous, D.; Capron, D.; Abella, A.; Massard, J.; Ngo, Y.; et al. The diagnostic value of biomarkers (SteatoTest) for the prediction of liver steatosis. Comp. Hepatol. 2005, 4, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Hadizadeh, F.; Faghihimani, E.; Adibi, P. Nonalcoholic fatty liver disease: Diagnostic biomarkers. World J. Gastrointest. Pathophysiol. 2017, 8, 11. [Google Scholar] [CrossRef]
- Pirola, C.J.; Gianotti, T.F.; Castaño, G.O.; Mallardi, P.; Martino, J.S.; Ledesma, M.M.G.L.; Flichman, D.; Mirshahi, F.; Sanyal, A.J.; Sookoian, S. Circulating microRNA signature in non-alcoholic fatty liver disease: From serum non-coding RNAs to liver histology and disease pathogenesis. Gut 2015, 64, 800–812. [Google Scholar] [CrossRef] [Green Version]
- Kim, R.D.; Sarker, D.; Meyer, T.; Yau, T.; Macarulla, T.; Park, J.W.; Choo, S.P.; Hollebecque, A.; Sung, M.W.; Lim, H.Y.; et al. First-in-human phase i study of fisogatinib (BLU-554) validates aberrant FGF19 signaling as a driver event in hepatocellular carcinoma. Cancer Discov. 2019, 9, 1696–1707. [Google Scholar] [CrossRef] [Green Version]
- Ho, H.K.; Pok, S.; Streit, S.; Ruhe, J.E.; Hart, S.; Lim, K.S.; Loo, H.L.; Aung, M.O.; Lim, S.G.; Ullrich, A. Fibroblast growth factor receptor 4 regulates proliferation, anti-apoptosis and alpha-fetoprotein secretion during hepatocellular carcinoma progression and represents a potential target for therapeutic intervention. J. Hepatol. 2009, 50, 118–127. [Google Scholar] [CrossRef]
- Nicholes, K.; Guillet, S.; Tomlinson, E.; Hillan, K.; Wright, B.; Frantz, G.D.; Pham, T.A.; Dillard-Telm, L.; Tsai, S.P.; Stephan, J.P.; et al. A mouse model of hepatocellular carcinoma: Ectopic expression of fibroblast growth factor 19 in skeletal muscle of transgenic mice. Am. J. Pathol. 2002, 160, 2295–2307. [Google Scholar] [CrossRef]
- George, J.; Rao, K.R.; Stern, R.; Chandrakasan, G. Dimethylnitrosamine-induced liver injury in rats: The early deposition of collagen. Toxicology 2001, 156, 129–138. [Google Scholar] [CrossRef]
- Dong, S.; Chen, Q.L.; Song, Y.N.; Sun, Y.; Wei, B.; Li, X.Y.; Hu, Y.Y.; Liu, P.; Su, S.B. Mechanisms of CCl4-induced liver fibrosis with combined transcriptomic and proteomic analysis. J. Toxicol. Sci. 2016, 41, 561–572. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.X.; Hu, Y.; French, S.W.; Gonzalez, F.J.; Wan, Y.J.Y. Forced expression of fibroblast growth factor 21 reverses the sustained impairment of liver regeneration in PPARαPAC mice due to dysregulated bile acid synthesis. Oncotarget 2015, 6, 9686–9700. [Google Scholar] [CrossRef] [Green Version]
- Guagnano, V.; Kauffmann, A.; Wöhrle, S.; Stamm, C.; Ito, M.; Barys, L.; Pornon, A.; Yao, Y.; Li, F.; Zhang, Y.; et al. FGFR genetic alterations predict for sensitivity to NVP-BGJ398, a selective Pan-FGFR inhibitor. Cancer Discov. 2012, 2, 1118–1133. [Google Scholar] [CrossRef] [Green Version]
- French, D.M.; Lin, B.C.; Wang, M.; Adams, C.; Shek, T.; Hötzel, K.; Bolon, B.; Ferrando, R.; Blackmore, C.; Schroeder, K.; et al. Targeting FGFR4 inhibits hepatocellular carcinoma in preclinical mouse models. PLoS ONE 2012, 7, e0036713. [Google Scholar] [CrossRef]
- Luo, Y.; Yang, C.; Ye, M.; Jin, C.; Abbruzzese, J.L.; Lee, M.-H.; Yeung, S.-C.J.; McKeehan, W.L. Deficiency of metabolic regulator FGFR4 delays breast cancer progression through systemic and microenvironmental metabolic alterations. Cancer Metab. 2013, 1, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Uriarte, I.; Latasa, M.U.; Carotti, S.; Fernandez-Barrena, M.G.; Garcia-Irigoyen, O.; Elizalde, M.; Urtasun, R.; Vespasiani-Gentilucci, U.; Morini, S.; Mingo, A.D.; et al. Ileal FGF15 contributes to fibrosis-associated hepatocellular carcinoma development. Int. J. Cancer 2015, 136, 2469–2475. [Google Scholar] [CrossRef] [Green Version]
