
Cardiovascular Inflammaging: Mechanisms and Translational Aspects
by
 , , ,
, , ,
Maria Luisa Barcena
1,2,* ,
,
Muhammad Aslam
3,4,5 ,
,
Sofya Pozdniakova
1,6,
Kristina Norman
1,7,8 and
Yury Ladilov
1,9 1
Department of Geriatrics and Medical Gerontology, Charité—Universitätsmedizin Berlin, Hindenburgdamm 30, 12203 Berlin, Germany
2
DZHK (German Centre for Cardiovascular Research), Partner Site Berlin, 10785 Berlin, Germany
3
Experimental Cardiology, Department of Internal Medicine I, Justus Liebig University, Aulweg 129, 35392 Giessen, Germany
4
Department of Cardiology, Kerckhoff Clinic GmbH, 61231 Bad Nauheim, Germany
5
DZHK (German Centre for Cardiovascular Research), Partner Site Rhein-Main, 61231 Bad Nauheim, Germany
6
Barcelona Biomedical Research Park (PRBB), Barcelona Institute for Global Health (ISGlobal), Doctor Aiguader, 88, 08003 Barcelona, Spain
7
Department of Nutrition and Gerontology, German Institute of Human Nutrition Potsdam-Rehbrücke, Arthur-Scheunert-Allee 114-116, 14558 Nuthetal, Germany
8
Department of Nutrition & Gerontology, Institute of Nutritional Science, University of Potsdam, Arthur-Scheunert-Allee 114-116, 14558 Nuthetal, Germany
9
Department of Cardiovascular Surgery, Heart Center Brandenburg, Brandenburg Medical School Theodor Fontane, University Hospital, Ladeburger Str. 17, 16321 Bernau, Germany
*
Author to whom correspondence should be addressed.
Cells 2022, 11(6), 1010; https://doi.org/10.3390/cells11061010
Submission received: 2 February 2022
/
Revised: 7 March 2022
/
Accepted: 15 March 2022
/
Published: 16 March 2022
(This article belongs to the Topic Inflammaging: The Immunology of Aging)
Abstract
:Aging is one of the major non-reversible risk factors for several chronic diseases, including cancer, type 2 diabetes, dementia, and cardiovascular diseases (CVD), and it is a key cause of multimorbidity, disability, and frailty (decreased physical activity, fatigue, and weight loss). The underlying cellular mechanisms are complex and consist of multifactorial processes, such as telomere shortening, chronic low-grade inflammation, oxidative stress, mitochondrial dysfunction, accumulation of senescent cells, and reduced autophagy. In this review, we focused on the molecular mechanisms and translational aspects of cardiovascular aging-related inflammation, i.e., inflammaging.
{kind=link}
{kind=link}
1. Inflammaging
Chronic low-grade systemic inflammation is a well-established hallmark of aging [1,2]. It is characterized by high levels of circulating cytokines in the serum of older [3] but apparently healthy individuals [1,4,5], in the absence of general pathophysiological stress or acute infection. Chronic inflammation is involved in accelerated biological aging and age-related diseases, particularly cardiovascular diseases (CVD), type 2 diabetes, or cancer [6,7,8,9]. Accumulating data demonstrate an age-related increase in the levels of blood inflammatory markers such as tumor necrosis factor alpha (TNF-α), interleukin 1 beta (IL-1β), IL-6, and C-reactive protein (CRP). This condition is known as inflammaging [9]. Inflammaging is a sign and a cause of accelerated aging and a comprehensive marker of multimorbidity, disability, frailty, and premature death in old adults [9,10]. Moreover, it is associated with the failure of the immune system to clean pathogens and dysfunctional cells [6]. Inflammaging is driven by a variety of molecular age-related mechanisms, leading to cellular senescence [11], including impaired mitochondrial function, oxidative stress, DNA damage, inflammasome activation, and telomere shortening [11,12,13,14]. However, upregulation or activation of pro-inflammatory mediators plays a central role in inflammaging.
NF-κB is a central mediator of pro-inflammatory gene induction, and the upregulation of the NF-κB pathway has been documented in aging-related inflammatory disorders [15,16,17,18,19]. The well-established driver of the NF-κB-mediated inflammatory reaction is the formation of a Nod-like receptor pyrin domain containing 3 (NLRP3) inflammasome. NLRP3 is the most extensively studied inflammasome and is composed of NLRP3, apoptosis-associated speck-like protein containing a CARD (ASC), and pro-caspase 1. The NLRP3 inflammasome seems to play a pivotal role in several mechanisms related to inflammaging since inhibition of the NLRP3 inflammasome extends healthspan and diminishes age-dependent degenerative changes [20,21]. Increased NLRP3 inflammasome activity is associated with age-related pathological manifestations, including atherosclerosis, type 2 diabetes, and Alzheimer’s disease [20,22,23]. Within several downstream pro-inflammatory pathways activated by the NLRP3 inflammasome, IL-1β and IL-18 have a potential role for inflammaging [24]. Of note, IL-18 expression increases during the aging processes in humans [21,25,26]. In accordance with this, IL-18 is increased in aged healthy hearts in a sex-dependent manner [27].
Altogether, inflammaging is associated with several age-related diseases, including CVD, due to the aberrant release of several pro-inflammatory factors.
2. Cardiac Inflammaging
2.1. Low-Grade Systemic Inflammation in Aging Heart, Sex-Related Differences
Increased levels of circulating pro-inflammatory factors, e.g., CRP and IL-6, are closely associated with cardiovascular pathologies, including myocardial infarction or coronary heart disease [28,29,30,31].
Obesity, hypertension, diabetes, smoking, and atherosclerosis are risk factors for cardiac inflammaging. Recent research has also emphasized an essential role of biological sex in inflammaging-associated CVD. Loss of estrogen in older females promotes the activation of inflammatory pathways in cardiac aging [32] that are accompanied by a decline in antioxidative defense mechanisms and mitochondrial biogenesis and function [33,34]. Furthermore, non-diseased hearts from old women show reduced anti-inflammatory protection that is reflected by high levels of the pro-inflammatory cytokine IL-12 and low levels of the anti-inflammatory cytokine IL-10 (IL-12high/IL-10low) [27]. Interestingly, while cardiac inflammation becomes more prominent in aging female hearts, male hearts seem to be less prone to inflammation due to reduced activation of NF-κB in male hearts during aging [27]. In contrast to non-diseased hearts, NF-κB seems to be directly related to inflammatory processes in male myocardial tissue in age-related diseases such as dilated cardiomyopathy or end-stage myocarditis, since it is strongly upregulated in the diseased hearts solely in males [6,35,36,37].
In addition to the circulating pro-inflammatory cytokines contributing to cardiac aging, there is an increased amount of pro-inflammatory macrophages infiltrated into the myocardium of healthy older women [27], which are the most abundant resident immune cells in the heart [38]. We and others observed an age-related increase in the number of pro-inflammatory M1 macrophages that is accompanied by a decrease in the anti-inflammatory M2 macrophage phenotype in both healthy women and female mice [1,27]. In contrast to the healthy myocardium of old individuals, hearts from male aged patients with end-stage dilated cardiomyopathy and myocarditis show more CD68-positive macrophages ([35], unpublished data).
2.2. Role of AMPK and Sirtuins in Aging Heart
Cardiomyocytes are high energy-consuming cells, and a sufficient supply of ATP is essential for their contractile function. Involved in numerous signaling pathways controlling cellular metabolism, 5’ AMP-activated protein kinase (AMPK) is a key energy sensor and regulator of energy metabolism and mitochondrial homeostasis. ATP depletion activates AMPK, which leads to stimulation of catabolic processes to re-establish the energy balance [39]. In particular, AMPK switches off many energy-consuming processes, such as protein and lipid syntheses, and it activates energy-releasing processes, such as lipo- and glycolysis, autophagy, and fatty acid oxidation [40].
The mammalian heterotrimeric AMPK complex consists of a catalytic (alpha) subunit and regulatory (beta and gamma) subunits. The main, well-characterized mechanisms of AMPK activation are phosphorylation at Thr172 of the alpha subunit and allosteric modification by AMP and/or ADP binding to the gamma subunit. Under energy-depleted conditions, high levels of AMP and ADP bind to the AMPK gamma subunit, which prevents the phosphatases from accessing Thr172 of the AMPK alpha-subunit, thus increasing its phosphorylation. There are three upstream activating kinases that phosphorylate AMPK at Thr172: liver kinase B1 (LKB1), Ca2+/calmodulin-dependent protein kinase kinase 2 (CaMKK2), and TGF-β-activated kinase 1 (TAK1). The phosphorylated Thr172 can be dephosphorylated by the phosphatases protein phosphatase 1 (PP1), PP2A, and PP2C, resulting in AMPK inhibition. Moreover, there are several upstream Ser/Thr kinases, including PKA, AKT, and ERK1/2, that inactivate AMPK (for a review see [41,42]).
Increasing evidence suggests that the basal activity of cardiac AMPK, as well as its stress-related activation, decreases with age, although the total AMPK content remains mainly unchanged [27,42]. Although the precise underlying molecular mechanisms of age-related AMPK inactivation are not completely understood, the role of protein phosphatases, disturbance of the Ca2+ homeostasis, and activity of the inhibitory upstream kinases, e.g., PKA or ERK1/2, has been considered as a potential cause [42].
