
Neuroprotective Effects of PARP Inhibitors in Drosophila Models of Alzheimer’s Disease

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Fly Stocks
2.2. Pharmacological Inhibition of PARP-1
2.3. Climbing Assay
2.4. Lifespan Assay
2.5. NAD+ Measurement
2.6. Western and Slot Blot
2.7. RNA Isolation and qRT-PCR Analysis
2.8. Drosophila Adult Brain Immunofluorescence
2.9. Statistical Analysis
3. Results
3.1. Pharmacological Inhibition or RNAi-Mediated Genetic Knockdown of PARP-1 Ameliorates Aβ42-Induced Locomotor Defects in Aβ42 Model of AD
3.2. PARP-1 Inhibitors Improve NAD+ Content
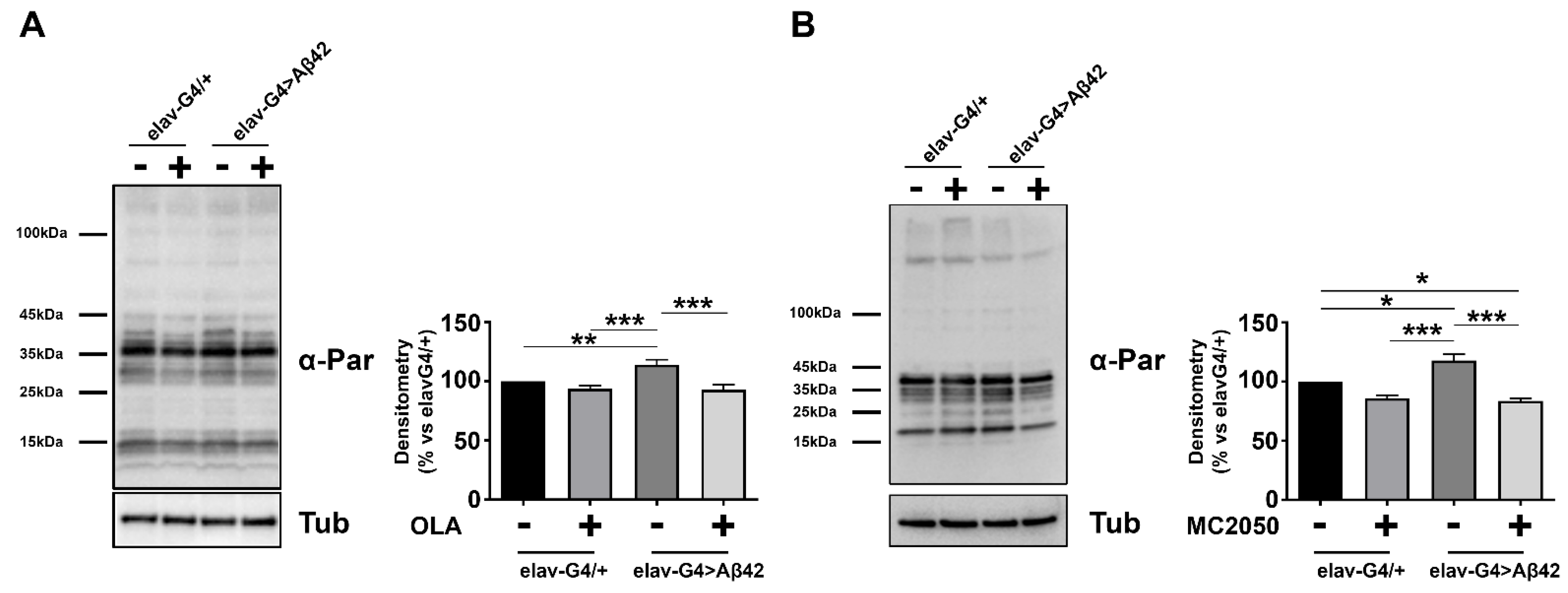
3.3. PARP-1 Inhibitors Impair PARylation without Affecting Aβ42 or PARP-1 Protein Expression
3.4. Olaparib and MC2050 Inhibits Aggregation of Aβ42 Peptides in AD Adult Brains
3.5. Effects of PARP-1 Inhibitors on the Transposable Elements’ Expression
3.6. PARP-1 Inhibition Restores Histone Modifications in the Aβ42 Model
3.7. RNAi-Mediated Gene Silencing of PARP-1 Rescues Motor Dysfunction and Improves the Life Expectancy of a Transgenic Model Expressing Human APP and BACE1 (APP/BACE1 Model)
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Braak, H.; Del Tredici, K. Neuroanatomy and pathology of sporadic Alzheimer’s disease. Adv. Anat. Embryol. Cell Biology 2015, 215, 1–162. [Google Scholar]
- Butterfield, D.A. Amyloid beta-peptide (1-42)-induced oxidative stress and neurotoxicity: Implications for neurodegeneration in Alzheimer’s disease brain. A review. Free Radic. Res. 2002, 36, 1307–1313. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, S.; Sato, N.; Yamamoto, A.; Ikegame, Y.; Nakashima, S.; Ogihara, T.; Morishita, R. Alzheimer disease-associated peptide, amyloid beta40, inhibits vascular regeneration with induction of endothelial autophagy. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1909–1915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Jiao, B.; Shen, L. The Epigenetics of Alzheimer’s Disease: Factors and Therapeutic Implications. Front. Genet. 2018, 9, 579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luscher, B.; Ahel, I.; Altmeyer, M.; Ashworth, A.; Bai, P.; Chang, P.; Cohen, M.; Corda, D.; Dantzer, F.; Daugherty, M.D.; et al. ADP-ribosyltransferases, an update on function and nomenclature. FEBS J 2021. [Google Scholar] [CrossRef]
- Love, S.; Barber, R.; Wilcock, G.K. Increased poly(ADP-ribosyl)ation of nuclear proteins in Alzheimer’s disease. Brain 1999, 122 Pt 2, 247–253. [Google Scholar] [CrossRef] [Green Version]
- Strosznajder, J.B.; Jesko, H.; Strosznajder, R.P. Effect of amyloid beta peptide on poly(ADP-ribose) polymerase activity in adult and aged rat hippocampus. Acta Biochim. Pol. 2000, 47, 847–854. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.P.; Lin, W.Y.; Wu, B.T.; Liu, S.H.; Wang, W.F.; Tsai, C.H.; Lee, C.C.; Tsai, F.J. Evaluation of the poly(ADP-ribose) polymerase-1 gene variants in Alzheimer’s disease. J. Clin. Lab. Anal. 2010, 24, 182–186. [Google Scholar] [CrossRef]