- Heinzle, C.; Gsur, A.; Hunjadi, M.; Erdem, Z.; Gauglhofer, C.; Staẗtner, S.; Karner, J.; Klimpfinger, M.; Wrba, F.; Reti, A.; et al. Differential effects of polymorphic alleles of FGF receptor 4 on colon cancer growth and metastasis. Cancer Res. 2012, 72, 5767–5777. [Google Scholar] [CrossRef] [Green Version]
- Sugiyama, N.; Varjosalo, M.; Meller, P.; Lohi, J.; Chan, K.M.; Zhou, Z.; Alitalo, K.; Taipale, J.; Keski-Oja, J.; Lehti, K. FGF receptor-4 (FGFR4) polymorphism acts as an activity switch of a membrane type 1 matrix metalloproteinase—FGFR4 complex. Proc. Natl. Acad. Sci. USA 2010, 107, 15786–15791. [Google Scholar] [CrossRef] [Green Version]
- Stadler, C.R.; Knyazev, P.; Bange, J.; Ullrich, A. FGFR4 GLY388 isotype suppresses motility of MDA-MB-231 breast cancer cells by EDG-2 gene repression. Cell. Signal. 2006, 18, 783–794. [Google Scholar] [CrossRef]
- Presta, M.; Chiodelli, P.; Giacomini, A.; Rusnati, M.; Ronca, R. Fibroblast growth factors (FGFs) in cancer: FGF traps as a new therapeutic approach. Pharmacol. Ther. 2017, 179, 171–187. [Google Scholar] [CrossRef]
- Singh, D.; Chan, J.M.; Zoppoli, P.; Niola, F.; Sullivan, R.; Castano, A.; Liu, E.M.; Reichel, J.; Porrati, P.; Pellegatta, S.; et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 2012, 337, 1231–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polyak, K.; Haviv, I.; Campbell, I.G. Co-evolution of tumor cells and their microenvironment. Trends Genet. 2009, 25, 30–38. [Google Scholar] [CrossRef]
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Edkins, S.; et al. Europe PMC Funders Group Patterns of somatic mutation in human cancer genomes. Nature 2009, 446, 153–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greulich, H.; Pollock, P.M. Targeting mutant fibroblast growth factor receptors in cancer. Trends Mol. Med. 2011, 17, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauglhofer, C.; Paur, J.; Schrottmaier, W.C.; Wingelhofer, B.; Huber, D.; Naegelen, I.; Pirker, C.; Mohr, T.; Heinzle, C.; Holzmann, K.; et al. Fibroblast growth factor receptor 4: A putative key driver for the aggressive phenotype of hepatocellular carcinoma. Carcinogenesis 2014, 35, 2331–2338. [Google Scholar] [CrossRef] [Green Version]
- Larrieu-Lahargue, F.; Welm, A.L.; Bouchecareilh, M.; Alitalo, K.; Li, D.Y.; Bikfalvi, A.; Auguste, P. Blocking fibroblast growth factor receptor signaling inhibits tumor growth, lymphangiogenesis, and metastasis. PLoS ONE 2012, 7, e39540. [Google Scholar] [CrossRef] [Green Version]
- Taeger, J.; Moser, C.; Hellerbrand, C.; Mycielska, M.E.; Glockzin, G.; Schlitt, H.J.; Geissler, E.K.; Stoeltzing, O.; Lang, S.A. Targeting FGFR/PDGFR/VEGFR impairs tumor growth, angiogenesis, and metastasis by effects on tumor cells, endothelial cells, and pericytes in pancreatic cancer. Mol. Cancer Ther. 2011, 10, 2157–2167. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.P.; Shang, K.; Chen, H.; Ding, F.; Wang, Z.; Liang, C.; Xu, Y.; Sun, M.H.; Li, Y.Y. FGF-1/-3/FGFR4 signaling in cancer-associated fibroblasts promotes tumor progression in colon cancer through Erk and MMP-7. Cancer Sci. 2015, 106, 1278–1287. [Google Scholar] [CrossRef]
- Whittaker, S.; Marais, R.; Zhu, A.X. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene 2010, 29, 4989–5005. [Google Scholar] [CrossRef]