Another key cellular metabolic regulator is the deacetylase Sirt1, a member of the deacetylase family that utilizes NAD+ as a co-substrate to remove acetyl groups from lysine residues of a target protein. The protein deacetylation promotes enzyme-substrate binding and, therefore, enzyme activity. Notably, Sirt1 promotes AMPK activity via deacetylation and activation of AMPK’s upstream kinase LKB1 [43]. Several studies demonstrated that Sirt1 expression and activity declines with age in various tissues [44,45], which may also contribute to the impairment of AMPK signaling. Interestingly, we recently found that Sirt1 expression is significantly reduced in aged female but not aged male myocardium [27]. Accordingly, the acetylation of nuclear protein Ku70, a marker of the nuclear deacetylase activity, was significantly increased. Similar sex-dependent downregulation of another key cellular sirtuin, mitochondrial Sirt3, was also observed in this study. Sirt3 is a key mitochondrial deacetylase involved in regulating numerous mitochondrial enzymes, including superoxide dismutase 2 and isocitrate dehydrogenase 2 [46,47]. The net contribution of Sirt3 activity comprises the regulation of mitochondrial dynamics [48] and function [49]. The age-related downregulation of cellular deacetylase expression is further worsened by the impairment of sirtuin activity due to the reduced availability of NAD+ in aged cells. This phenomenon has been particularly attributed to the enhanced expression of the NAD+-consuming NADase CD38 in numerous tissues of old mice [50].
Age-related inactivation of key metabolic regulators such as AMPK and sirtuins has a significant negative impact on the cellular energy balance. In particular, mitochondrial function, biogenesis, and clearance control (mitophagy) are strongly dependent on the activity of AMPK, Sirt1, and Sirt3 [51,52,53]. Thus, the age-related decline in AMPK and sirtuin activity may lead to accumulation of dysfunctional and damaged mitochondria, which in turn leads to excessive ROS formation, the release of mitochondrial DNA (mtDNA) in the cytosol, and, as a result, inflammation [54,55] (see also below).
In addition, age-related downregulation of AMPK and sirtuin activity may promote cardiac inflammation in other ways. AMPK suppresses NF-κB activity via phosphorylation (at Ser177 and Ser181) and inactivation of IκB kinase (IKK), which attenuates activation of NF-κB signaling and the expression of pro-inflammatory genes [56]. Similarly, Sirt1 directly represses NF-κB gene expression by deacetylating RelA/p65 at lysine 310 [37]. Furthermore, AMPK suppresses NF-κB signaling indirectly via several downstream mediators, e.g., Sirt1 or PGC-1α, which can subsequently reduce the expression of pro-inflammatory factors [57]. Our recent report [27] demonstrated the association of age-related AMPK inactivation and cardiac inflammation in human heart. In agreement, activation of AMPK attenuates inflammation [58,59].
Similarly, downregulation of Sirt1 activity may promote cardiac inflammation. Indeed, Sirt1 promotes mitochondrial biogenesis and autophagy via deacetylation of PGC-1α [60], a key transcription factor regulating the expression of mitochondrial proteins, and Sirt1 increases AMPK activity via LKB1 deacetylation [43]. Thus, age-related Sirt1 downregulation may impair mitochondrial function, leading to mitochondrial ROS formation and mtDNA release.
Finally, downregulation of AMPK and Sirt1 leads to a metabolic shift that arises from the downregulation of mitochondrial oxidation of substrates and upregulation of glycolytic pathways. This shift to glycolysis may promote an inflammatory reaction [61,62] and, thus, may additionally contribute to disease development and age-related inflammation.
Taken together, the current knowledge argues for a significant role of the age-related downregulation of key metabolic regulators (AMPK, Sirt1, and Sirt3) in cardiac inflammation.
2.3. Mitochondria and Cardiac Inflammaging
Mitochondrial dysfunction is viewed as one of the major hallmarks of aging [6]. Cardiac aging is associated with the general decline in mitochondrial function and accumulation of dysfunctional mitochondria, mainly due to the dysregulation of quality control processes [63,64]. Mitochondria are a key source of the reactive oxygen species (ROS), which not only serve as signaling molecules but may also be destructive: accumulation of ROS promotes enhanced oxidative stress that gives rise to subsequent accumulation of damaged DNA, proteins, and lipids as well as mtDNA damage and release [65]. Although ROS are key signaling messengers required for proper cell functioning, when their exaggerated production level exceed the capacity of ROS scavenger systems that neutralize them, ROS become harmful to the cell. Indeed, we and others have clearly seen diminished antioxidant expression in aging individuals [27,66], thus explaining age-associated damage caused by ROS.
In old individuals, ROS can significantly increase the release of mtDNA from the mitochondrial matrix into the cytosol of a cell [67], which produces damage-associated molecular patterns (DAMPs) and is considered to be a driver of inflammatory responses [68]. Released mtDNA may stimulate many pattern recognition receptors, leading to activation of the cyclic GMP–AMP synthase-stimulator of interferon genes protein (cGAS-STING) pathway and hyperactivation of the innate immune response through Toll-like receptor (TLR) signaling; here, activation of the NLRP3 inflammasome play a central role (detailed review [69,70]).
Due to its proximity to a major source of ROS, mtDNA frequently harbors numerous mutations (for mtDNA mutation refer to a comprehensive review [71]). Thus, as each mitochondrion has several mtDNA copies, some of them may be mutated, while others are intact—the so-called heteroplasmy phenomenon that is typical for mitochondria [72]. The level of the heteroplasmy is increased during the aging processes, and mitochondria bearing mtDNA heteroplasmy is a hallmark of aging [73]. Moreover, aging-related mutations in mtDNA cause disruption of cellular homeostatic mechanisms and mitochondrial dysfunction via impaired oxidative phosphorylation [74].
Mitochondria are dynamic organelles whose function is intimately linked to their morphology; this is regulated by opposing fusion and fission, which are essential processes not only for the division but also for the preservation of functional mitochondrial integrity [75]. A disturbance of the balance between fusion and fission promotes the accumulation of damaged mitochondria and a fragmented mitochondrial network. Thus, cardiac aging is accompanied by disrupted mitochondrial structure and expanded mitochondrial size [76].
3. Vascular Inflammaging
CVD is the main cause of death worldwide [9], with increasing incidence in aged individuals. Enhanced vascular aging is associated with more severe atherosclerosis and microvascular dysfunction [77,78], and it is characterized by pathological vascular remodeling and vascular stiffness [79].
Inflammation is typical for aging hearts [27]. Within numerous factors promoting cardiac inflammation, circulating pro-inflammatory factors, which are highly elevated in old individuals play a substantial role [21].
Accumulating data demonstrate that vascular aging starts as early as childhood and is characterized by gradual changes in the vascular structure (e.g., luminal dilation and intimal and medial thickening) [80] and function (e.g., endothelial dysfunction), resulting in reduced vascular compliance and increased arterial stiffness [81]. The major hallmarks of vascular aging include impaired endothelium-dependent vasodilation and defective vessel repair capacity. Understanding the mechanisms mediating vascular aging may allow specific pathways to be targeted in order to delay the progression and adverse outcome of vascular aging.
The endothelium maintains normal vascular tone via releasing several vascular protective factors, such as endothelial-derived hyperpolarizing factor (EDHF) and nitric oxide (NO), under normal homeostatic conditions [82]. Endothelial dysfunction is the major hallmark of cardiovascular aging and is defined as the failure of endothelium to mediate an adequate vasodilatory response to hemodynamic stimuli such as shear stress [83,84]. Under these conditions, there is a reduction in the bioavailability of vasodilators (particularly NO) but an increase in endothelial-derived contracting factors [85,86]. Moreover, a dysfunctional endothelium is associated with a pro-inflammatory and pro-thrombotic state as well as with an increased risk of cardiovascular events [87,88,89].
Even in the absence of other cardiovascular risk factors, aging progressively causes reduced NO bioavailability [85,90] and endothelial dysfunction [91,92]. In endothelial cells, NO is produced via conversion of L-arginine to L-citrulline catalyzed by endothelial NO synthase (eNOS) in the presence of nicotinamide adenine dinucleotide phosphate (NADPH), tetrahydrobiopterin (BH4), and other cofactors [93,94]. In endothelial dysfunction, the reduced bioavailability of NO may be the result of reduced expression or activity of eNOS, reduced supply of eNOS substrate (L-arginine), increased endogenous eNOS inhibitors, or increased NO scavenging. Indeed, reduced eNOS activity accompanied by an impaired vasodilatory response to acetylcholine and bradykinin was observed in the vessels of aging rats [95,96]. Likewise, flow-mediated vasodilation was impaired in aged rats, which was ameliorated by hydralazine treatment [97].
Arginase, an important enzyme of the urea cycle, competes with eNOS for L-arginine as substrate and, thus, may limit the availability of L-arginine for NO production, particularly under conditions of increased arginase activity. An upregulation of arginase expression as well as activity that was accompanied by reduced vasodilation has been demonstrated in aged rat aortas; pharmacological inhibition of arginase improved the vasodilatory response [98]. Similarly, in rabbit cavernous, the carbachol-induced vasodilatory response was impaired in aged animals along with upregulation of arginase activity, and this impaired vasodilation was normalized by treatment with arginase inhibitors or excessive supply of L-arginine [99]. Indeed, arginase inhibition reverses endothelial cell aging phenotype in vitro [100]. Excessive production of ROS in the vascular wall may also reduce NO bioavailability by converting it to peroxynitrite [101,102,103]. This was verified by the observation that aging-induced reduction in NO bioavailability in rat vasculature was improved by administering the ROS scavenger TEMPOL [104]. Reduced vascular NO bioavailability may also result from a deficiency in BH4, an important cofactor for enzymatic NO production. BH4 depletion leads to eNOS uncoupling, resulting in increased ROS instead of NO production [105]. Indeed, reduced levels of BH4 accompanied by impaired endothelium-dependent vasodilation has been observed in arterioles of aged rats [106]. Supplementation of BH4 in these rats improved flow-mediated vasodilatation. Likewise, BH4 supplementation improved flow-dependent vasodilation in sedentary but not exercising older human adults [107].
