
Development and Experimental Validation of Regularized Machine Learning Models Detecting New, Structurally Distinct Activators of PXR

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Sets
2.2. Data Preprocessing
2.3. Feature Calculation
2.4. Experimental Approaches
2.4.1. Chemicals and Reagents
2.4.2. Compound Purity Checks
2.4.3. Cell Culture
2.4.4. Cell Viability
2.4.5. Plasmid Constructs
2.4.6. Transient Transfections, Promoter Reporter Gene, and Mammalian Two-Hybrid Assays
2.4.7. RNA Preparation and Reverse Transcription Quantitative Real-Time PCR Analysis
3. Results and Discussion
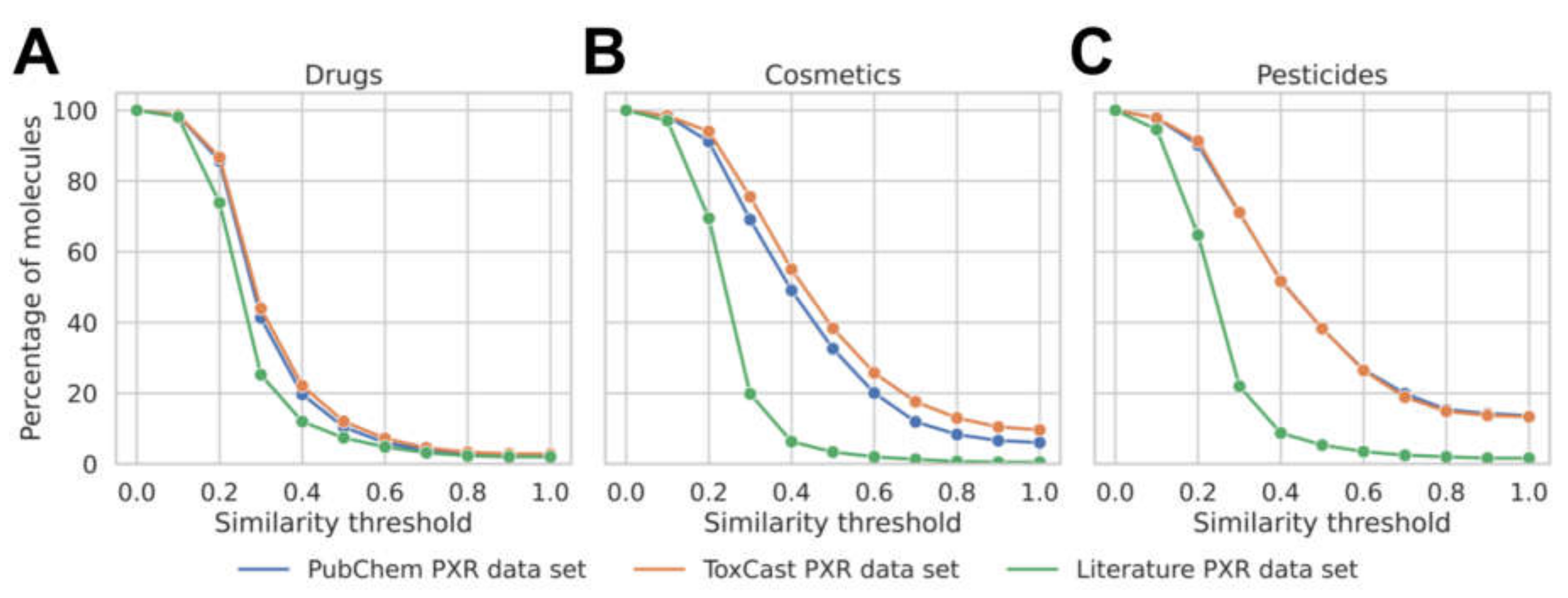
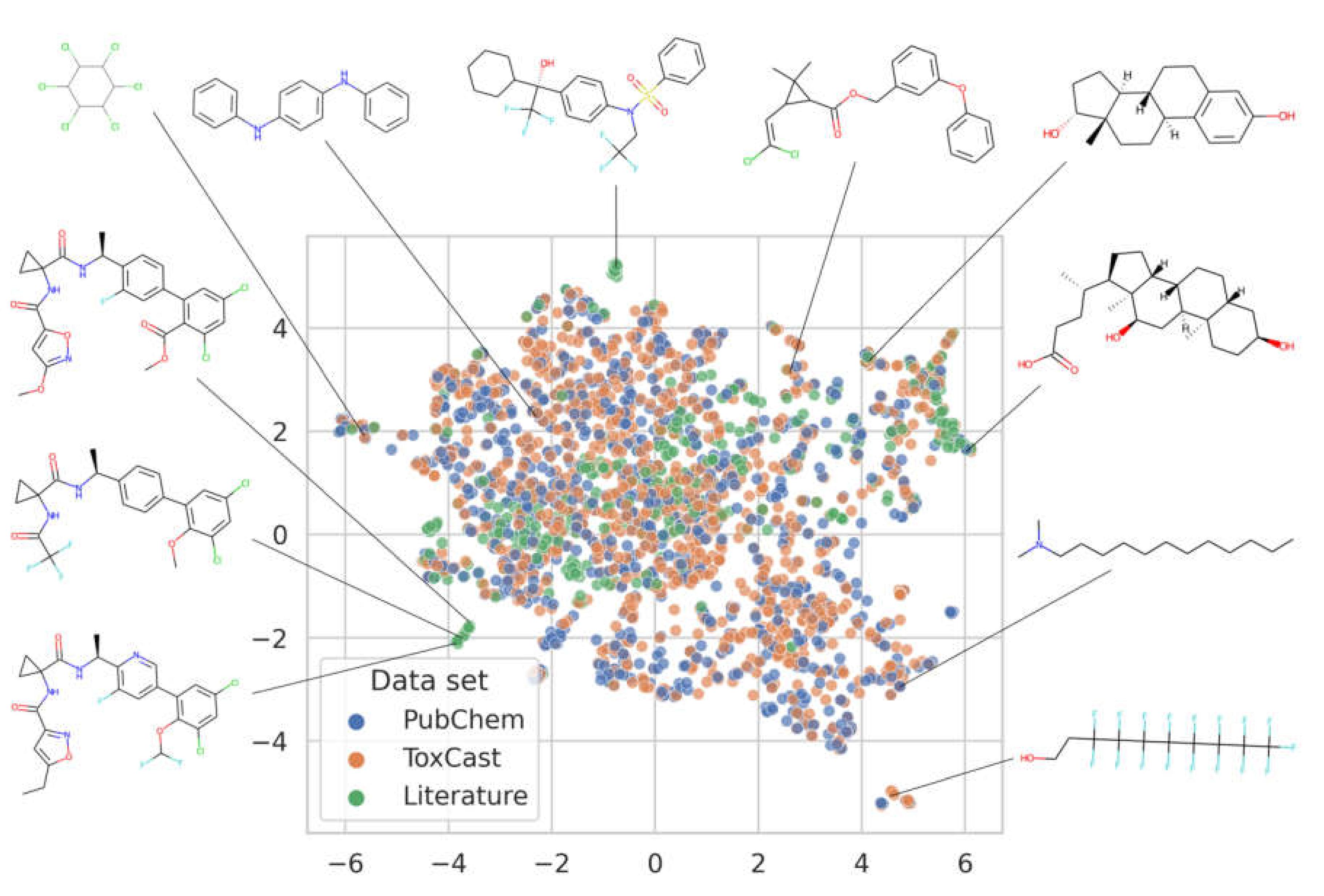
3.1. Analysis of the Data Available for Model Development
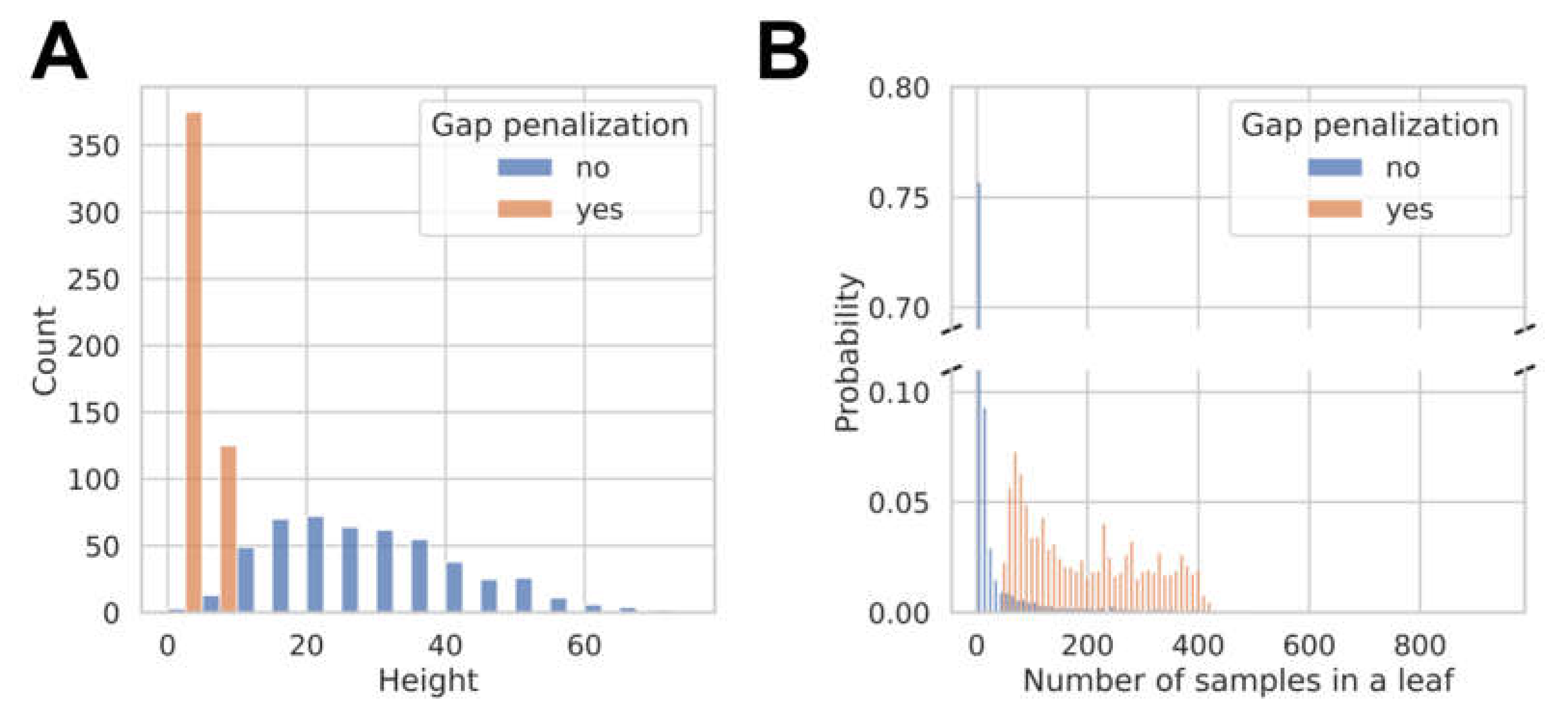
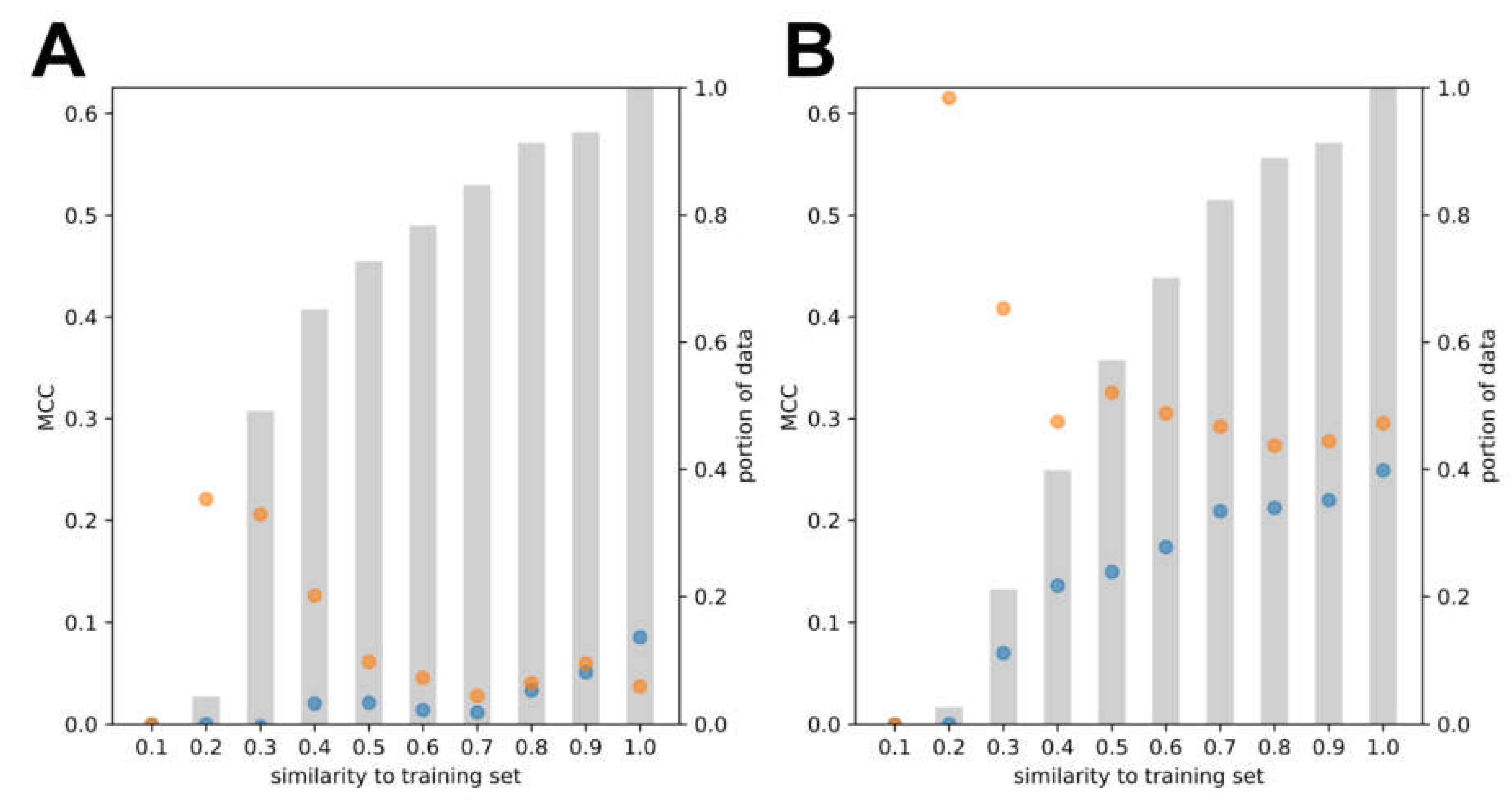
3.2. Model Development and Internal Validation
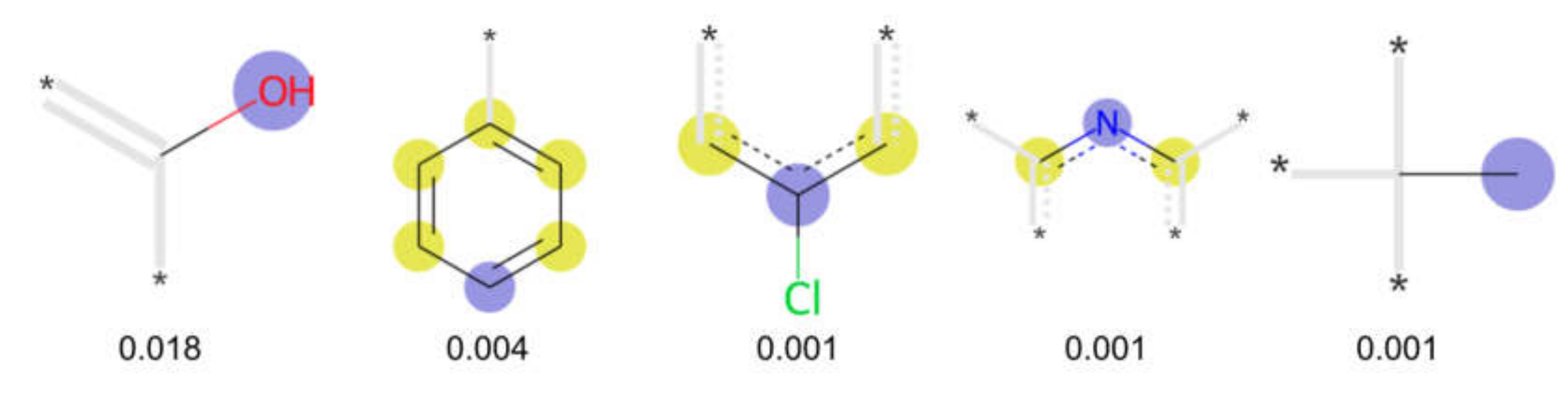
3.3. Analysis of Feature Importance
3.4. Prospective Screening for PXR Activators
- High confidence in predictions: Any selected compound must be predicted as a PXR activator by all of the three RF and three SVM models.
- Novelty: The selected compounds must be structurally distinct to any known PXR ligands. This means that, at the time of selection, chemical structure similarity searches with CAS Scifinder did not result in the retrieval of any known, structurally related PXR ligands. More specifically, a minimum similarity threshold of 70 was used for the searches in CAS Scifinder, meaning that the platform would report literature even for rather distantly related PXR agonists.
- Purchasability: The selected compounds must be available from MolPort in sufficient quantities (5 mg) and at moderate costs.