- Hansen, S.M.; Berezin, V.; Bock, E. Signaling mechanisms of neurite outgrowth induced by the cell adhesion molecules NCAM and N-Cadherin. Cell. Mol. Life Sci. 2008, 65, 3809–3821. [Google Scholar] [CrossRef]
- Cavallaro, U.; Niedermeyer, J.; Fuxa, M.; Christofori, G. N-CAM modulates tumour-cell adhesion to matrix by inducing FGF-receptor signalling. Nat. Cell Biol. 2001, 3, 650–657. [Google Scholar] [CrossRef]
- Padrissa-Altés, S.; Bachofner, M.; Bogorad, R.L.; Pohlmeier, L.; Rossolini, T.; Böhm, F.; Liebisch, G.; Hellerbrand, C.; Koteliansky, V.; Speicher, T.; et al. Control of hepatocyte proliferation and survival by Fgf receptors is essential for liver regeneration in mice. Gut 2015, 64, 1444–1453. [Google Scholar] [CrossRef]
- Hansen, A.M.K.; Vienberg, S.G.; Lykkegaard, K.; Zhao, X.; Tingqing, G.; Han, D.; Zhang, X.; Thøgersen, H.; Sass-Ørum, K.; Tagmose, T.; et al. Differential receptor selectivity of the FGF15/FGF19 orthologues determines distinct metabolic activities in db/db mice. Biochem. J. 2018, 475, 2985–2996. [Google Scholar] [CrossRef]
- Kong, B.; Sun, R.; Huang, M.; Chow, M.D.; Zhong, X.; Xie, W.; Lee, Y.; Guo, G.L. Fibroblast growth factor 15-dependent and bile acid-independent promotion of liver regeneration in mice. Hepatology 2018, 68, 1961–1976. [Google Scholar] [CrossRef] [Green Version]
- Baier, P.; Wolf-Vorbeck, G.; Hempel, S.; Hopt, U.T.; Von Dobschuetz, E. Effect of liver regeneration after partial hepatectomy and ischemia-reperfusion on expression of growth factor receptors. World J. Gastroenterol. 2006, 12, 3835–3840. [Google Scholar] [CrossRef]
- Kong, B.; Huang, J.; Zhu, Y.; Li, G.; Williams, J.; Shen, S.; Aleksunes, L.M.; Richardson, J.R.; Apte, U.; Rudnick, D.A.; et al. Fibroblast growth factor 15 deficiency impairs liver regeneration in mice. Am. J. Physiol. Liver Physiol. 2014, 306, G893–G902. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wang, Y.-D.; Chen, W.; Wang, X.; Lou, G.; Liu, N.; Lin, M.; Forman, B.M.; Huang, W. Promotion of liver regeneration/repair by farnesoid X receptor in both liver and intestine in mice. Hepatology 2012, 56, 2336–2343. [Google Scholar] [CrossRef] [Green Version]
- Naugler, W.E. Bile acid flux is necessary for normal liver regeneration. PLoS ONE 2014, 9, e0097426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Yang, C.; Chang, J.Y.F.; You, P.; Li, Y.; Jin, C.; Luo, Y.; Li, X.; McKeehan, W.L.; Wang, F. Hepatocyte FRS2α is essential for the endocrine fibroblast growth factor to limit the amplitude of bile acid production induced by prandial activity. Curr. Mol. Med. 2014, 14, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Uriarte, I.; Fernandez-Barrena, M.G.; Monte, M.J.; Latasa, M.U.; Chang, H.C.Y.; Carotti, S.; Vespasiani-Gentilucci, U.; Morini, S.; Vicente, E.; Concepcion, A.R.; et al. Identification of fibroblast growth factor 15 as a novel mediator of liver regeneration and its application in the prevention of post-resection liver failure in mice. Gut 2013, 62, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Fausto, N.; Campbell, J.S.; Riehle, K.J. Liver regeneration. Hepatology 2006, 43, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Riehle, K.J.; Dan, Y.Y.; Campbell, J.S.; Fausto, N. New concepts in liver regeneration. J. Gastroenterol. Hepatol. 2011, 26, 203–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalopoulos, G.K. Liver regeneration. J. Cell. Physiol. 2007, 213, 286–300. [Google Scholar] [CrossRef]