The factors discussed above create a pro-inflammatory environment in the vessel wall and perivascular region that causes of infiltration of inflammatory cells mainly monocytes and macrophages [108]. For example, in aged Ldlr(−/−) mice on a high fat diet, the infiltration of monocytes and macrophages in aortic tissue was much higher compared to young littermates [109]. Likewise, number of perivascular infiltrating monocytes and macrophages in aging hypertensive rats was much higher compared young rats [110,111]. In addition to monocytes and macrophages, some recent reports indicate the involvement of other immune cells in eliciting aging-related tissue inflammation. For example, it has been reported that aging in mice is associated with increased accumulation of CD4+-T cells with dysfunctional mitochondria in mediastinal lymph nodes where they secrete massive amounts of IFN-γ [112,113], which plays crucial role in myocardial aging [114].
4. Inflammaging and the Microbiome
The microbiome contributes substantially to health, and as such, is also an important modifier of disease. Although definitions differ slightly [115], the microbiome is understood to be the community of all microorganisms such bacteria, viruses, fungi, and protozoa living in or on the human body and interacting with it. While most of the bacteria colonize the gastrointestinal tract, making the gut microbiome the most established and studied microbiota, bacteria also reside in the oral cavity, the skin, the vagina, and the urinary tract [116]. The effects of aging on the gut microbiota, as discussed below, have been well established, but the non-gut microbiome is also known to change with aging, which in turn has implications for organ health and may result in various diseases [117,118].
The microbiome is affected by various factors in higher age, including poly-medication and certain lifestyle factors, such as decreased physical activity and impaired dietary intake. Aging itself is known to impact many aspects of the gut and may also impair the functionality of the gastrointestinal tract. Disturbed motility, a decrease in digestive enzymes, and a prolongation of the colonic transit time, as well as increased intestinal permeability, lead to a functional decline of the aging gut [119].
Immune barriers have been reported to be compromised in higher age. For example, decreased production of antimicrobial mucin in the epithelial cells, which constitute a first important barrier, may lead to higher permeability and facilitate bacterial translocation [120]. The increased uptake of lipopolysaccharides into the bloodstream and further extra-intestinal sites may in turn trigger pro-inflammatory processes [121].
Concomitantly, intestinal microbiota can be drastically altered in advanced age, resulting in an aged-type microbiota [122] that exhibits a loss of biodiversity, an increase in opportunistic pathogens, and a reduction of health-associated species, such as short chain fatty acid (SCFA)-producing species [123]. In addition to their role in energy production, SCFA such as acetate, propionate, and butyrate have many health-promoting and protective properties, such as anti-inflammatory [124] and immunomodulatory properties [125,126]. Their role in the regulation of inflammation is well established [124]. SCFAs are produced by the intestinal tract during anaerobic fermentation of indigestible fibers and resistant starch. They, in particular butyrate, can trigger signaling cascades via activation of G-protein-coupled receptors (GPCRs) such as GPR41, GPR43, and GPR109A, and thus, exert an important immunomodulatory role, which has been documented in intestinal inflammation [127]. Due to these immunomodulatory functions and the impact of dysbiosis on low-grade systemic inflammation, recent evidence also points to a close relationship between the microbiome and inflammaging [128]. Animal studies have shown that, typically, age-related changes in the gut microbiome result in a pro-inflammatory status, with increased levels of IL-6, IL-10, TNF-α, and TGF-β as well as activation of NF-κB and mTOR and decreased levels of cyclin E and CDK2. In older humans, an increase in proteobacteria and a decrease in butyrate-producing species were associated with the increased levels of IL-6 and IL-8 [129]. A recent systematic review summarizing the evidence also revealed associations between certain bacteria (such as Parabacteroides, Mucispirillum, Clostridium, and Sarcina) and the pro-inflammatory cytokine MCP-1, whereas Lactobacillus, Akkermansia, Oscillospira, and Blautia are negatively associated with MCP-1 [130]. Moreover, transfer of aged type microbiota from old mice to germ-free mice resulted in increased systemic inflammation, as inflammation in the gut was promoted with subsequent transfer of lipopolysaccharides and increased T cell activation [131], which implies a causal role of the gut microbiome in inflammaging.
Age-related changes in the microbiome composition (age-related gut dysbiosis) have also been associated with the development of the frailty syndrome and anorexia of aging, which in turn may trigger further catabolic processes, although more studies are needed in this context. Gut microbiome dysbiosis has, moreover, been implicated in the pathophysiology of many diseases, and emerging evidence has also clearly established an association between the gut microbiome and the development of cardiovascular disease [132,133,134,135], especially via inflammatory pathways triggered by the interaction between host and microbiota [136], so that microbiome-based strategies for prevention of cardiovascular disease have been proposed [137]. However, causative mechanisms have not thus far been fully elucidated [137]. A role has been suggested for the gut microbiota-derived marker trimethylamine N-oxide (TMAO), a pro-atherogenic metabolite. It is an oxidation product of trimethylamine derived from bacterial metabolism of dietary L-carnitine, choline, and betaine [138]. High plasma concentrations of TMAO have previously been associated with a higher risk of cardiovascular disease [139] and cardiovascular mortality in peripheral artery disease [140].
While causality regarding the role of the microbiome in disease development is still a matter of debate, studies with fecal transplantation have shown that transplanted microbiota of adults with diseases such as cardiometabolic syndrome [141], hypertension [142], or obesity [143] have the potential to impact metabolism, inflammation, and body composition phenotypes in their host environments, which clearly implies a causal relationship between the microbiome and disease development. Similarly, in preclinical research, fecal microbiota transplantation decreased inflammation and, thus, alleviated myocardial injury in an experimental autoimmune myocarditis mouse model [144]. The use of nanomedicine in order to modify gut microbiota for the prevention of coronary artery disease is currently under investigation; it has been shown that nanoparticles can be used to transfer specific gut microbiota that are associated with a decrease in inflammation as well as an increase in SCFA and HDL [137].
Dietary quality, i.e., the quantity and quality of nutrients, also has a direct effect on the gastrointestinal microbiome, which in turn partly explains the beneficial effect of certain dietary patterns. As such, recent approaches in nutritional intervention have specifically targeted the aged-type microbiome. A one-year intervention with an individualized Mediterranean diet in older adults has been shown to successfully modify the microbiome and subsequently decrease inflammation and improve the aging phenotype frailty, clearly outlining the potential of dietary modification of aged-type microbiota [145].
5. Translational Aspects of Cardiovascular Inflammaging
Several large-scale studies, including those of the MARK-AGE group and NU-AGE consortium, the Leiden Longevity Study, and the “Berliner Alterstudie II”, have identified biomarkers of aging and inflammaging [31]. These studies revealed that CRP, TNF-α, and IL-6 are tentative candidate parameters of systemic inflammation related to aging [146,147,148,149].
Low-grade systemic inflammation is associated with mitochondrial dysfunction and biogenesis. Preservation of mitochondrial morphology, dynamics, and function may be a therapeutic approach to prevent cardiac aging. There are several potential drugs that may improve age-related mitochondrial dysfunction and, thus, attenuate cardiac inflammaging. One of them that has been tested in large clinical trials to combat age-related disorders and improve healthspan is metformin, which has a therapeutic effect by lowering oxidative stress via mitochondrial complex I inhibition, followed by the increase in cytoplasmatic AMP:ATP and ADP:ATP ratios, which in turns leads to activation of AMPK [150].
Similar to metformin, some nutraceuticals, i.e., agents derived from natural sources, possess a potential to interfere inflammaging via activation of cAMP-AMPK signaling [151]. Particularly, resveratrol, a stilbenoid produced by several plants, has been intensively investigated in numerous clinical trials related to various diseases [152]. A pioneer study by Park et al. [153] reported in skeletal muscle cells that resveratrol acts as a PDE inhibitor, enhancing cAMP levels and leading to activation of AMPK in an EPAC1-dependent manner. The authors showed that activation of EPAC1 increases intracellular Ca2+ levels and promotes CaMKK2 activity, which phosphorylates and activates AMPK. In line with this study, resveratrol attenuates endothelial inflammation through the activation of the cAMP-PKA-AMPK-SIRT1 signaling pathway [154].
Another potential approach to protect heart against inflammaging is attenuation of oxidative stress. Supplementation with coenzyme Q10, which naturally decreases with age [155], protects the heart from aging-related oxidative stress and improves mitochondrial function [156] by inhibition of mtDNA release and its accumulation in cytosol [157]. In a double-blind trial, long-term coenzyme Q10 treatment reduced major adverse cardiovascular events in heart failure patients [158]. There is an ongoing clinical trial in a large study population designed to confirm clinical benefits of long-term treatment with coenzyme Q10 in patients with cardiovascular disorders (NCT03133793). While these treatments are not mitochondria specific, there are some approaches targeting mitochondria that utilize triphenylphosphonium as a mitochondria-targeted vehicle to deliver antioxidants such as mitoquinone, SkQ1, or Mito-Tempo, to mitochondria [159]. The preclinical studies demonstrated promising beneficial results in patients with cardiac disorders, and there are ongoing clinical trials of these treatments (for a comprehensive review, see [159]).
In conclusion, cardiac inflammaging leads to several dysfunctional processes, including energy imbalance, mitochondrial dysfunction, changes in the microbiome, and vascular senescence. The prevention of cardiac aging and age-related CVD might be focus by the modulation of inflammaging and its related mitochondrial dysfunction (Figure 1).