- Abeti, R.; Duchen, M.R. Activation of PARP by oxidative stress induced by beta-amyloid: Implications for Alzheimer’s disease. Neurochem. Res. 2012, 37, 2589–2596. [Google Scholar] [CrossRef]
- Strosznajder, J.B.; Czapski, G.A.; Adamczyk, A.; Strosznajder, R.P. Poly(ADP-ribose) polymerase-1 in amyloid beta toxicity and Alzheimer’s disease. Mol. Neurobiol. 2012, 46, 78–84. [Google Scholar] [CrossRef]
- Zeng, J.; Libien, J.; Shaik, F.; Wolk, J.; Hernandez, A.I. Nucleolar PARP-1 Expression Is Decreased in Alzheimer’s Disease: Consequences for Epigenetic Regulation of rDNA and Cognition. Neural Plast. 2016, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Narne, P.; Pandey, V.; Simhadri, P.K.; Phanithi, P.B. Poly(ADP-ribose)polymerase-1 hyperactivation in neurodegenerative diseases: The death knell tolls for neurons. Semin. Cell Dev. Biol. 2017, 63, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Regier, M.; Liang, J.; Choi, A.; Verma, K.; Libien, J.; Hernandez, A.I. Evidence for Decreased Nucleolar PARP-1 as an Early Marker of Cognitive Impairment. Neural Plast. 2019, 2019, 4383258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Fang, Y. New insights of poly(ADP-ribosylation) in neurodegenerative diseases: A focus on protein phase separation and pathologic aggregation. Biochem. Pharmacol. 2019, 167, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Salech, F.; Ponce, D.P.; Paula-Lima, A.C.; SanMartin, C.D.; Behrens, M.I. Nicotinamide, a Poly [ADP-Ribose] Polymerase 1 (PARP-1) Inhibitor, as an Adjunctive Therapy for the Treatment of Alzheimer’s Disease. Front. Aging Neurosci. 2020, 12, 255. [Google Scholar] [CrossRef]
- D’Amours, D.; Desnoyers, S.; D’Silva, I.; Poirier, G.G. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 1999, 342 Pt 2, 249–268. [Google Scholar] [CrossRef]
- Kraus, W.L. Transcriptional control by PARP-1: Chromatin modulation, enhancer-binding, coregulation, and insulation. Curr. Opin. Cell. Biol. 2008, 20, 294–302. [Google Scholar] [CrossRef] [Green Version]
- Poirier, G.G.; de Murcia, G.; Jongstra-Bilen, J.; Niedergang, C.; Mandel, P. Poly(ADP-ribosyl)ation of polynucleosomes causes relaxation of chromatin structure. Proc. Natl. Acad. Sci. USA 1982, 79, 3423–3427. [Google Scholar] [CrossRef] [Green Version]
- d’Erme, M.; Yang, G.; Sheagly, E.; Palitti, F.; Bustamante, C. Effect of poly(ADP-ribosyl)ation and Mg2+ ions on chromatin structure revealed by scanning force microscopy. Biochemistry 2001, 40, 10947–10955. [Google Scholar] [CrossRef]