- Böhm, F.; Köhler, U.A.; Speicher, T.; Werner, S. Regulation of liver regeneration by growth factors and cytokines. EMBO Mol. Med. 2010, 2, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Gingras, A.C.; Raught, B.; Sonenberg, N. eIF4 initiation factors: Effectors of mRNA recruitment to ribosomes and regulators of translation. Annu. Rev. Biochem. 1999, 68, 913–963. [Google Scholar] [CrossRef] [PubMed]
- Cicione, C.; Degirolamo, C.; Moschetta, A. Emerging role of fibroblast growth factors 15/19 and 21 as metabolic integrators in the liver. Hepatology 2012, 56, 2404–2411. [Google Scholar] [CrossRef]
- Mao, S.A.; Glorioso, J.M.; Nyberg, S.L. Liver regeneration. Transl. Res. 2014, 163, 352–362. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.D.; Wang, Y.D.; Meng, Z.; Zhang, L.; Huang, W. Nuclear bile acid receptor FXR in the hepatic regeneration. Biochim. Biophys. Acta Mol. Basis Dis. 2011, 1812, 888–892. [Google Scholar] [CrossRef] [Green Version]
- Chiang, J.Y.L. Bile acid regulation of gene expression: Roles of nuclear hormone receptors. Endocr. Rev. 2002, 23, 443–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Huang, X.; Meng, Z.; Dong, B.; Shiah, S.; Moore, D.D.; Huang, W. Significance and mechanism of CYP7a1 gene regulation during the acute phase of liver regeneration. Mol. Endocrinol. 2009, 23, 137–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, M.; Wang, X.; Xu, G.; Yan, Q.; Huang, W. Bile acid signaling and liver regeneration. Biochim. Biophys. Acta Gene Regul. Mech. 2015, 1849, 196–200. [Google Scholar] [CrossRef] [Green Version]
- Kir, S.; Zhang, Y.; Gerard, R.D.; Kliewer, S.A.; Mangelsdorf, D.J. Nuclear receptors HNF4α and LRH-1 cooperate in regulating Cyp7a1 in vivo. J. Biol. Chem. 2012, 287, 41334–41341. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Ma, K.; Zhang, J.; Qatanani, M.; Cuvillier, J.; Liu, J.; Dong, B.; Huang, X.; Moore, D.D. Nuclear receptor-dependent bile acid signaling is required for normal liver regeneration. Science 2006, 312, 233–236. [Google Scholar] [CrossRef]
- Kim, I.; Ahn, S.H.; Inagaki, T.; Choi, M.; Ito, S.; Guo, G.L.; Kliewer, S.A.; Gonzalez, F.J. Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. J. Lipid Res. 2007, 48, 2664–2672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borude, P.; Edwards, G.; Walesky, C.; Li, F.; Ma, X.; Kong, B.; Guo, G.L.; Apte, U. Hepatocyte-specific deletion of farnesoid X receptor delays but does not inhibit liver regeneration after partial hepatectomy in mice. Hepatology 2012, 56, 2344–2352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gumbiner, B.M.; Kim, N.G. The Hippo-YAP signaling pathway and contact inhibition of growth. J. Cell Sci. 2014, 127, 709–717. [Google Scholar] [CrossRef] [Green Version]
- Xin, M.; Kim, Y.; Sutherland, L.B.; Murakami, M.; Qi, X.; McAnally, J.; Porrello, E.R.; Mahmoud, A.I.; Tan, W.; Shelton, J.M.; et al. Hippo pathway effector Yap promotes cardiac regeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 13839–13844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, D. Hippo signaling in organ size control. Genes Dev. 2007, 21, 886–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koziczak, M.; Hynes, N.E. Cooperation between fibroblast growth factor receptor-4 and ErbB2 in regulation of cyclin D1 translation. J. Biol. Chem. 2004, 279, 50004–50011. [Google Scholar] [CrossRef] [Green Version]