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
We acknowledge support from the German Research Foundation (DFG) and the Open Access Publication Fund of Charité—Universitätsmedizin Berlin.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Toba, H.; de Castro Bras, L.E.; Baicu, C.F.; Zile, M.R.; Lindsey, M.L.; Bradshaw, A.D. Secreted protein acidic and rich in cysteine facilitates age-related cardiac inflammation and macrophage M1 polarization. Am. J. Physiol. Cell Physiol. 2015, 308, C972–C982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinn, P.M.; Holdren, G.O.; Westermeyer, B.A.; Abuissa, M.; Fischer, C.L.; Fairley, J.A.; Brogden, K.A.; Brogden, N.K. Age-dependent variation in cytokines, chemokines, and biologic analytes rinsed from the surface of healthy human skin. Sci. Rep. 2015, 5, 10472. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, E.; An, Y.; Zoli, M.; Simonsick, E.M.; Guralnik, J.M.; Bandinelli, S.; Boyd, C.M. Aging and the burden of multimorbidity: Associations with inflammatory and anabolic hormonal biomarkers. J. Gerontol. A Biol. Sci. Med. Sci. 2015, 70, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Ballou, S.P.; Lozanski, F.B.; Hodder, S.; Rzewnicki, D.L.; Mion, L.C.; Sipe, J.D.; Ford, A.B.; Kushner, I. Quantitative and qualitative alterations of acute-phase proteins in healthy elderly persons. Age Ageing 1996, 25, 224–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, J.; Xu, H.; Davies, J.L.; Hemmings, G.P. Increase of plasma IL-6 concentration with age in healthy subjects. Life Sci. 1992, 51, 1953–1956. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howcroft, T.K.; Campisi, J.; Louis, G.B.; Smith, M.T.; Wise, B.; Wyss-Coray, T.; Augustine, A.D.; McElhaney, J.E.; Kohanski, R.; Sierra, F. The role of inflammation in age-related disease. Aging 2013, 5, 84–93. [Google Scholar] [CrossRef] [Green Version]
- Salminen, A.; Kauppinen, A.; Suuronen, T.; Kaarniranta, K. SIRT1 longevity factor suppresses NF-kappaB -driven immune responses: Regulation of aging via NF-kappaB acetylation? Bioessays 2008, 30, 939–942. [Google Scholar] [CrossRef]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef]
- Ruparelia, N.; Chai, J.T.; Fisher, E.A.; Choudhury, R.P. Inflammatory processes in cardiovascular disease: A route to targeted therapies. Nat. Rev. Cardiol. 2017, 14, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Garagnani, P.; Vitale, G.; Capri, M.; Salvioli, S. Inflammaging and ‘Garb-aging’. Trends Endocrinol. Metab. 2017, 28, 199–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passos, J.F.; Nelson, G.; Wang, C.; Richter, T.; Simillion, C.; Proctor, C.J.; Miwa, S.; Olijslagers, S.; Hallinan, J.; Wipat, A.; et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 2010, 6, 347. [Google Scholar] [CrossRef] [PubMed]
- Jezek, J.; Cooper, K.F.; Strich, R. Reactive Oxygen Species and Mitochondrial Dynamics: The Yin and Yang of Mitochondrial Dysfunction and Cancer Progression. Antioxidants 2018, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Tavenier, J.; Rasmussen, L.J.H.; Houlind, M.B.; Andersen, A.L.; Panum, I.; Andersen, O.; Petersen, J.; Langkilde, A.; Nehlin, J.O. Alterations of monocyte NF-kappaB p65/RelA signaling in a cohort of older medical patients, age-matched controls, and healthy young adults. Immun. Ageing 2020, 17, 25. [Google Scholar] [CrossRef]
- Salminen, A.; Huuskonen, J.; Ojala, J.; Kauppinen, A.; Kaarniranta, K.; Suuronen, T. Activation of innate immunity system during aging: NF-kB signaling is the molecular culprit of inflamm-aging. Ageing Res. Rev. 2008, 7, 83–105. [Google Scholar] [CrossRef]
- Jurk, D.; Wilson, C.; Passos, J.F.; Oakley, F.; Correia-Melo, C.; Greaves, L.; Saretzki, G.; Fox, C.; Lawless, C.; Anderson, R.; et al. Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat. Commun. 2014, 2, 4172. [Google Scholar] [CrossRef]
- Bernal, G.M.; Wahlstrom, J.S.; Crawley, C.D.; Cahill, K.E.; Pytel, P.; Liang, H.; Kang, S.; Weichselbaum, R.R.; Yamini, B. Loss of Nfkb1 leads to early onset aging. Aging 2014, 6, 931–943. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Garcia, V.A.; Alameda, J.P.; Page, A.; Casanova, M.L. Role of NF-kappaB in Ageing and Age-Related Diseases: Lessons from Genetically Modified Mouse Models. Cells 2021, 10, 1906. [Google Scholar] [CrossRef]
- Youm, Y.H.; Grant, R.W.; McCabe, L.R.; Albarado, D.C.; Nguyen, K.Y.; Ravussin, A.; Pistell, P.; Newman, S.; Carter, R.; Laque, A.; et al. Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab. 2013, 18, 519–532. [Google Scholar] [CrossRef] [Green Version]
- Ferrucci, L.; Corsi, A.; Lauretani, F.; Bandinelli, S.; Bartali, B.; Taub, D.D.; Guralnik, J.M.; Longo, D.L. The origins of age-related proinflammatory state. Blood 2005, 105, 2294–2299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nunez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinon, F.; Mayor, A.; Tschopp, J. The inflammasomes: Guardians of the body. Annu. Rev. Immunol. 2009, 27, 229–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franceschi, C.; Capri, M.; Monti, D.; Giunta, S.; Olivieri, F.; Sevini, F.; Panourgia, M.P.; Invidia, L.; Celani, L.; Scurti, M.; et al. Inflammaging and anti-inflammaging: A systemic perspective on aging and longevity emerged from studies in humans. Mech. Ageing Dev. 2007, 128, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef]
- Barcena de Arellano, M.L.; Pozdniakova, S.; Kuhl, A.A.; Baczko, I.; Ladilov, Y.; Regitz-Zagrosek, V. Sex differences in the aging human heart: Decreased sirtuins, pro-inflammatory shift and reduced anti-oxidative defense. Aging 2019, 11, 1918–1933. [Google Scholar] [CrossRef]
- Ridker, P.M.; Cushman, M.; Stampfer, M.J.; Tracy, R.P.; Hennekens, C.H. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N. Engl. J. Med. 1997, 336, 973–979. [Google Scholar] [CrossRef]
- Ikonomidis, I.; Andreotti, F.; Economou, E.; Stefanadis, C.; Toutouzas, P.; Nihoyannopoulos, P. Increased proinflammatory cytokines in patients with chronic stable angina and their reduction by aspirin. Circulation 1999, 100, 793–798. [Google Scholar] [CrossRef] [Green Version]
- Liuzzo, G.; Biasucci, L.M.; Gallimore, J.R.; Caligiuri, G.; Buffon, A.; Rebuzzi, A.G.; Pepys, M.B.; Maseri, A. Enhanced inflammatory response in patients with preinfarction unstable angina. J. Am. Coll. Cardiol. 1999, 34, 1696–1703. [Google Scholar] [CrossRef] [Green Version]
- Muller-Werdan, U.; Nuding, S.; Ost, M. Assessing inflammageing. Curr. Opin. Clin. Nutr. Metab. Care 2017, 20, 346–348. [Google Scholar] [CrossRef] [PubMed]
- Stice, J.P.; Chen, L.; Kim, S.C.; Jung, J.S.; Tran, A.L.; Liu, T.T.; Knowlton, A.A. 17β-Estradiol, aging, inflammation, and the stress response in the female heart. Endocrinology 2011, 152, 1589–1598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Duckles, S.P.; Weiss, J.H.; Li, X.; Krause, D.N. 17β-Estradiol prevents cell death and mitochondrial dysfunction by an estrogen receptor-dependent mechanism in astrocytes after oxygen-glucose deprivation/reperfusion. Free Radic. Biol. Med. 2012, 52, 2151–2160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klinge, C.M. Estrogenic control of mitochondrial function and biogenesis. J. Cell Biochem. 2008, 105, 1342–1351. [Google Scholar] [CrossRef] [Green Version]
- Barcena, M.L.; Pozdniakova, S.; Haritonow, N.; Breiter, P.; Kuhl, A.A.; Milting, H.; Baczko, I.; Ladilov, Y.; Regitz-Zagrosek, V. Dilated cardiomyopathy impairs mitochondrial biogenesis and promotes inflammation in an age- and sex-dependent manner. Aging 2020, 12, 24117–24133. [Google Scholar] [CrossRef]
- Ghisays, F.; Brace, C.S.; Yackly, S.M.; Kwon, H.J.; Mills, K.F.; Kashentseva, E.; Dmitriev, I.P.; Curiel, D.T.; Imai, S.I.; Ellenberger, T. The N-Terminal Domain of SIRT1 Is a Positive Regulator of Endogenous SIRT1-Dependent Deacetylation and Transcriptional Outputs. Cell Rep. 2015, 10, 1665–1673. [Google Scholar] [CrossRef] [Green Version]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef] [Green Version]