Experimental Validation
3.5. Hit Follow-Up and SAR Analysis
3.6. Characterization of Prototypical Compounds
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lehmann, J.M.; McKee, D.D.; A Watson, M.; Willson, T.M.; Moore, J.T.; A Kliewer, S. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J. Clin. Investig. 1998, 102, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Ihunnah, C.A.; Jiang, M.; Xie, W. Nuclear receptor PXR, transcriptional circuits and metabolic relevance. Biochim. et Biophys. Acta (BBA) - Mol. Basis Dis. 2011, 1812, 956–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khandelwal, A.; Krasowski, M.; Reschly, E.J.; Sinz, M.W.; Swaan, P.; Ekins, S. Machine learning methods and docking for predicting human pregnane X receptor activation. Chem. Res. Toxicol. 2008, 21, 1457–1467. [Google Scholar] [CrossRef] [Green Version]
- Kliewer, S.A.; Willson, T.M. Regulation of xenobiotic and bile acid metabolism by the nuclear pregnane X receptor. J. Lipid Res. 2002, 43, 359–364. [Google Scholar] [CrossRef]
- Willson, T.M.; Kliewer, S.A. PXR, CAR and drug metabolism. Nat. Rev. Drug Discov. 2002, 1, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, J.; Chong, L.W.; Downes, M.; Barish, G.D.; Coulter, S.; Liddle, C.; Lee, C.-H.; Evans, R.M. Pregnane X receptor pre-vents hepatorenal toxicity from cholesterol metabolites. Proc. Natl. Acad. Sci. USA 2005, 102, 2198–2203. [Google Scholar] [CrossRef] [Green Version]
- Teotico, D.G.; Bischof, J.J.; Peng, L.; Kliewer, S.A.; Redinbo, M.R. Structural basis of human pregnane X receptor activation by the hops constituent colupulone. Mol. Pharmacol. 2008, 74, 1512–1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeshita, A.; Taguchi, M.; Koibuchi, N.; Ozawa, Y. Putative role of the orphan nuclear receptor SXR (steroid and xenobiotic receptor) in the mechanism of CYP3A4 inhibition by xenobiotics. J. Biol. Chem. 2002, 277, 32453–32458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coumoul, X.; Diry, M.; Barouki, R. PXR-Dependent induction of human CYP3A4 gene expression by organochlorine pesticides. Biochem. Pharmacol. 2002, 64, 1513–1519. [Google Scholar] [CrossRef]
- Lemaire, G.; Mnif, W.; Pascussi, J.-M.; Pillon, A.; Rabenoelina, F.; Fenet, H.; Gomez, E.; Casellas, C.; Nicolas, J.-C.; Cavaillès, V.; et al. Identification of new human pregnane X receptor ligands among pesticides using a stable reporter cell system. Toxicol. Sci. 2006, 91, 501–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abass, K.; Lämsä, V.; Reponen, P.; Küblbeck, J.; Honkakoski, P.; Mattila, S.; Pelkonen, O.; Hakkola, J. Characterization of human cytochrome P450 induction by pesticides. Toxicology 2012, 294, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Petrovic, V.; Teng, S.; Piquette-Miller, M. Regulation of drug transporters during infection and inflammation. Mol. Interv. 2007, 7, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Shah, Y.M.; Ma, X.; Morimura, K.; Kim, I.; Gonzalez, F.J. Pregnane X receptor activation ameliorates DSS-induced inflammatory bowel disease via inhibition of NF-κB target gene expression. Am. J. Physiol.-Gastrointest. Liver Physiol. 2007, 292, G1114–G1122. [Google Scholar] [CrossRef]
- Langmade, S.J.; Gale, S.E.; Frolov, A. Pregnane X receptor (PXR) activation: A mechanism for neuroprotection in a mouse model of Niemann–Pick C disease. Proc. Natl. Acad. Sci. USA 2006, 103, 13807–13812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinz, M.; Kim, S.; Zhu, Z.; Chen, T.; Anthony, M.; Dickinson, K.; Rodrigues, A.D. Evaluation of 170 xenobiotics as transactivators of human pregnane X receptor (hPXR) and correlation to known CYP3A4 drug interactions. Curr. Drug Metab. 2006, 7, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.-D.; Olson, S.H.; Balkovec, J.M.; Zhu, Y.; Royo, I.; Yabut, J.; Evers, R.; Tan, E.Y.; Tang, W.; Hartley, D.P.; et al. Attenuating pregnane X receptor (PXR) activation: A molecular modelling approach. Xenobiotica 2007, 37, 124–138. [Google Scholar] [CrossRef]
- Pascussi, J.M.; Drocourt, L.; Fabre, J.M.; Maurel, P.; Vilarem, M.J. Dexamethasone induces pregnane X receptor and retinoid X receptor-alpha expression in human hepatocytes: Synergistic increase of CYP3A4 induction by pregnane X receptor activators. Mol. Pharmacol. 2000, 58, 361–372. [Google Scholar] [CrossRef] [Green Version]
- Synold, T.; Dussault, I.; Forman, B.M. The orphan nuclear receptor SXR coordinately regulates drug metabolism and efflux. Nat. Med. 2001, 7, 584–590. [Google Scholar] [CrossRef]
- Shukla, S.J.; Sakamuru, S.; Huang, R.; Moeller, T.A.; Shinn, P.; Vanleer, D.; Auld, D.S.; Austin, C.P.; Xia, M. Identification of clinically used drugs that activate pregnane X receptors. Drug Metab. Dispos. 2011, 39, 151–159. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, B.; Hodgson, E.; Liddle, C. The orphan human pregnane X receptor mediates the transcriptional activation of CYP3A4 by rifampicin through a distal enhancer module. Mol. Pharmacol. 1999, 56, 1329–1339. [Google Scholar] [CrossRef] [Green Version]
- Luo, G.; Cunningham, M.; Kim, S.; Burn, T.; Lin, J.; Sinz, M.; Hamilton, G.; Rizzo, C.; Jolley, S.; Gilbert, D.; et al. CYP3A4 induction by drugs: Correlation between a pregnane X receptor reporter gene assay and CYP3A4 expression in human hepatocytes. Drug Metab. Dispos. 2002, 30, 795–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahi, J.; Milad, M.A.; Zheng, X.; Rose, K.A.; Wang, H.; Stilgenbauer, L.; Gilbert, D.; Jolley, S.; Stern, R.H.; LeCluyse, E.L. Avasimibe induces CYP3A4 and multiple drug resistance protein 1 gene expression through activation of the pregnane X receptor. J. Pharmacol. Exp. Ther. 2003, 306, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.B.; Goodwin, B.; Jones, S.A.; Wisely, G.B.; Serabjit-Singh, C.J.; Willson, T.M.; Collins, J.L.; Kliewer, S.A. St. John’s wort induces hepatic drug metabolism through activation of the pregnane X receptor. Proc. Natl. Acad. Sci. USA 2000, 97, 7500–7502. [Google Scholar] [CrossRef] [Green Version]
- Piscitelli, S.C.; Burstein, A.H.; Chaitt, D.; Alfaro, R.M.; Falloon, J. Indinavir concentrations and St John’s wort. Lancet 2000, 355, 547–548. [Google Scholar] [CrossRef]
- Mathijssen, R.H.J.; Verweij, J.; de Bruijn, P.; Loos, W.J.; Sparreboom, A. Effects of St. John’s wort on irinotecan metabolism. J. Natl. Cancer Inst. 2002, 94, 1247–1249. [Google Scholar] [CrossRef] [Green Version]
- Watkins, R.E.; Wisely, G.B.; Moore, L.B.; Collins, J.L.; Lambert, M.H.; Williams, S.P.; Willson, T.M.; Kliewer, S.A.; Redinbo, M.R. The human nuclear xenobiotic receptor PXR: Structural determinants of directed promiscuity. Science 2001, 292, 2329–2333. [Google Scholar] [CrossRef]
- Hall, A.; Chanteux, H.; Ménochet, K.; Ledecq, M.; Schulze, M.-S.E.D. Designing out PXR activity on drug discovery projects: A review of structure-based methods, empirical and computational approaches. J. Med. Chem. 2021, 64, 6413–6522. [Google Scholar] [CrossRef]