- Crose, L.E.S.; Etheridge, K.T.; Chen, C.; Belyea, B.; Talbot, L.J.; Bentley, R.C.; Linardic, C.M. FGFR4 blockade exerts distinct antitumorigenic effects in human embryonal versus alveolar rhabdomyosarcoma. Clin. Cancer Res. 2012, 18, 3780–3790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, S.A.; Neff, G.; Guy, C.D.; Bashir, M.R.; Paredes, A.H.; Frias, J.P.; Younes, Z.; Trotter, J.F.; Gunn, N.T.; Moussa, S.E.; et al. Efficacy and safety of aldafermin, an engineered FGF19 analog, in a randomized, double-blind, placebo-controlled trial of patients with nonalcoholic steatohepatitis. Gastroenterology 2021, 160, 219–231.e1. [Google Scholar] [CrossRef]
- Hirschfield, G.M.; Chazouillères, O.; Drenth, J.P.; Thorburn, D.; Harrison, S.A.; Landis, C.S.; Mayo, M.J.; Muir, A.J.; Trotter, J.F.; Leeming, D.J.; et al. Effect of NGM282, an FGF19 analogue, in primary sclerosing cholangitis: A multicenter, randomized, double-blind, placebo-controlled phase II trial. J. Hepatol. 2019, 70, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, P.; Hochrath, K.; Horvath, A.; Chen, P.; Seebauer, C.T.; Llorente, C.; Wang, L.; Alnouti, Y.; Fouts, D.E.; Stärkel, P.; et al. Modulation of the intestinal bile acid/farnesoid X receptor/fibroblast growth factor 15 axis improves alcoholic liver disease in mice. Hepatology 2018, 67, 2150–2166. [Google Scholar] [CrossRef]
- Kim, Y.C.; Seok, S.; Zhang, Y.; Ma, J.; Kong, B.; Guo, G.; Kemper, B.; Kemper, J.K. Intestinal FGF15/19 physiologically repress hepatic lipogenesis in the late fed-state by activating SHP and DNMT3A. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Sola, G.; Uriarte, I.; Latasa, M.U.; Fernandez-Barrena, M.G.; Urtasun, R.; Elizalde, M.; Barcena-Varela, M.; Jiménez, M.; Chang, H.C.; Barbero, R.; et al. Fibroblast growth factor 15/19 (FGF15/19) protects from diet-induced hepatic steatosis: Development of an FGF19-based chimeric molecule to promote fatty liver regeneration. Gut 2017, 66, 1818–1828. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Learned, R.M.; Rossi, S.J.; DePaoli, A.M.; Tian, H.; Ling, L. Engineered FGF19 eliminates bile acid toxicity and lipotoxicity leading to resolution of steatohepatitis and fibrosis in mice. Hepatol. Commun. 2017, 1, 1024–1042. [Google Scholar] [CrossRef]
- Hu, Z.; Han, Y.; Liu, Y.; Zhao, Z.; Ma, F.; Cui, A.; Zhang, F.; Liu, Z.; Xue, Y.; Bai, J.; et al. CREBZF as a key regulator of STAT3 pathway in the control of liver regeneration in mice. Hepatology 2020, 71, 1421–1436. [Google Scholar] [CrossRef] [PubMed]
- Debonera, F.; Aldeguer, X.; Shen, X.; Gelman, A.E.; Gao, F.; Que, X.; Greenbaum, L.E.; Furth, E.E.; Taub, R.; Olthoff, K.M. Activation of interleukin-6/STAT3 and liver regeneration following transplantation. J. Surg. Res. 2001, 96, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Abe, M.; Yoshida, T.; Akiba, J.; Ikezono, Y.; Wada, F.; Masuda, A.; Sakaue, T.; Tanaka, T.; Iwamoto, H.; Nakamura, T.; et al. STAT3 defciency prevents hepatocarcinogenesis and promotes biliary proliferation in thioacetamide-induced liver injury. World J. Gastroenterol. 2017, 23, 6833–6844. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B.; Worden, F.; Kudo, M. Sorafenib: Key lessons from over 10 years of experience. Expert Rev. Anticancer Ther. 2019, 19, 177–189. [Google Scholar] [CrossRef]
- Garten, A.; Grohmann, T.; Kluckova, K.; Lavery, G.G.; Kiess, W.; Penke, M. Sorafenib-induced apoptosis in hepatocellular carcinoma is reversed by SIRT1. Int. J. Mol. Sci. 2019, 20, 4048. [Google Scholar] [CrossRef] [Green Version]
- Gordan, J.D.; Kennedy, E.B.; Abou-Alfa, G.K.; Beg, M.S.; Brower, S.T.; Gade, T.P.; Goff, L.; Gupta, S.; Guy, J.; Harris, W.P.; et al. Systemic therapy for advanced hepatocellula carcinoma: ASCO guideline. J. Clin. Oncol. 2020, 38, 4317–4345. [Google Scholar] [CrossRef] [PubMed]