- Messner, S.; Altmeyer, M.; Zhao, H.; Pozivil, A.; Roschitzki, B.; Gehrig, P.; Rutishauser, D.; Huang, D.; Caflisch, A.; Hottiger, M.O. PARP1 ADP-ribosylates lysine residues of the core histone tails. Nucleic Acids Res. 2010, 38, 6350–6362. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Chen, Y.; Wu, J.; Chen, S.H.; Liu, X.; Singh, A.K.; Yu, X. Poly(ADP-ribosyl)ation mediates early phase histone eviction at DNA lesions. Nucleic Acids Res. 2020, 48, 3001–3013. [Google Scholar] [CrossRef] [PubMed]
- Tulin, A.; Spradling, A. Chromatin loosening by poly(ADP)-ribose polymerase (PARP) at Drosophila puff loci. Science 2003, 299, 560–562. [Google Scholar] [CrossRef] [PubMed]
- Happel, N.; Doenecke, D. Histone H1 and its isoforms: Contribution to chromatin structure and function. Genes 2009, 431, 1–12. [Google Scholar] [CrossRef]
- Krishnakumar, R.; Kraus, W.L. PARP-1 regulates chromatin structure and transcription through a KDM5B-dependent pathway. Mol. Cell 2010, 39, 736–749. [Google Scholar] [CrossRef] [Green Version]
- Krishnakumar, R.; Gamble, M.J.; Frizzell, K.M.; Berrocal, J.G.; Kininis, M.; Kraus, W.L. Reciprocal binding of PARP-1 and histone H1 at promoters specifies transcriptional outcomes. Science 2008, 319, 819–821. [Google Scholar] [CrossRef] [PubMed]
- Halappanavar, S.S.; Shah, G.M. Defective control of mitotic and post-mitotic checkpoints in poly(ADP-ribose) polymerase-1(-/-) fibroblasts after mitotic spindle disruption. Cell Cycle 2004, 3, 335–342. [Google Scholar] [CrossRef] [Green Version]
- Bai, P.; Canto, C. The role of PARP-1 and PARP-2 enzymes in metabolic regulation and disease. Cell Metab. 2012, 16, 290–295. [Google Scholar] [CrossRef] [Green Version]
- De Vos, M.; Schreiber, V.; Dantzer, F. The diverse roles and clinical relevance of PARPs in DNA damage repair: Current state of the art. Biochem. Pharmacol. 2012, 84, 137–146. [Google Scholar] [CrossRef]
- Burkle, A.; Virag, L. Poly(ADP-ribose): PARadigms and PARadoxes. Mol. Asp. Med. 2013, 34, 1046–1065. [Google Scholar] [CrossRef] [Green Version]
- Cho-Park, P.F.; Steller, H. Proteasome regulation by ADP-ribosylation. Cell 2013, 153, 614–627. [Google Scholar] [CrossRef] [Green Version]
- Gupte, R.; Liu, Z.; Kraus, W.L. PARPs and ADP-ribosylation: Recent advances linking molecular functions to biological outcomes. Genes Dev. 2017, 31, 101–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascal, J.M. The comings and goings of PARP-1 in response to DNA damage. DNA Repair 2018, 71, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Abeti, R.; Abramov, A.Y.; Duchen, M.R. Beta-amyloid activates PARP causing astrocytic metabolic failure and neuronal death. Brain 2011, 134, 1658–1672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kauppinen, T.M.; Suh, S.W.; Higashi, Y.; Berman, A.E.; Escartin, C.; Won, S.J.; Wang, C.; Cho, S.H.; Gan, L.; Swanson, R.A. Poly(ADP-ribose)polymerase-1 