- Ekins, S.; Erickson, J.A. A pharmacophore for human pregnane X receptor ligands. Drug Metab. Dispos. 2002, 30, 96–99. [Google Scholar] [CrossRef] [Green Version]
- Bachmann, K.; Patel, H.; Batayneh, Z.; Slama, J.; White, D.; Posey, J.; Ekins, S.; Gold, D.; Sambucetti, L. PXR and the regulation of apoA1 and HDL-cholesterol in rodents. Pharmacol. Res. 2004, 50, 237–246. [Google Scholar] [CrossRef]
- Schuster, D.; Langer, T. The identification of ligand features essential for PXR activation by pharmacophore modeling. J. Chem. Inf. Model. 2005, 36, 431–439. [Google Scholar] [CrossRef]
- Lemaire, G.; Benod, C.; Nahoum, V.; Pillon, A.; Boussioux, A.-M.; Guichou, J.-F.; Subra, G.; Pascussi, J.-M.; Bourguet, W.; Chavanieu, A.; et al. Discovery of a highly active ligand of human pregnane X receptor: A case study from pharmacophore modeling and virtual screening to “in vivo” biological activity. Mol. Pharmacol. 2007, 72, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, K.; Ranade, A.; Venkataramanan, R.; Strom, S.; Chupka, J.; Ekins, S.; Schuetz, E.G.; Bachmann, K. A comprehensive in vitro and in silico analysis of antibiotics that activate pregnane X receptor and induce CYP3A4 in liver and intestine. Drug Metab. Dispos. 2008, 36, 1689–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.-N.; Shih, Y.-H.; Ding, Y.-L.; Leong, M.K. Predicting activation of the promiscuous human pregnane X receptor by pharmacophore ensemble/support vector machine approach. Chem. Res. Toxicol. 2011, 24, 1765–1778. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Kang, H.; Tang, K.; Liu, Q.; Cao, Z.; Zhu, R. Screening ingredients from herbs against pregnane X receptor in the study of inductive herb-drug interactions: Combining pharmacophore and docking-based rank aggregation. BioMed Res. Int. 2015, 657159. [Google Scholar] [CrossRef]
- Ekins, S.; Chang, C.; Mani, S.; Krasowski, M.D.; Reschly, E.J.; Iyer, M.; Kholodovych, V.; Ai, N.; Welsh, W.J.; Sinz, M.; et al. Human pregnane X receptor antagonists and agonists define molecular requirements for different binding sites. Mol. Pharmacol. 2007, 72, 592–603. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, M.; Chen, T. Differential regulation of CYP3A4 promoter activity by a new class of natural product derivatives binding to pregnane X receptor. Biochem. Pharmacol. 2013, 86, 824–835. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Li, L.; Kim, G.; Ekins, S.; Wang, H.; Swaan, P.W. Identification and validation of novel human pregnane X receptor activators among prescribed drugs via ligand-based virtual screening. Drug Metab. Dispos. 2010, 39, 337–344. [Google Scholar] [CrossRef] [Green Version]
- Ekins, S.; Kortagere, S.; Iyer, M.; Reschly, E.J.; Lill, M.A.; Redinbo, M.R.; Krasowski, M.D. Challenges predicting ligand-receptor interactions of promiscuous proteins: The nuclear receptor PXR. PLoS Comput. Biol. 2009, 5, e1000594. [Google Scholar] [CrossRef] [Green Version]
- Kortagere, S.; Chekmarev, D.; Welsh, W.J.; Ekins, S. Hybrid scoring and classification approaches to predict human pregnane X receptor activators. Pharm. Res. 2009, 26, 1001–1011. [Google Scholar] [CrossRef] [Green Version]
- Yin, C.; Yang, X.; Wei, M.; Liu, H. Predictive models for identifying the binding activity of structurally diverse chemicals to human pregnane X receptor. Environ. Sci. Pollut. Res. 2017, 24, 20063–20071. [Google Scholar] [CrossRef] [PubMed]
- Dybdahl, M.; Nikolov, N.G.; Wedebye, E.B.; Jónsdóttir, S.; Niemelä, J.R. QSAR model for human pregnane X receptor (PXR) binding: Screening of environmental chemicals and correlations with genotoxicity, endocrine disruption and teratogenicity. Toxicol. Appl. Pharmacol. 2012, 262, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Xia, M.; Huang, R.; Nikolov, N.G.; Wedebye, E.B.; Dybdahl, M. QSAR development and profiling of 72,524 REACH substances for PXR activation and CYP3A4 induction. Comput. Toxicol. 2017, 1, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, M. In silico tools to aid risk assessment of endocrine disrupting chemicals. Toxicology 2004, 205, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Matter, H.; Anger, L.T.; Giegerich, C.; Güssregen, S.; Hessler, G.; Baringhaus, K.-H. Development of in silico filters to predict activation of the pregnane X receptor (PXR) by structurally diverse drug-like molecules. Bioorg. Med. Chem. 2012, 20, 5352–5365. [Google Scholar] [CrossRef]
- Gadaleta, D.; Manganelli, S.; Roncaglioni, A.; Toma, C.; Benfenati, E.; Mombelli, E. QSAR modeling of ToxCast assays relevant to the molecular initiating events of AOPs leading to hepatic steatosis. J. Chem. Inf. Model. 2018, 58, 1501–1517. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-M.; Chang, M.-J.; Yang, X.-S.; Han, X. In silico investigation of agonist activity of a structurally diverse set of drugs to hPXR using HM-BSM and HM-PNN. J. Huazhong Univ. Sci. Technol. 2016, 36, 463–468. [Google Scholar] [CrossRef]
- Rathod, V.; Belekar, V.; Garg, P.; Sangamwar, A.T. Classification of human pregnane X receptor (hPXR) activators and non-activators by machine learning techniques: A multifaceted approach. Comb. Chem. High Throughput Screen. 2016, 19, 307–318. [Google Scholar] [CrossRef]
- Yoshida, S.; Yamashita, F.; Itoh, T.; Hashida, M. Structure-activity relationship modeling for predicting interactions with pregnane X receptor by recursive partitioning. Drug Metab. Pharmacokinet. 2012, 27, 506–512. [Google Scholar] [CrossRef]
- Ung, C.Y.; Li, H.; Yap, C.W.; Chen, Y.Z. In silico prediction of pregnane X receptor activators by machine learning ap-proaches. Mol. Pharmacol. 2007, 71, 158–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, H.; Wang, Y.; Zeng, X.; Wang, X.; Liu, Y.; Yin, J.; He, H.; Zhu, F.; Li, Z. In silico identification of human pregnane X receptor activators from molecular descriptors by machine learning approaches. Chemom. Intell. Lab. Syst. 2012, 118, 271–279. [Google Scholar] [CrossRef]
- AbdulHameed, M.D.M.; Ippolito, D.L.; Wallqvist, A. Predicting rat and human pregnane X receptor activators using Bayesian classification models. Chem. Res. Toxicol. 2016, 29, 1729–1740. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Tian, S.; Li, Y.; Li, D.; Yu, H.; Zhen, X.; Hou, T. Absorption, distribution, metabolism, excretion, and toxicity evaluation in drug discovery. 14. Prediction of human pregnane X receptor activators by using naive Bayesian classification technique. Chem. Res. Toxicol. 2015, 28, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395, PMCID:PMC7778930. [Google Scholar] [CrossRef] [PubMed]
- PubChem Open Chemistry Database at the National Institutes of Health (NIH), U.S. National Library of Medicine. Available online: https://pubchem.ncbi.nlm.nih.gov/ (accessed on 7 January 2021).