- Epelman, S.; Liu, P.P.; Mann, D.L. Role of innate and adaptive immune mechanisms in cardiac injury and repair. Nat. Rev. Immunol. 2015, 15, 117–129. [Google Scholar] [CrossRef]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G. AMP-activated protein kinase: Maintaining energy homeostasis at the cellular and whole-body levels. Annu. Rev. Nutr. 2014, 34, 31–55. [Google Scholar] [CrossRef] [Green Version]
- Jeon, S.M. Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 2016, 48, e245. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Age-related changes in AMPK activation: Role for AMPK phosphatases and inhibitory phosphorylation by upstream signaling pathways. Ageing Res. Rev. 2016, 28, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Lan, F.; Cacicedo, J.M.; Ruderman, N.; Ido, Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J. Biol. Chem. 2008, 283, 27628–27635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, H.; Pang, J.; Han, Y.; Dai, Y.; Dai, D.; Cai, J.; Zhang, T.M. Age-dependent tissue expression patterns of Sirt1 in senescence-accelerated mice. Mol. Med. Rep. 2014, 10, 3296–3302. [Google Scholar] [CrossRef] [Green Version]
- Braidy, N.; Guillemin, G.J.; Mansour, H.; Chan-Ling, T.; Poljak, A.; Grant, R. Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PLoS ONE 2011, 6, e19194. [Google Scholar] [CrossRef]
- Xu, H.; Gan, C.; Gao, Z.; Huang, Y.; Wu, S.; Zhang, D.; Wang, X.; Sheng, J. Caffeine Targets SIRT3 to Enhance SOD2 Activity in Mitochondria. Front. Cell Dev. Biol. 2020, 8, 822. [Google Scholar] [CrossRef]
- Yu, W.; Dittenhafer-Reed, K.E.; Denu, J.M. SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status. J. Biol. Chem. 2012, 287, 14078–14086. [Google Scholar] [CrossRef] [Green Version]
- Morigi, M.; Perico, L.; Rota, C.; Longaretti, L.; Conti, S.; Rottoli, D.; Novelli, R.; Remuzzi, G.; Benigni, A. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J. Clin. Investig. 2015, 125, 715–726. [Google Scholar] [CrossRef] [Green Version]
- Parihar, P.; Solanki, I.; Mansuri, M.L.; Parihar, M.S. Mitochondrial sirtuins: Emerging roles in metabolic regulations, energy homeostasis and diseases. Exp. Gerontol. 2015, 61, 130–141. [Google Scholar] [CrossRef]
- Camacho-Pereira, J.; Tarrago, M.G.; Chini, C.C.S.; Nin, V.; Escande, C.; Warner, G.M.; Puranik, A.S.; Schoon, R.A.; Reid, J.M.; Galina, A.; et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016, 23, 1127–1139. [Google Scholar] [CrossRef] [Green Version]
- Jayarajan, V.; Appukuttan, A.; Aslam, M.; Reusch, P.; Regitz-Zagrosek, V.; Ladilov, Y. Regulation of AMPK activity by type 10 adenylyl cyclase: Contribution to the mitochondrial biology, cellular redox and energy homeostasis. Cell Mol. Life Sci. 2019, 76, 4945–4959. [Google Scholar] [CrossRef] [PubMed]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenmoehl, J.; Hoeflich, A. Dual control of mitochondrial biogenesis by sirtuin 1 and sirtuin 3. Mitochondrion 2013, 13, 755–761. [Google Scholar] [CrossRef] [PubMed]
- El Assar, M.; Angulo, J.; Rodriguez-Manas, L. Oxidative stress and vascular inflammation in aging. Free Radic. Biol. Med. 2013, 65, 380–401. [Google Scholar] [CrossRef]
- Oka, T.; Hikoso, S.; Yamaguchi, O.; Taneike, M.; Takeda, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; Nakayama, H.; Nishida, K.; et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012, 485, 251–255. [Google Scholar] [CrossRef]
- Bess, E.; Fisslthaler, B.; Fromel, T.; Fleming, I. Nitric oxide-induced activation of the AMP-activated protein kinase alpha2 subunit attenuates IkappaB kinase activity and inflammatory responses in endothelial cells. PLoS ONE 2011, 6, e20848. [Google Scholar] [CrossRef] [Green Version]
- Salminen, A.; Hyttinen, J.M.; Kaarniranta, K. AMP-activated protein kinase inhibits NF-kappaB signaling and inflammation: Impact on healthspan and lifespan. J. Mol. Med. 2011, 89, 667–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peairs, A.; Radjavi, A.; Davis, S.; Li, L.; Ahmed, A.; Giri, S.; Reilly, C.M. Activation of AMPK inhibits inflammation in MRL/lpr mouse mesangial cells. Clin. Exp. Immunol. 2009, 156, 542–551. [Google Scholar] [CrossRef]
- Hoogendijk, A.J.; Pinhancos, S.S.; van der Poll, T.; Wieland, C.W. AMP-activated protein kinase activation by 5-aminoimidazole-4-carbox-amide-1-beta-D-ribofuranoside (AICAR) reduces lipoteichoic acid-induced lung inflammation. J. Biol. Chem. 2013, 288, 7047–7052. [Google Scholar] [CrossRef] [Green Version]
- Gurd, B.J. Deacetylation of PGC-1alpha by SIRT1: Importance for skeletal muscle function and exercise-induced mitochondrial biogenesis. Appl Physiol. Nutr. Metab. 2011, 36, 589–597. [Google Scholar] [CrossRef]
- Soto-Heredero, G.; Gomez de Las Heras, M.M.; Gabande-Rodriguez, E.; Oller, J.; Mittelbrunn, M. Glycolysis—A key player in the inflammatory response. FEBS J. 2020, 287, 3350–3369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palsson-McDermott, E.M.; O’Neill, L.A.J. Targeting immunometabolism as an anti-inflammatory strategy. Cell Res. 2020, 30, 300–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhutia, S.K.; Naik, P.P.; Panigrahi, D.P.; Bhol, C.S.; Mahapatra, K.K. Mitophagy, Diseases, and Aging. In Models, Molecules and Mechanisms in Biogerontology; Rath, P.C., Ed.; Springer Nature: Singapore, 2019; pp. 177–191. [Google Scholar]
- Chen, G.; Kroemer, G.; Kepp, O. Mitophagy: An Emerging Role in Aging and Age-Associated Diseases. Front. Cell Dev. Biol. 2020, 8, 200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizvi, F.; Preston, C.C.; Emelyanova, L.; Yousufuddin, M.; Viqar, M.; Dakwar, O.; Ross, G.R.; Faustino, R.S.; Holmuhamedov, E.L.; Jahangir, A. Effects of Aging on Cardiac Oxidative Stress and Transcriptional Changes in Pathways of Reactive Oxygen Species Generation and Clearance. J. Am. Heart Assoc. 2021, 10, e019948. [Google Scholar] [CrossRef] [PubMed]
- Pinti, M.; Cevenini, E.; Nasi, M.; De Biasi, S.; Salvioli, S.; Monti, D.; Benatti, S.; Gibellini, L.; Cotichini, R.; Stazi, M.A.; et al. Circulating mitochondrial DNA increases with age and is a familiar trait: Implications for “inflamm-aging”. Eur. J. Immunol. 2014, 44, 1552–1562. [Google Scholar] [CrossRef]
- Dela Cruz, C.S.; Kang, M.J. Mitochondrial dysfunction and damage associated molecular patterns (DAMPs) in chronic inflammatory diseases. Mitochondrion 2018, 41, 37–44. [Google Scholar] [CrossRef]
- Riley, J.S.; Tait, S.W. Mitochondrial DNA in inflammation and immunity. EMBO Rep. 2020, 21, e49799. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef]
- Kauppila, T.E.S.; Kauppila, J.H.K.; Larsson, N.G. Mammalian Mitochondria and Aging: An Update. Cell Metab. 2017, 25, 57–71. [Google Scholar] [CrossRef] [Green Version]
- Nissanka, N.; Moraes, C.T. Mitochondrial DNA heteroplasmy in disease and targeted nuclease-based therapeutic approaches. EMBO Rep. 2020, 21, e49612. [Google Scholar] [CrossRef] [PubMed]
- Elorza, A.A.; Soffia, J.P. mtDNA Heteroplasmy at the Core of Aging-Associated Heart Failure. An Integrative View of OXPHOS and Mitochondrial Life Cycle in Cardiac Mitochondrial Physiology. Front. Cell Dev. Biol. 2021, 9, 625020. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.L.; Whitehall, J.C.; Bradshaw, C.; Gay, D.; Robertson, F.; Blain, A.P.; Hudson, G.; Pyle, A.; Houghton, D.; Hunt, M.; et al. Age-associated mitochondrial DNA mutations cause metabolic remodelling that contributes to accelerated intestinal tumorigenesis. Nat. Cancer 2020, 1, 976–989. [Google Scholar] [CrossRef] [PubMed]