modulates microglial responses to amyloid beta. J. Neuroinflammation 2011, 8, 152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martire, S.; Fuso, A.; Rotili, D.; Tempera, I.; Giordano, C.; De Zottis, I.; Muzi, A.; Vernole, P.; Graziani, G.; Lococo, E.; et al. PARP-1 modulates amyloid beta peptide-induced neuronal damage. PLoS ONE 2013, 8, e72169. [Google Scholar] [CrossRef] [Green Version]
- Bayrakdar, E.T.; Armagan, G.; Uyanikgil, Y.; Kanit, L.; Koylu, E.; Yalcin, A. Ex vivo protective effects of nicotinamide and 3-aminobenzamide on rat synaptosomes treated with Abeta(1-42). Cell Biochem. Funct. 2014, 32, 557–564. [Google Scholar] [CrossRef]
- Wencel, P.L.; Lukiw, W.J.; Strosznajder, J.B.; Strosznajder, R.P. Inhibition of Poly(ADP-ribose) Polymerase-1 Enhances Gene Expression of Selected Sirtuins and APP Cleaving Enzymes in Amyloid Beta Cytotoxicity. Mol. Neurobiol. 2018, 55, 4612–4623. [Google Scholar] [CrossRef] [Green Version]
- Alano, C.C.; Garnier, P.; Ying, W.; Higashi, Y.; Kauppinen, T.M.; Swanson, R.A. NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J. Neurosci. 2010, 30, 2967–2978. [Google Scholar] [CrossRef]
- Correani, V.; Martire, S.; Mignogna, G.; Caruso, L.B.; Tempera, I.; Giorgi, A.; Grieco, M.; Mosca, L.; Schinina, M.E.; Maras, B.; et al. Poly(ADP-ribosylated) proteins in beta-amyloid peptide-stimulated microglial cells. Biochem. Pharmacol. 2019, 167, 50–57. [Google Scholar] [CrossRef]
- Fehr, A.R.; Singh, S.A.; Kerr, C.M.; Mukai, S.; Higashi, H.; Aikawa, M. The impact of PARPs and ADP-ribosylation on inflammation and host-pathogen interactions. Genes. Dev. 2020, 34, 341–359. [Google Scholar] [CrossRef]
- Jang, S.; Kim, E.W.; Zhang, Y.; Lee, J.; Cho, S.Y.; Ha, J.; Kim, H.; Kim, E. Particulate matter increases beta-amyloid and activated glial cells in hippocampal tissues of transgenic Alzheimer’s mouse: Involvement of PARP-1. Biochem. Biophys. Res. Commun. 2018, 500, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Wang, C.; Zhang, J.; Zhong, X.; Wang, R.; Zeng, X.; Ba, X. The Role of PARPs in Inflammation-and Metabolic-Related Diseases: Molecular Mechanisms and Beyond. Cells 2019, 8, 1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiuri, T.; Bowie, L.E.; Truant, R. DNA Repair Signaling of Huntingtin: The Next Link Between Late-Onset Neurodegenerative Disease and Oxidative DNA Damage. DNA Cell Biol. 2019, 38, 1–6. [Google Scholar] [CrossRef] [PubMed]