- U.S. EPA ToxCast & Tox21 Summary Files from Invitrodb_v2. Available online: http://www2.epa.gov/chemical-research/toxicity-forecaster-toxcasttm-data (accessed on 19 July 2018).
- Chemical Identifier Resolver Beta 3. Available online: http://cactus.nci.nih.gov/chemical/structure (accessed on 1 December 2018).
- The DrugBank Database. Available online: https://www.drugbank.ca (accessed on 25 July 2018).
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank Database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Lowe, C.N.; Williams, A.J. Enabling high-throughput searches for multiple chemical data using the U.S.-EPA CompTox Chemicals Dashboard. J. Chem. Inf. Model. 2021, 61, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Cosmos DB. Available online: https://comptox.epa.gov/dashboard/chemical_lists/COSMOSDB (accessed on 2 February 2021).
- Pesticide Chemical Search Database. Available online: https://comptox.epa.gov/dashboard/chemical_lists/EPAPCS (accessed on 2 February 2021).
- MolPort in-Stock Compounds. Available online: https://www.molport.com (accessed on 6 August 2018).
- Swain, M. MolVS: Molecule Validation and Standardization, Version 0.1.1; 2018. Available online: https://github.com/mcs07/MolVS (accessed on 26 February 2022).
- Landrum, G. RDKit: Open-Source Cheminformatics Software, Version 2018_03; 2018. Available online: https://www.rdkit.org/ (accessed on 26 February 2022).
- Bitter, A.; Rümmele, P.; Klein, K.; Kandel, B.A.; Rieger, J.K.; Nüssler, A.K.; Zanger, U.M.; Trauner, M.; Schwab, M.; Burk, O. Pregnane X receptor activation and silencing promote steatosis of human hepatic cells by distinct lipogenic mechanisms. Arch. Toxicol. 2015, 89, 2089–2103. [Google Scholar] [CrossRef] [PubMed]
- Antherieu, S.; Chesne, C.; Li, R.; Camus, S.; Lahoz, A.; Picazo, L.; Turpeinen, M.; Tolonen, A.; Uusitalo, J.; Guguen-Guillouzo, C.; et al. Stable expression, activity, and inducibility of cytochromes P450 in differentiated HepaRG Cells. Drug Metab. Dispos. 2009, 38, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Burk, O.; Tegude, H.; Koch, I.; Hustert, E.; Wolbold, R.; Glaeser, H.; Klein, K.; Fromm, M.F.; Nuessler, A.K.; Neuhaus, P.; et al. Molecular mechanisms of polymorphic CYP3A7 expression in adult human liver and intestine. J. Biol. Chem. 2002, 277, 24280–24288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathäs, M.; Burk, O.; Qiu, H.; Nußhag, C.; Gödtel-Armbrust, U.; Baranyai, D.; Deng, S.; Römer, K.; Nem, D.; Windshügel, B.; et al. Evolutionary history and functional characterization of the amphibian xenosensor CAR. Mol. Endocrinol. 2012, 26, 14–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burk, O.; Arnold, K.A.; Nussler, A.K.; Schaeffeler, E.; Efimova, E.; Avery, B.A.; Avery, M.A.; Fromm, M.F.; Eichelbaum, M. Antimalarial artemisinin drugs induce cytochrome P450 and MDR1 expression by activation of xenosensors pregnane X receptor and constitutive androstane receptor. Mol. Pharmacol. 2005, 67, 1954–1965. [Google Scholar] [CrossRef] [Green Version]
- Arnold, K.A.; Eichelbaum, M.; Burk, O. Alternative splicing affects the function and tissue-specific expression of the human constitutive androstane receptor. Nucl. Recept. 2004, 2, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burk, O.; Kuzikov, M.; Kronenberger, T.; Jeske, J.; Keminer, O.; Thasler, W.E.; Schwab, M.; Wrenger, C.; Windshügel, B. Identification of approved drugs as potent inhibitors of pregnane X receptor activation with differential receptor interaction profiles. Arch. Toxicol. 2018, 92, 1435–1451. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Faucette, S.; Sueyoshi, T.; Moore, R.; Ferguson, S.; Negishi, M.; LeCluyse, E. A novel distal enhancer module regulated by pregnane X receptor/constitutive androstane receptor is essential for the maximal induction of CYP2B6 gene expression. J. Biol. Chem. 2003, 278, 14146–14152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hustert, E.; Zibat, A.; Presecan-Siedel, E.; Eiselt, R.; Mueller, R.; Fuss, C.; Brehm, I.; Brinkmann, U.; Eichelbaum, M.; Wojnowski, L.; et al. Natural protein variants of pregnane X receptor with altered transactivation activity toward CYP3A4. Drug Metab. Dispos. 2001, 29, 1454–1459. [Google Scholar]
- Burk, O.; Kronenberger, T.; Keminer, O.; Lee, S.M.L.; Schiergens, T.S.; Schwab, M.; Windshügel, B. Nelfinavir and its active metabolite M8 are partial agonists and competitive antagonists of the human pregnane X receptor. Mol. Pharmacol. 2021, 99, 184–196. [Google Scholar] [CrossRef] [PubMed]
- Jeske, J.; Windshügel, B.; Thasler, W.E.; Schwab, M.; Burk, O. Human pregnane X receptor is activated by dibenzazepine carbamate-based inhibitors of constitutive androstane receptor. Arch. Toxicol. 2017, 2, 2375–2390. [Google Scholar] [CrossRef]