- Tabara, L.C.; Morris, J.L.; Prudent, J. The Complex Dance of Organelles during Mitochondrial Division. Trends Cell Biol. 2021, 31, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Duicu, O.M.; Mirica, S.N.; Gheorgheosu, D.E.; Privistirescu, A.I.; Fira-Mladinescu, O.; Muntean, D.M. Ageing-induced decrease in cardiac mitochondrial function in healthy rats. Can. J. Physiol. Pharmacol. 2013, 91, 593–600. [Google Scholar] [CrossRef]
- Assar, M.E.; Angulo, J.; Rodriguez-Manas, L. Diabetes and ageing-induced vascular inflammation. J. Physiol. 2016, 594, 2125–2146. [Google Scholar] [CrossRef] [Green Version]
- Scuteri, A.; Cunha, P.G.; Agabiti Rosei, E.; Badariere, J.; Bekaert, S.; Cockcroft, J.R.; Cotter, J.; Cucca, F.; De Buyzere, M.L.; De Meyer, T.; et al. Arterial stiffness and influences of the metabolic syndrome: A cross-countries study. Atherosclerosis 2014, 233, 654–660. [Google Scholar] [CrossRef] [Green Version]
- Guzik, T.J.; Cosentino, F. Epigenetics and Immunometabolism in Diabetes and Aging. Antioxid. Redox Signal. 2018, 29, 257–274. [Google Scholar] [CrossRef] [Green Version]
- Lakatta, E.G. Arterial and cardiac aging: Major shareholders in cardiovascular disease enterprises: Part III: Cellular and molecular clues to heart and arterial aging. Circulation 2003, 107, 490–497. [Google Scholar] [CrossRef] [Green Version]
- Laina, A.; Stellos, K.; Stamatelopoulos, K. Vascular ageing: Underlying mechanisms and clinical implications. Exp. Gerontol. 2018, 109, 16–30. [Google Scholar] [CrossRef]
- Long, D.A.; Newaz, M.A.; Prabhakar, S.S.; Price, K.L.; Truong, L.D.; Feng, L.; Mu, W.; Oyekan, A.O.; Johnson, R.J. Loss of nitric oxide and endothelial-derived hyperpolarizing factor-mediated responses in aging. Kidney Int. 2005, 68, 2154–2163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luscher, T.F.; Corti, R. Flow: The signal of life. Circ. Res. 2004, 95, 749–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godo, S.; Shimokawa, H. Endothelial Functions. Arter. Thromb. Vasc. Biol. 2017, 37, e108–e114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trinity, J.D.; Groot, H.J.; Layec, G.; Rossman, M.J.; Ives, S.J.; Morgan, D.E.; Gmelch, B.S.; Bledsoe, A.; Richardson, R.S. Passive leg movement and nitric oxide-mediated vascular function: The impact of age. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H672–H679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerman, A.; Burnett, J.C., Jr. Intact and altered endothelium in regulation of vasomotion. Circulation 1992, 86 (Suppl. 6), III12–III19. [Google Scholar] [PubMed]
- Kitta, Y.; Obata, J.E.; Nakamura, T.; Hirano, M.; Kodama, Y.; Fujioka, D.; Saito, Y.; Kawabata, K.; Sano, K.; Kobayashi, T.; et al. Persistent impairment of endothelial vasomotor function has a negative impact on outcome in patients with coronary artery disease. J. Am. Coll. Cardiol. 2009, 53, 323–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, T.J. Assessment and treatment of endothelial dysfunction in humans. J. Am. Coll. Cardiol. 1999, 34, 631–638. [Google Scholar] [CrossRef] [Green Version]
- Bonetti, P.O.; Pumper, G.M.; Higano, S.T.; Holmes, D.R.; Kuvin, J.T., Jr.; Lerman, A. Noninvasive identification of patients with early coronary atherosclerosis by assessment of digital reactive hyperemia. J. Am. Coll. Cardiol. 2004, 44, 2137–2141. [Google Scholar] [CrossRef] [Green Version]
- Sverdlov, A.L.; Ngo, D.T.; Chan, W.P.; Chirkov, Y.Y.; Horowitz, J.D. Aging of the nitric oxide system: Are we as old as our NO? J. Am. Heart Assoc. 2014, 3, e000973. [Google Scholar] [CrossRef] [Green Version]
- Celermajer, D.S.; Sorensen, K.E.; Spiegelhalter, D.J.; Georgakopoulos, D.; Robinson, J.; Deanfield, J.E. Aging is associated with endothelial dysfunction in healthy men years before the age-related decline in women. J. Am. Coll. Cardiol. 1994, 24, 471–476. [Google Scholar] [CrossRef] [Green Version]
- Eskurza, I.; Monahan, K.D.; Robinson, J.A.; Seals, D.R. Effect of acute and chronic ascorbic acid on flow-mediated dilatation with sedentary and physically active human ageing. J. Physiol. 2004, 556, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Knowles, R.G.; Moncada, S. Nitric oxide synthases in mammals. Biochem. J. 1994, 298, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Andrew, P.J.; Mayer, B. Enzymatic function of nitric oxide synthases. Cardiovasc. Res. 1999, 43, 521–531. [Google Scholar] [CrossRef]
- Cernadas, M.R.; Sanchez de Miguel, L.; Garcia-Duran, M.; Gonzalez-Fernandez, F.; Millas, I.; Monton, M.; Rodrigo, J.; Rico, L.; Fernandez, P.; de Frutos, T.; et al. Expression of constitutive and inducible nitric oxide synthases in the vascular wall of young and aging rats. Circ. Res. 1998, 83, 279–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.R.; Visioli, F.; Hagen, T.M. Plasma membrane-associated endothelial nitric oxide synthase and activity in aging rat aortic vascular endothelia markedly decline with age. Arch. Biochem. Biophys. 2006, 454, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Dumont, O.; Pinaud, F.; Guihot, A.L.; Baufreton, C.; Loufrani, L.; Henrion, D. Alteration in flow (shear stress)-induced remodelling in rat resistance arteries with aging: Improvement by a treatment with hydralazine. Cardiovasc. Res. 2008, 77, 600–608. [Google Scholar] [CrossRef] [Green Version]
- Berkowitz, D.E.; White, R.; Li, D.; Minhas, K.M.; Cernetich, A.; Kim, S.; Burke, S.; Shoukas, A.A.; Nyhan, D.; Champion, H.C.; et al. Arginase reciprocally regulates nitric oxide synthase activity and contributes to endothelial dysfunction in aging blood vessels. Circulation 2003, 108, 2000–2006. [Google Scholar] [CrossRef] [Green Version]
- Sakai, Y.; Masuda, H.; Kihara, K.; Kurosaki, E.; Yamauchi, Y.; Azuma, H. Involvement of increased arginase activity in impaired cavernous relaxation with aging in the rabbit. J. Urol. 2004, 172, 369–373. [Google Scholar] [CrossRef]
- Garate-Carrillo, A.; Navarrete-Yanez, V.; Ortiz-Vilchis, P.; Guevara, G.; Castillo, C.; Mendoza-Lorenzo, P.; Ceballos, G.; Ortiz-Flores, M.; Najera, N.; Bustamante-Pozo, M.M.; et al. Arginase inhibition by (-)-Epicatechin reverses endothelial cell aging. Eur. J. Pharmacol. 2020, 885, 173442. [Google Scholar] [CrossRef]
- van der Loo, B.; Labugger, R.; Skepper, J.N.; Bachschmid, M.; Kilo, J.; Powell, J.M.; Palacios-Callender, M.; Erusalimsky, J.D.; Quaschning, T.; Malinski, T.; et al. Enhanced peroxynitrite formation is associated with vascular aging. J. Exp. Med. 2000, 192, 1731–1744. [Google Scholar] [CrossRef] [Green Version]
- Taddei, S.; Virdis, A.; Ghiadoni, L.; Salvetti, G.; Bernini, G.; Magagna, A.; Salvetti, A. Age-related reduction of NO availability and oxidative stress in humans. Hypertension 2001, 38, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, C.A.; Brosnan, M.J.; McIntyre, M.; Graham, D.; Dominiczak, A.F. Superoxide excess in hypertension and aging: A common cause of endothelial dysfunction. Hypertension 2001, 37, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Fleenor, B.S.; Seals, D.R.; Zigler, M.L.; Sindler, A.L. Superoxide-lowering therapy with TEMPOL reverses arterial dysfunction with aging in mice. Aging Cell 2012, 11, 269–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alp, N.J.; Channon, K.M. Regulation of endothelial nitric oxide synthase by tetrahydrobiopterin in vascular disease. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Delp, M.D.; Behnke, B.J.; Spier, S.A.; Wu, G.; Muller-Delp, J.M. Ageing diminishes endothelium-dependent vasodilatation and tetrahydrobiopterin content in rat skeletal muscle arterioles. J. Physiol. 2008, 586, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Eskurza, I.; Myerburgh, L.A.; Kahn, Z.D.; Seals, D.R. Tetrahydrobiopterin augments endothelium-dependent dilatation in sedentary but not in habitually exercising older adults. J. Physiol. 2005, 568 Pt 3, 1057–1065. [Google Scholar] [CrossRef]