- Al-Salama, Z.T.; Syed, Y.Y.; Scott, L.J. Lenvatinib: A review in hepatocellular carcinoma. Drugs 2019, 79, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Johnson, P.J.; Qin, S.; Park, J.W.; Poon, R.T.P.; Raoul, J.L.; Philip, P.A.; Hsu, C.H.; Hu, T.H.; Heo, J.; Xu, J.; et al. Brivanib versus sorafenib as first-line therapy in patients with unresectable, advanced hepatocellular carcinoma: Results from the randomized phase III BRISK-FL study. J. Clin. Oncol. 2013, 31, 3517–3524. [Google Scholar] [CrossRef] [Green Version]
- Foà, R.; Bassan, R.; Vitale, A.; Elia, L.; Piciocchi, A.; Puzzolo, M.-C.; Canichella, M.; Viero, P.; Ferrara, F.; Lunghi, M.; et al. Dasatinib—Blinatumomab for Ph-positive acute lymphoblastic leukemia in adults. N. Engl. J. Med. 2020, 383, 1613–1623. [Google Scholar] [CrossRef]
- Motzer, R.J.; Hutson, T.E.; Glen, H.; Michaelson, M.D.; Molina, A.; Eisen, T.; Jassem, J.; Zolnierek, J.; Maroto, J.P.; Mellado, B.; et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: A randomised, phase 2, open-label, multicentre trial. Lancet Oncol. 2015, 16, 1473–1482. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.C.; Fazio, N.; Singh, S.; Buzzoni, R.; Carnaghi, C.; Wolin, E.; Tomasek, J.; Raderer, M.; Lahner, H.; Voi, M.; et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): A randomised, placebo-controlled, phase 3 study. Lancet 2016, 387, 968–977. [Google Scholar] [CrossRef]
- Cholongitas, E.; Goulis, I.; Theocharidou, E.; Antoniadis, N.; Fouzas, I.; Imvrios, G.; Giouleme, O.; Angelaki, A.; Vasiliadis, T.; Papanikolaou, V.; et al. Everolimus with or without mycophenolate mofetil in a liver transplantation setting: A single-center experience. Ann. Gastroenterol. 2018, 31, 613–620. [Google Scholar] [CrossRef]
- Huynh, H.; Ngo, V.C.; Fargnoli, J.; Ayers, M.; Khee, C.S.; Heng, N.K.; Choon, H.T.; Hock, S.O.; Chung, A.; Chow, P.; et al. Brivanib alaninate, a dual inhibitor of vascular endothelial growth factor receptor and fibroblast growth factor receptor tyrosine kinases, induces growth inhibition in mouse models of human hepatocellular carcinoma. Clin. Cancer Res. 2008, 14, 6146–6153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, J.Y.L. Bile acids: Regulation of synthesis. J. Lipid Res. 2009, 50, 1955–1966. [Google Scholar] [CrossRef] [Green Version]
- Kuipers, F.; Havinga, R.; Huijsmans, C.M.; Vonk, R.J.; Princen, H.M. Inhibition and induction of bile acid synthesis by ketoconazole. Effects on bile formation in the rat. Lipids 1989, 24, 759–764. [Google Scholar] [CrossRef]
- Princen, H.M.G.; Huijsmans, C.M.G.; Kuipers, F.; Vonk, R.J.; Kempen, H.J. Ketoconazole blocks bile acid synthesis in hepatocyte monolayer cultures and in vivo in rat by inhibiting cholesterol 7α-hydroxylase. J. Clin. Investig. 1986, 78, 1064–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gbaguidi, G.F.; Agellon, L.B. The inhibition of the human cholesterol 7α-hydroxylase gene (CYP7A1) promoter by fibrates in cultured cells is mediated via the liver x receptor α and peroxisome proliferator-activated receptor α heterodimer. Nucleic Acids Res. 2004, 32, 1113–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baghdasaryan, A.; Fuchs, C.D.; Österreicher, C.H.; Lemberger, U.J.; Halilbasic, E.; Påhlman, I.; Graffner, H.; Krones, E.; Fickert, P.; Wahlström, A.; et al. Inhibition of intestinal bile acid absorption improves cholestatic liver and bile duct injury in a mouse model of sclerosing cholangitis. J. Hepatol. 2016, 64, 674–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clayton, P.T. Disorders of bile acid synthesis. J. Inherit. Metab. Dis. 2011, 34, 593–604. [Google Scholar] [CrossRef]