- Hoffart, E.; Ghebreghiorghis, L.; Nussler, A.K.; Thasler, W.E.; Weiss, T.S.; Schwab, M.; Burk, O. Effects of atorvastatin metabolites on induction of drug-metabolizing enzymes and membrane transporters through human pregnane X receptor. Br. J. Pharmacol. 2012, 165, 1595–1608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McInnes, L.; Healy, J.; Melville, J. UMAP: Uniform manifold approximation and projection for dimension reduction. arXiv 2018, arXiv:1802.03426. [Google Scholar] [CrossRef]
- Küblbeck, J.; Jyrkkärinne, J.; Poso, A.; Turpeinen, M.; Sippl, W.; Honkakoski, P.; Windshügel, B. Discovery of substituted sulfonamides and thiazolidin-4-one derivatives as agonists of human constitutive androstane receptor. Biochem. Pharmacol. 2008, 76, 1288–1297. [Google Scholar] [CrossRef] [PubMed]
- Pissios, P.; Tzameli, I.; Kushner, P.J.; Moore, D.D. Dynamic stabilization of nuclear receptor ligand binding domains by hormone or corepressor binding. Mol. Cell 2000, 6, 245–253. [Google Scholar] [CrossRef]
- Lin, W.; Wang, Y.-M.; Chai, S.C.; Lv, L.; Zheng, J.; Wu, J.; Zhang, Q.; Wang, Y.-D.; Griffin, P.R.; Chen, T. SPA70 is a potent antagonist of human pregnane X receptor. Nat. Commun. 2017, 8, 741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sueyoshi, T.; Kawamoto, T.; Zelko, I.; Honkakoski, P.; Negishi, M. The repressed nuclear receptor CAR responds to Phenobarbital in activating the human CYP2B6 Gene. J. Biol. Chem. 1999, 274, 6043–6046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drocourt, L.; Ourlin, J.-C.; Pascussi, J.-M.; Maurel, P.; Vilarem, M.-J. Expression of CYP3A4, CYP2B6, and CYP2C9 is regulated by the vitamin D receptor pathway in primary human hepatocytes. J. Biol. Chem. 2002, 277, 25125–25132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, J.; Plummer, S.M.; Rode, A.; Scheer, N.; Bower, C.C.; Vogel, O.; Henderson, C.J.; Wolf, C.R.; Elcombe, C.R. Human constitutive androstane receptor (CAR) and pregnane X receptor (PXR) support the hypertrophic but not the hyperplastic response to the murine nongenotoxic hepatocarcinogens phenobarbital and chlordane in vivo. Toxicol. Sci. 2010, 116, 452–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PubChem PXR Data Set | ToxCast PXR Data Set | Literature PXR Data Set | |
|---|---|---|---|
| Initial data set | 2864 | 3626 | 434 |
| Missing identifier (CID/CAS number) or SMILES | 14 | 308 | 0 |
| Inconclusive activity | 488 | 776 | 0 |
| MW ≤ 200 Da | 1219 | 1339 | 16 |
| Presence of inorganic elements | 23 | 2 | 0 |
| Contradicting class labels | 24 | 5 | 0 |
| Duplicates | 155 | 17 | 9 |
| Final PXR data set | 941 (A: 202, N: 739) 1 | 1179 (A: 642, N: 537) | 409 (A: 250, N: 159) |
| PubChem PXR Data Set | ToxCast PXR Data Set | Literature PXR Data Set | |
|---|---|---|---|
| No. compounds | 941 | 1179 | 409 |
| No. scaffolds | 387 | 470 | 259 |
| No. drugs scaffolds | 187 (4%) 1 | 224 (4%) | 145 (3%) |
| No. cosmetics scaffolds | 108 (15%) | 154 (22%) | 36 (5%) |
| No. pesticides scaffolds | 170 (25%) | 171 (25%) | 51 (7%) |
| Model | Cross-Validation on the PubChem PXR Data Set | Testing on the | ||||||
|---|---|---|---|---|---|---|---|---|
| ToxCast PXR Data Set | Literature PXR Data Set | |||||||
| ML Algorithm | Feature Set(s) | Gap-Penalization 2 | MCC 3 | AUC 3 | MCC | AUC | MCC | AUC |
| RF | PC | no | 0.43 (±0.04) | 0.83 (±0.04) | 0.41 | 0.82 | 0.10 | 0.59 |
| yes | 0.43 (±0.05) | 0.80 (±0.04) | 0.46 | 0.81 | 0.21 | 0.57 | ||
| FP | no | 0.45 (±0.07) | 0.82 (±0.03) | 0.25 | 0.75 | 0.09 | 0.55 | |
| yes | 0.31 (±0.04) | 0.76 (±0.01) | 0.28 | 0.72 | 0.02 | 0.55 | ||
| PC + FP | no | 0.48 (±0.06) | 0.83 (±0.03) | 0.35 | 0.81 | 0.03 | 0.56 | |
| yes | 0.42 (±0.06) | 0.81 (±0.03) | 0.47 | 0.82 | 0.24 | 0.59 | ||
| SVM | PC | no | 0.41 (±0.06) | 0.80 (±0.03) | 0.40 | 0.77 | 0.14 | 0.56 |
| yes | 0.41 (±0.06) | 0.80 (±0.02) | 0.44 | 0.81 | 0.13 | 0.56 | ||
| FP | no | 0.46 (±0.07) | 0.82 (±0.03) | 0.31 | 0.77 | 0.00 | 0.54 | |
| yes | 0.32 (±0.06) | 0.77 (±0.03) | 0.34 | 0.74 | 0.06 | 0.52 | ||
| PC + FP | no | 0.48 (±0.10) | 0.86 (±0.03) | 0.44 | 0.82 | 0.00 | 0.55 | |
| yes | 0.42 (±0.03) | 0.81 (±0.02) | 0.45 | 0.82 | 0.15 | 0.55 | ||
| ID | Hit Compounds | Nearest Neighbor in Training Set | Similarity of Hit Compound to Nearest Neighbor | Cluster ID | Measured Activity 1 | S.D. 2 |
|---|---|---|---|---|---|---|
| 1 |  MolPort-001-946-370 |  PubChem | 0.57 | A | 0.15 | 1.09 |
| 2 |  MolPort-009-220-213 |  ToxCast | 0.52 | A | 1.41 | 1.59 |
| 3 |  MolPort-003-820-268 |  PubChem | 0.47 | A | 9.41 | 4.72 |
| 4 |  MolPort-001-823-879 |  ToxCast | 0.51 | A | 1.59 | 1.66 |