- Guzik, T.J.; Touyz, R.M. Oxidative Stress, Inflammation, and Vascular Aging in Hypertension. Hypertension 2017, 70, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Wong, C.; Song, Y.; Shen, H.; Mori, D.; Rotllan, N.; Price, N.; Dobrian, A.D.; Meng, H.; Kleinstein, S.H.; et al. Age-associated vascular inflammation promotes monocytosis during atherogenesis. Aging Cell 2016, 15, 766–777. [Google Scholar] [CrossRef]
- Liu, Y.; Jacobowitz, D.M.; Barone, F.; McCarron, R.; Spatz, M.; Feuerstein, G.; Hallenbeck, J.M.; Siren, A.L. Quantitation of perivascular monocytes and macrophages around cerebral blood vessels of hypertensive and aged rats. J. Cereb. Blood Flow Metab. 1994, 14, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Nosalski, R.; Mikolajczyk, T.; Siedlinski, M.; Saju, B.; Koziol, J.; Maffia, P.; Guzik, T.J. Nox1/4 inhibition exacerbates age dependent perivascular inflammation and fibrosis in a model of spontaneous hypertension. Pharmacol. Res. 2020, 161, 105235. [Google Scholar] [CrossRef]
- Desdin-Mico, G.; Soto-Heredero, G.; Aranda, J.F.; Oller, J.; Carrasco, E.; Gabande-Rodriguez, E.; Blanco, E.M.; Alfranca, A.; Cusso, L.; Desco, M.; et al. T cells with dysfunctional mitochondria induce multimorbidity and premature senescence. Science 2020, 368, 1371–1376. [Google Scholar] [CrossRef] [PubMed]
- Ramos, G.C.; van den Berg, A.; Nunes-Silva, V.; Weirather, J.; Peters, L.; Burkard, M.; Friedrich, M.; Pinnecker, J.; Abesser, M.; Heinze, K.G.; et al. Myocardial aging as a T-cell-mediated phenomenon. Proc. Natl. Acad. Sci. USA 2017, 114, E2420–E2429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgobo, M.; Heinrichs, M.; Hapke, N.; Ashour, D.; Appel, M.; Srivastava, M.; Heckel, T.; Spyridopoulos, I.; Hofmann, U.; Frantz, S.; et al. Terminally Differentiated CD4+ T Cells Promote Myocardial Inflammaging. Front. Immunol. 2021, 12, 584538. [Google Scholar] [CrossRef] [PubMed]
- Berg, G.; Rybakova, D.; Fischer, D.; Cernava, T.; Verges, M.C.; Charles, T.; Chen, X.; Cocolin, L.; Eversole, K.; Corral, G.H.; et al. Microbiome definition re-visited: Old concepts and new challenges. Microbiome 2020, 8, 103. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.; Zhao, J.; Wu, L.; Carru, C.; Biagi, E.; Franceschi, C. Microbiomes other than the gut: Inflammaging and age-related diseases. Semin. Immunopathol. 2020, 42, 589–605. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.; Zeng, D.N.; Chi, L.; Tan, Y.; Galzote, C.; Cardona, C.; Lax, S.; Gilbert, J.; Quan, Z.X. The Influence of Age and Gender on Skin-Associated Microbial Communities in Urban and Rural Human Populations. PLoS ONE 2015, 10, e0141842. [Google Scholar]
- Kim, H.J.; Kim, J.J.; Myeong, N.R.; Kim, T.; Kim, D.; An, S.; Kim, H.; Park, T.; Jang, S.I.; Yeon, J.H.; et al. Segregation of age-related skin microbiome characteristics by functionality. Sci. Rep. 2019, 9, 16748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remond, D.; Shahar, D.R.; Gille, D.; Pinto, P.; Kachal, J.; Peyron, M.A.; Dos Santos, C.N.; Walther, B.; Bordoni, A.; Dupont, D.; et al. Understanding the gastrointestinal tract of the elderly to develop dietary solutions that prevent malnutrition. Oncotarget 2015, 6, 13858–13898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagpal, R.; Yadav, H. Bacterial Translocation from the Gut to the Distant Organs: An Overview. Ann. Nutr. Metab. 2017, 71 (Suppl. 1), 11–16. [Google Scholar] [CrossRef]
- Berg, R.D. Bacterial Translocation from the Gastrointestinal Tract. In Mechanisms in the Pathogenesis of Enteric Diseases 2. Advances in Experimental Medicine and Biology; Prem, S.P., David, H.F., Eds.; Springer: Boston, MA, USA, 1999; Volume 473, pp. 11–30. [Google Scholar]
- Biagi, E.; Candela, M.; Franceschi, C.; Brigidi, P. The aging gut microbiota: New perspectives. Ageing Res. Rev. 2011, 10, 428–429. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, R.; Mainali, R.; Ahmadi, S.; Wang, S.; Singh, R.; Kavanagh, K.; Kitzman, D.W.; Kushugulova, A.; Marotta, F.; Yadav, H. Gut microbiome and aging: Physiological and mechanistic insights. Nutr. Healthy Aging 2018, 4, 267–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinolo, M.A.; Rodrigues, H.G.; Nachbar, R.T.; Curi, R. Regulation of inflammation by short chain fatty acids. Nutrients 2011, 3, 858–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inan, M.S.; Rasoulpour, R.J.; Yin, L.; Hubbard, A.K.; Rosenberg, D.W.; Giardina, C. The luminal short-chain fatty acid butyrate modulates NF-kappaB activity in a human colonic epithelial cell line. Gastroenterology 2000, 118, 724–734. [Google Scholar] [CrossRef]
- Parada Venegas, D.; De la Fuente, M.K.; Landskron, G.; Gonzalez, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short Chain Fatty Acids (SCFAs)-Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, G.; Chen, S.; Deng, B.; Tan, C.; Deng, J.; Zhu, G.; Yin, Y.; Ren, W. Implication of G Protein-Coupled Receptor 43 in Intestinal Inflammation: A Mini-Review. Front. Immunol. 2018, 9, 1434. [Google Scholar] [CrossRef] [PubMed]
- Ragonnaud, E.; Biragyn, A. Gut microbiota as the key controllers of “healthy” aging of elderly people. Immun. Ageing 2021, 18, 2. [Google Scholar] [CrossRef]
- Biagi, E.; Nylund, L.; Candela, M.; Ostan, R.; Bucci, L.; Pini, E.; Nikkila, J.; Monti, D.; Satokari, R.; Franceschi, C.; et al. Through ageing, and beyond: Gut microbiota and inflammatory status in seniors and centenarians. PLoS ONE 2010, 5, e10667. [Google Scholar] [CrossRef]
- Shintouo, C.M.; Mets, T.; Beckwee, D.; Bautmans, I.; Ghogomu, S.M.; Souopgui, J.; Leemans, L.; Meriki, H.D.; Njemini, R. Is inflammageing influenced by the microbiota in the aged gut? A systematic review. Exp. Gerontol. 2020, 141, 111079. [Google Scholar] [CrossRef]
- Fransen, F.; van Beek, A.A.; Borghuis, T.; Aidy, S.E.; Hugenholtz, F.; van der Gaast-de Jongh, C.; Savelkoul, H.F.J.; De Jonge, M.I.; Boekschoten, M.V.; Smidt, H.; et al. Aged Gut Microbiota Contributes to Systemical Inflammaging after Transfer to Germ-Free Mice. Front. Immunol. 2017, 8, 1385. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, A.F.; Dwivedi, G.; O’Gara, F.; Caparros-Martin, J.; Ward, N.C. The gut microbiome and cardiovascular disease: Current knowledge and clinical potential. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H923–H938. [Google Scholar] [CrossRef]
- Yoshida, N.; Yamashita, T.; Hirata, K.I. Gut Microbiome and Cardiovascular Diseases. Diseases 2018, 6, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ettinger, G.; MacDonald, K.; Reid, G.; Burton, J.P. The influence of the human microbiome and probiotics on cardiovascular health. Gut Microbes 2014, 5, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Ahmadmehrabi, S.; Tang, W.H.W. Gut microbiome and its role in cardiovascular diseases. Curr. Opin. Cardiol. 2017, 32, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Troseid, M.; Andersen, G.O.; Broch, K.; Hov, J.R. The gut microbiome in coronary artery disease and heart failure: Current knowledge and future directions. EBioMedicine 2020, 52, 102649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazemian, N.; Mahmoudi, M.; Halperin, F.; Wu, J.C.; Pakpour, S. Gut microbiota and cardiovascular disease: Opportunities and challenges. Microbiome 2020, 8, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Li, X.; Yang, F.; Zhao, R.; Pan, X.; Liang, J.; Tian, L.; Li, X.; Liu, L.; Xing, Y.; et al. Gut Microbiota-Dependent Marker TMAO in Promoting Cardiovascular Disease: Inflammation Mechanism, Clinical Prognostic, and Potential as a Therapeutic Target. Front. Pharmacol. 2019, 10, 1360. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Luo, Y.; Liu, J.P.; Sun, N.; Guo, D.; Cui, L.L.; Zheng, P.P.; Yao, S.M.; Yang, J.F.; Wang, H. Trimethylamine N-Oxide, a Gut Microbiota-Dependent Metabolite, is Associated with Frailty in Older Adults with Cardiovascular Disease. Clin. Interv. Aging 2020, 15, 1809–1820. [Google Scholar] [CrossRef] [PubMed]