- Jia, W.; Wei, M.; Rajani, C.; Zheng, X. Targeting the alternative bile acid synthetic pathway for metabolic diseases. Protein Cell 2020, 12, 411–425. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Type of Model | Type of Sample | Treatments Applied Based on FGF15/19-FGR4 Axis Modulation | Cancer Induction | Purpose to Evaluate | Reference |
|---|---|---|---|---|---|
| Human BD donors | Human biopsy | Any treatment | No | Cell types expressing FGF15 and FGFR4 | [20] |
| Mouse immortalized hepatocytes (AML12), Hepa1-6 hepatoma cells, and C2C12 myoblasts | Cell culture | Administration of lipid nanoparticles carrying FGFR4 siRNA for gen expression blockage | No | FGFR function in liver regeneration | [101] |
| JHH4, HEP3B, JHH7, HUH7, PLC/PRF/5, and JHH5 HCC cells | Human cell culture | Incubation with anti-FGFR4 monoclonal antibody (LD1) | Yes | FGFRs expression in liver cancer, FGFR4 participation in colony formation, and evaluation of therapeutic potential of LD1 to inhibit FGFR4 function in cancer | [83] |
| BaF3 pro-B cells IL-3-dependent | Human cell culture | FGFR4 chimeric construct transfection and incubation in the absence of IL-3 | No | FGFR4 pro-mitogenic capabilities | [83] |
| Primary rat hepatocytes | Cell culture | Incubation with human FGF21 and FGF19 and mouse FGF15 | No | To determine FGF19 and FGF15 functions | [102] |
| Normal and FGFR4−/− mouse liver tissue, DEN-initiated hepatomas and derived hepatoma cells | Cell culture from hepatoma samples | Transfection with full-length murine βKL and incubation with FGF19 or FGF1 | Yes | βKL role in FGF19 or FGF1-mediated FGFR4 function | [31] |
| Hepatocytes isolated from 70% PH non-steatotic mice without ischemic period | Cell culture | Incubation whit siRNA of FGFR4 and recombinant human FGF19 | No | FGF19 role in LR after PH | [22] |
| LT of steatotic and non-steatotic BD donor rat liver grafts | SD and ZKob/ob male rats | Administration of FGF15 alone or combined with BA or YAP inhibitor | No | Effects and signaling pathways implication of FGF15 | [20] |
| LT of steatotic and non-steatotic CDD rat liver grafts | ZKob/ob and ZKob/− male rats | FGFR4 inhibitor in donors | No | Role of FGF15 and signaling pathways | [21] |
| Tissue-specific inducible FGF15 Tg mice undergoing 70% PH without ischemia | C57BL/6J mice | Administration of Dox to fgf15 transgene inhibition | No | Actions of FGF15 on LR | [103] |
| Whole-body Fgf15 KO mice undergoing 70% PH without ischemia | C57BL/6J mice | AAV-FGF15 overexpressing FGF15 | No | Effects of FGF15 on LR | [103] |
| Rats subjected to 80% PH with IR | SD male rats | Any treatment | No | Effects of warm IR ** caused by LT on LR | [104] |
| FGF15 KO mice undergoing 70% PH without ischemia | 75% C57BL/6J and 25% 129SvJ mice * | Any treatment | No | FGF15 actions on LR | [105] |
| FGF15−/− mice subjected to HCC induction by DEN + CCl4 administration | C57BL/6129/Sv mice * | Any treatment | Yes | FGF15 role in HCC | [85] |
| Whole-body FXR KO, intestine-specific and liver-specific FXR null in mice undergoing 70% PH or CCl4 liver injury induction | - | Any treatment | No | Restorative mechanisms of FGF15 in LR | [106] |
| Hepatocyte-specific FGFR1 and FGFR2 KO mice undergoing 70% PH | - | FGFR4 siRNA | No | Impact of FGFR4 loss in LR | [101] |
| Wild-type mice | FVB strain female mice | ID1 antibody and FGF19 administration | Yes | Effects of FGFR4 inhibition on HCC | [83] |