| 5 |  MolPort-001-529-219 |  PubChem | 0.48 | A | 1.10 | 0.02 |
| 6 |  MolPort-001-545-599 |  PubChem | 0.45 | A | 2.78 | 0.80 |
| 7 |  MolPort-000-993-714 |  PubChem | 0.54 | A | 8.65 | 7.57 |
| 8 |  MolPort-000-993-856 |  PubChem | 0.59 | A | 4.25 | 1.97 |
| 9 |  MolPort-001-838-155 |  PubChem | 0.71 | A | 32.77 | 13.01 |
| 10 |  MolPort-002-238-843 |  PubChem | 0.52 | A | 5.88 | 0.38 |
| 11 |  MolPort-000-279-714 |  ToxCast | 0.47 | A | 33.00 | 19.54 |
| 12 |  MolPort-020-102-538 |  ToxCast | 0.53 | B | 17.00 | 6.15 |
| 13 |  MolPort-027-674-395 |  ToxCast | 0.44 | B | 28.00 | 4.97 |
| 14 |  MolPort-027-691-387 |  ToxCast | 0.37 | B | 0.34 | 2.18 |
| 15 |  MolPort-027-933-109 |  ToxCast | 0.52 | B | 26.50 | 8.41 |
| 16 |  MolPort-035-741-775 |  ToxCast | 0.44 | B | 4.10 | 0.46 |
| 17 |  MolPort-003-881-027 |  ToxCast | 0.33 | B | 44.80 | 14.97 |
| 18 |  MolPort-003-881-070 |  ToxCast | 0.34 | B | 39.63 | 19.31 |
| 19 |  MolPort-003-881-625 |  ToxCast | 0.32 | B | 9.54 | 1.57 |
| 20 |  MolPort-002-237-349 |  ToxCast | 0.34 | B | 4.83 | 2.00 |
| 21 |  MolPort-000-196-136 |  ToxCast | 0.34 | B | 7.21 | 3.28 |
| 22 |  MolPort-000-431-925 |  ToxCast | 0.26 | singleton | 91.00 | 36.91 |
| 23 |  MolPort-000-690-666 |  PubChem | 0.30 | singleton | 11.08 | 5.40 |
| 24 |  MolPort-005-280-909 |  ToxCast | 0.58 | singleton | 1.34 | 0.71 |
| 25 |  MolPort-006-630-224 |  PubChem | 0.36 | singleton | 136.87 | 28.01 |
| 26 |  MolPort-001-619-443 |  ToxCast | 0.46 | singleton | 20.11 | 9.51 |
| 27 |  MolPort-002-547-842 |  PubChem | 0.39 | singleton | 10.14 | 1.55 |
| 28 |  MolPort-002-656-531 |  PubChem | 0.31 | singleton | 0.34 | 0.63 |
| 29 |  MolPort-001-991-071 |  PubChem | 0.47 | singleton | 4.49 | 1.11 |
| 30 |  MolPort-004-004-399 |  PubChem | 0.29 | singleton | 0.02 | 1.81 |
| 31 |  MolPort-007-570-291 |  ToxCast | 0.41 | singleton | 15.32 | 14.08 |
 | |||||
| ID | R1 | R2 | R3 | Measured Activity 1 | S.D. 2 |
| 32 MolPort-006-630-016 | CF3 | H |  | 13.19 | 4.02 |
| 33 MolPort-006-630-300 | CF3 | H |  | 37.32 | 5.57 |
| 34 MolPort-006-629-818 | CF3 | H |  | 100.91 | 15.25 |
| 35 MolPort-006-630-013 | CF3 | H |  | 56.85 | 7.23 |
| 36 MolPort-006-629-816 | CF3 | H |  | 12.59 | 4.53 |
| 37 MolPort-006-629-973 | CF3 | H |  | 4.79 | 1.33 |
| 38 MolPort-006-630-273 | H | OCH2CH3 |  | 14.11 | 2.52 |
| 39 MolPort-006-630-110 | H | F |  | 80.37 | 25.75 |
| 40 MolPort-006-630-237 | H |  |  | 6.43 | 1.57 |
 | |||||||
|---|---|---|---|---|---|---|---|
| ID | R1 | R2 | R3 | R4 | R5 | Measured Activity 1 | S.D. 2 |
| 41 MolPort-003-179-500 | H | H | H | COH | H | 6.27 | 0.87 |
| 42 MolPort-000-431-927 | H | H | Cl | Me | Cl | 115.84 | 17.76 |
| 43 MolPort-000-431-934 | H | H | Me | Me | Me | 71.67 | 15.93 |
| 44 * MolPort-020-176-525 | Me | Me | H | Me | H | 215.49 | 31.07 |
| 45 MolPort-004-827-215 | Me | Me | H | 4-BrC6H4 | H | 9.25 | 4.38 |
 | |||||||
|---|---|---|---|---|---|---|---|
| ID | R1 | R2 | R3 | R4 | Configuration | Measured Activity 1 | S.D. 2 |
| 46 MolPort-002-173-405 | S | H |  | (CH2)2OCH3 | E | 12.45 | 5.06 |
| 47 MolPort-003-881-006 | S | H |  |  | Z | 21.96 | 2.87 |
| 48 MolPort-003-881-001 | S | H |  |  | Z | 19.72 | 4.24 |
| 49 MolPort-000-419-900 | S | H |  |  | Z | 8.54 | 2.04 |
| 50 MolPort-003-880-748 | S | OCH3 |  |  | Z | 6.66 | 1.30 |
| 51 MolPort-003-881-514 | NH | OCH3 |  | H | Z | 4.10 | 1.63 |
| 52 MolPort-044-415-081 | S | OCH3 |  |  | E/Z | 15.73 | 4.05 |
| 53 MolPort-002-216-386 | S | OCH3 |  | Prop | Z | 84.18 | 23.30 |
| 54 MolPort-001-899-455 | S | OCH3 |  | Et | E | 19.46 | 8.00 |
| 55 MolPort-001-552-801 | S | OCH3 | Et | Et | Z | 18.65 | 6.09 |
| 56 MolPort-039-019-225 | S | OCH2CH3 | Prop |  | Z | 22.63 | 10.48 |
| 57 * MolPort-001-634-394 | S | OCH2CH3 |  | Et | Z | 95.59 | 45.14 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hirte, S.; Burk, O.; Tahir, A.; Schwab, M.; Windshügel, B.; Kirchmair, J. Development and Experimental Validation of Regularized Machine Learning Models Detecting New, Structurally Distinct Activators of PXR. Cells 2022, 11, 1253. https://doi.org/10.3390/cells11081253
Hirte S, Burk O, Tahir A, Schwab M, Windshügel B, Kirchmair J. Development and Experimental Validation of Regularized Machine Learning Models Detecting New, Structurally Distinct Activators of PXR. Cells. 2022; 11(8):1253. https://doi.org/10.3390/cells11081253
Chicago/Turabian StyleHirte, Steffen, Oliver Burk, Ammar Tahir, Matthias Schwab, Björn Windshügel, and Johannes Kirchmair. 2022. "Development and Experimental Validation of Regularized Machine Learning Models Detecting New, Structurally Distinct Activators of PXR" Cells 11, no. 8: 1253. https://doi.org/10.3390/cells11081253