- Roncal, C.; Martinez-Aguilar, E.; Orbe, J.; Ravassa, S.; Fernandez-Montero, A.; Saenz-Pipaon, G.; Ugarte, A.; Estrella-Hermoso de Mendoza, A.; Rodriguez, J.A.; Fernandez-Alonso, S.; et al. Trimethylamine-N-Oxide (TMAO) Predicts Cardiovascular Mortality in Peripheral Artery Disease. Sci. Rep. 2019, 9, 15580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leshem, A.; Horesh, N.; Elinav, E. Fecal Microbial Transplantation and Its Potential Application in Cardiometabolic Syndrome. Front. Immunol. 2019, 10, 1341. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhao, F.; Wang, Y.; Chen, J.; Tao, J.; Tian, G.; Wu, S.; Liu, W.; Cui, Q.; Geng, B.; et al. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome 2017, 5, 14. [Google Scholar] [CrossRef] [Green Version]
- Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Cheng, J.; Duncan, A.E.; Kau, A.L.; Griffin, N.W.; Lombard, V.; Henrissat, B.; Bain, J.R.; et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 2013, 341, 1241214. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.F.; Zhang, W.Y.; Wen, Q.; Chen, W.J.; Wang, Z.M.; Chen, J.; Zhu, F.; Liu, K.; Cheng, L.X.; Yang, J.; et al. Fecal microbiota transplantation alleviates myocardial damage in myocarditis by restoring the microbiota composition. Pharmacol. Res. 2019, 139, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, T.S.; Rampelli, S.; Jeffery, I.B.; Santoro, A.; Neto, M.; Capri, M.; Giampieri, E.; Jennings, A.; Candela, M.; Turroni, S.; et al. Mediterranean diet intervention alters the gut microbiome in older people reducing frailty and improving health status: The NU-AGE 1-year dietary intervention across five European countries. Gut 2020, 69, 1218–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berendsen, A.; Santoro, A.; Pini, E.; Cevenini, E.; Ostan, R.; Pietruszka, B.; Rolf, K.; Cano, N.; Caille, A.; Lyon-Belgy, N.; et al. Reprint of: A parallel randomized trial on the effect of a healthful diet on inflammageing and its consequences in European elderly people: Design of the NU-AGE dietary intervention study. Mech. Ageing Dev. 2014, 136–137, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Arai, Y.; Martin-Ruiz, C.M.; Takayama, M.; Abe, Y.; Takebayashi, T.; Koyasu, S.; Suematsu, M.; Hirose, N.; von Zglinicki, T. Inflammation, But Not Telomere Length, Predicts Successful Ageing at Extreme Old Age: A Longitudinal Study of Semi-supercentenarians. EBioMedicine 2015, 2, 1549–1558. [Google Scholar] [CrossRef] [Green Version]
- Goldeck, D.; Pawelec, G.; Norman, K.; Steinhagen-Thiessen, E.; Oettinger, L.; Haehnel, K.; Demuth, I. No strong correlations between serum cytokine levels, CMV serostatus and hand-grip strength in older subjects in the Berlin BASE-II cohort. Biogerontology 2016, 17, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Waaijer, M.E.; Westendorp, R.G.; Goldeck, D.; Gunn, D.A.; Pawelec, G.; Stijntjes, M.; Slagboom, P.E.; Maier, A.B. Assessment of health status by molecular measures in adults ranging from middle-aged to old: Ready for clinical use? Exp. Gerontol. 2017, 87, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.S.; Gubbi, S.; Barzilai, N. Benefits of Metformin in Attenuating the Hallmarks of Aging. Cell Metab. 2020, 32, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Aslam, M.; Ladilov, Y. Emerging Role of cAMP/AMPK Signaling. Cells 2022, 11, 308. [Google Scholar] [CrossRef]
- Gupta, S.C.; Kunnumakkara, A.B.; Aggarwal, S.; Aggarwal, B.B. Inflammation, a Double-Edge Sword for Cancer and Other Age-Related Diseases. Front. Immunol. 2018, 9, 2160. [Google Scholar] [CrossRef]
- Park, S.J.; Ahmad, F.; Philp, A.; Baar, K.; Williams, T.; Luo, H.; Ke, H.; Rehmann, H.; Taussig, R.; Brown, A.L.; et al. Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell 2012, 148, 421–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.L.; Yi, L.; Jin, X.; Liang, X.Y.; Zhou, Y.; Zhang, T.; Xie, Q.; Zhou, X.; Chan, H.; Fu, Y.J.; et al. Resveratrol attenuates vascular endothelial inflammation by inducing autophagy through the cAMP signaling pathway. Autophagy 2013, 9, 2033–2045. [Google Scholar] [CrossRef]
- Diaz-Casado, M.E.; Quiles, J.L.; Barriocanal-Casado, E.; Gonzalez-Garcia, P.; Battino, M.; Lopez, L.C.; Varela-Lopez, A. The Paradox of Coenzyme Q10 in Aging. Nutrients 2019, 11, 2221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochoa, J.J.; Quiles, J.L.; Huertas, J.R.; Mataix, J. Coenzyme Q10 protects from aging-related oxidative stress and improves mitochondrial function in heart of rats fed a polyunsaturated fatty acid (PUFA)-rich diet. J. Gerontol. A Biol. Sci. Med. Sci. 2005, 60, 970–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakazawa, H.; Ikeda, K.; Shinozaki, S.; Yasuhara, S.; Yu, Y.M.; Martyn, J.A.J.; Tompkins, R.G.; Yorozu, T.; Inoue, S.; Kaneki, M. Coenzyme Q10 protects against burn-induced mitochondrial dysfunction and impaired insulin signaling in mouse skeletal muscle. FEBS Open Biol. 2019, 9, 348–363. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, S.A.; Rosenfeldt, F.; Kumar, A.; Dolliner, P.; Filipiak, K.J.; Pella, D.; Alehagen, U.; Steuer, G.; Littarru, G.P.; Symbio, Q. Study Investigators.The effect of coenzyme Q10 on morbidity and mortality in chronic heart failure: Results from Q-SYMBIO: A randomized double-blind trial. JACC Heart Fail. 2014, 2, 641–649. [Google Scholar] [CrossRef]
- Forini, F.; Canale, P.; Nicolini, G.; Iervasi, G. Mitochondria-Targeted Drug Delivery in Cardiovascular Disease: A Long Road to Nano-Cardio Medicine. Pharmaceutics 2020, 12, 1122. [Google Scholar]
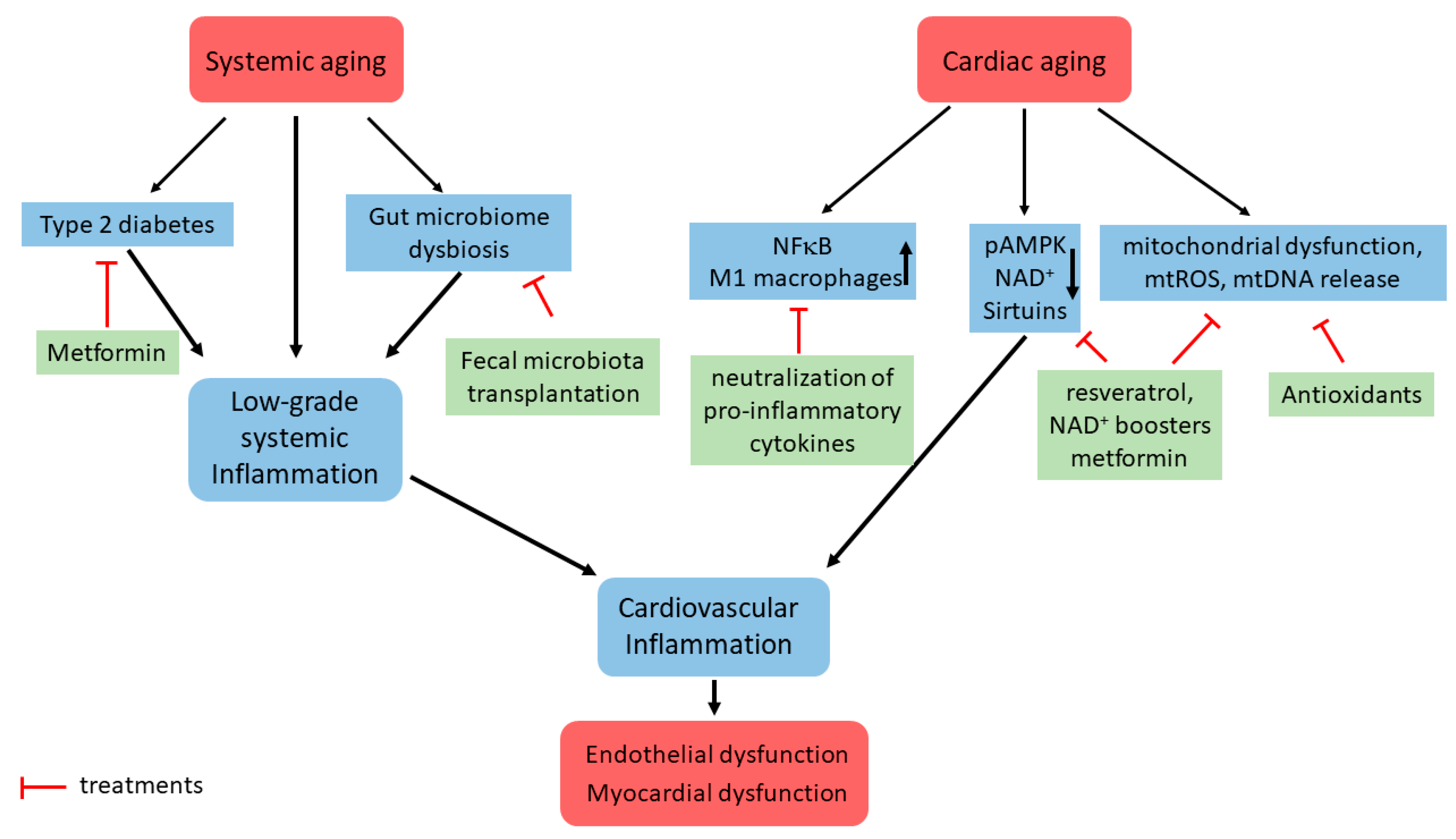
Figure 1.
Schematic representation of some signaling pathways involved in cardiac inflammaging development and potential treatment strategies. Systemic aging leads to low-grade systemic inflammation, which among numerous other mechanisms includes type 2 diabetes and gut microbiome dysbiosis. Cardiac aging is linked with (i) increased expression of pro-inflammatory factors and immune cell infiltration, (ii) downregulation of main energy regulating mechanisms, such as pAMPK and sirtuin, and (iii) mitochondrial dysfunction. Together with the low-grade systemic inflammation, it leads to cardiac inflammation and endothelial/myocardial dysfunction. The potential treatment strategies are shown.
Figure 1.
Schematic representation of some signaling pathways involved in cardiac inflammaging development and potential treatment strategies. Systemic aging leads to low-grade systemic inflammation, which among numerous other mechanisms includes type 2 diabetes and gut microbiome dysbiosis. Cardiac aging is linked with (i) increased expression of pro-inflammatory factors and immune cell infiltration, (ii) downregulation of main energy regulating mechanisms, such as pAMPK and sirtuin, and (iii) mitochondrial dysfunction. Together with the low-grade systemic inflammation, it leads to cardiac inflammation and endothelial/myocardial dysfunction. The potential treatment strategies are shown.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Barcena, M.L.; Aslam, M.; Pozdniakova, S.; Norman, K.; Ladilov, Y. Cardiovascular Inflammaging: Mechanisms and Translational Aspects. Cells 2022, 11, 1010. https://doi.org/10.3390/cells11061010
AMA Style
Barcena ML, Aslam M, Pozdniakova S, Norman K, Ladilov Y. Cardiovascular Inflammaging: Mechanisms and Translational Aspects. Cells. 2022; 11(6):1010. https://doi.org/10.3390/cells11061010
Chicago/Turabian StyleBarcena, Maria Luisa, Muhammad Aslam, Sofya Pozdniakova, Kristina Norman, and Yury Ladilov. 2022. "Cardiovascular Inflammaging: Mechanisms and Translational Aspects" Cells 11, no. 6: 1010. https://doi.org/10.3390/cells11061010
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.