| Xenograft mice | nu/nu female mice | 5 × 106 mice cancer cells inoculation and LD1 administration | Yes | Effects of FGFR4 inhibition on tumor growth | [83] |
| FGF19 Tg and simultaneously FGFR4 KO mice | Progeny of a breed FGF19 Tg with FGFR4 KO mice | FGF19 Tg mice | Yes | FGFR4 role in HCC development | [83] |
| Wild-type mice undergoing 70% PH | C57BL/6 male mice | Any treatment | No | BA flux | [107] |
| Rats with a biliary fistula with or without chemical compound administration (CCl4 and meloxicam) administration | SD male rats | Any treatment | No | Implication of enterohepatic circulation of BA in the outcome of LR | [107] |
| db/db mice | C57BL6/ JbomTac-KS male mice | Any treatment recombinant human FGF21, human FGF19 or mouse FGF15 | No | Actions of both mouse FGF15 and human FGF19 | [102] |
| C57BL/6 Tg mice | C57BL/6 hepatocyte-specific Fgfr4 KO and Frs2α floxed male mice | Conditional Frs2α ablation | No | Mechanisms by which FGFR4 regulates BAs | [108] |
| FGF15 KO mice undergoing 70% or 85% PH | C57BL/6/129/Sv * mice | AAV-Fgf15 injection and 2% cholestyramine resin dietary administration | No | FGF15 role in BA homeostasis | [109] |
| Wild-type mice undergoing 70% PH | C57BL/6 male mice | Any treatment | No | FGF15-FGFR4 axis role in LR | [22] |
| Drug | Action Mechanism | Disease |
|---|---|---|
| Aldafermin (NGM282) [130,131] | FGF15 variant. Activation of the FGFR1c-KLB receptor | Cholestatic liver disease and NASH (clinical trials) |
| Fibapo [134] | Interaction with scavenger receptor class B type I (SR-BI) | Fatty liver regeneration (Preclinical model FGF15−/− mice) |
| M70 [135] | Repression of Cyp7a1 expression but not STAT3 activation | Steatohepatitis and fibrosis |
| Brivanib [144] | Tyrosine kinase inhibitor and FGFRs inhibitor | HCC |
| Dasatinib [145] | Tyrosine kinase inhibitor | Some kind of leukemias |
| Sorafenib [139,140] | Multi-kinase inhibitor | HCC and other cancers |
| Everolimus [146,147,148] | mTOR inhibitor | Immunosuppressive drug or treatment of some cancers |
| Lenvatinib [141,142] | Inhibitor of multiple receptor tyrosine kinases | Unresectable HCC and some other cancers |
| Regorafenib [141,143] | Multi-kinase inhibitor that targets angiogenic, stromal (FGFR), and oncogenic receptor tyrosine kinases | Advanced HCC in patients previously treated with Sorafenib and metastatic colorectal cancer |
| Cabozantinib [141] | Multi-receptor tyrosine kinase (RTK) inhibitor | HCC resistant to sorafenib |
| LD-1 [83] | anti-FGFR4 monoclonal antibody | Preclinical model of liver cancer (mice) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caballeria-Casals, A.; Micó-Carnero, M.; Rojano-Alfonso, C.; Maroto-Serrat, C.; Casillas-Ramírez, A.; Álvarez-Mercado, A.I.; Gracia-Sancho, J.; Peralta, C. Role of FGF15 in Hepatic Surgery in the Presence of Tumorigenesis: Dr. Jekyll or Mr. Hyde? Cells 2021, 10, 1421. https://doi.org/10.3390/cells10061421
Caballeria-Casals A, Micó-Carnero M, Rojano-Alfonso C, Maroto-Serrat C, Casillas-Ramírez A, Álvarez-Mercado AI, Gracia-Sancho J, Peralta C. Role of FGF15 in Hepatic Surgery in the Presence of Tumorigenesis: Dr. Jekyll or Mr. Hyde? Cells. 2021; 10(6):1421. https://doi.org/10.3390/cells10061421
Chicago/Turabian StyleCaballeria-Casals, Albert, Marc Micó-Carnero, Carlos Rojano-Alfonso, Cristina Maroto-Serrat, Araní Casillas-Ramírez, Ana I. Álvarez-Mercado, Jordi Gracia-Sancho, and Carmen Peralta. 2021. "Role of FGF15 in Hepatic Surgery in the Presence of Tumorigenesis: Dr. Jekyll or Mr. Hyde?" Cells 10, no. 6: 1421. https://doi.org/10.3390/cells10061421