- McGurk, L.; Gomes, E.; Guo, L.; Mojsilovic-Petrovic, J.; Tran, V.; Kalb, R.G.; Shorter, J.; Bonini, N.M. Poly(ADP-Ribose) Prevents Pathological Phase Separation of TDP-43 by Promoting Liquid Demixing and Stress Granule Localization. Mol. Cell 2018, 71, 703–717.e9. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.; Walker, A.J.; Berk, M.; Maes, M.; Puri, B.K. Cell Death Pathways: A Novel Therapeutic Approach for Neuroscientists. Mol. Neurobiol. 2018, 55, 5767–5786. [Google Scholar] [CrossRef] [Green Version]
- Vida, A.; Marton, J.; Miko, E.; Bai, P. Metabolic roles of poly(ADP-ribose) polymerases. Semin. Cell. Dev. Biol. 2017, 63, 135–143. [Google Scholar] [CrossRef] [Green Version]
- Schneider, L.S. Rethinking the Food and Drug Administration’s 2013 guidance on developing drugs for early-stage Alzheimer’s disease. Alzheimer’s Dement. 2014, 10, 247–250. [Google Scholar] [CrossRef] [Green Version]
- Peskind, E.R.; Potkin, S.G.; Pomara, N.; Ott, B.R.; Graham, S.M.; Olin, J.T.; McDonald, S. Memantine treatment in mild to moderate Alzheimer disease: A 24-week randomized, controlled trial. Am. J. Geriatr. Psychiatry 2006, 14, 704–715. [Google Scholar] [CrossRef]
- Cummings, J.; Reiber, C.; Kumar, P. The price of progress: Funding and financing Alzheimer’s disease drug development. Alzheimer’s Dement. 2018, 4, 330–343. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussiere, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Martire, S.; Mosca, L.; d’Erme, M. PARP-1 involvement in neurodegeneration: A focus on Alzheimer’s and Parkinson’s diseases. Mech. Ageing Dev. 2015, 146–148, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Marin, E.C.; Jefferis, G.S.; Komiyama, T.; Zhu, H.; Luo, L. Representation of the glomerular olfactory map in the Drosophila brain. Cell 2002, 109, 243–255. [Google Scholar] [CrossRef] [Green Version]
- Rein, K.; Zockler, M.; Mader, M.T.; Grubel, C.; Heisenberg, M. The Drosophila standard brain. Curr. Biol. 2002, 12, 227–231. [Google Scholar] [CrossRef] [Green Version]
- Wong, A.M.; Wang, J.W.; Axel, R. Spatial representation of the glomerular map in the Drosophila protocerebrum. Cell 2002, 109, 229–241. [Google Scholar] [CrossRef] [Green Version]
- Jeon, Y.; Lee, J.H.; Choi, B.; Won, S.Y.; Cho, K.S. Genetic Dissection of Alzheimer’s Disease Using Drosophila Models. Int. J. Mol. Sci. 2020, 21, 884. [Google Scholar] [CrossRef] [Green Version]
- Tan, F.H.P.; Azzam, G. Drosophila melanogaster: Deciphering Alzheimer’s Disease. Malays. J. Med. Sci. 2017, 24, 6–20. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, L.; Lim, Y.M. Alzheimer’s Disease Model System Using Drosophila. Adv. Exp. Med. Biol. 2018, 1076, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Prussing, K.; Voigt, A.; Schulz, J.B. Drosophila melanogaster as a model organism for Alzheimer’s disease. Mol. Neurodegener. 2013, 8, 35. [Google Scholar] [CrossRef] [Green Version]
- Marsh, J.L.; Thompson, L.M. Drosophila in the study of neurodegenerative disease. Neuron 2006, 52, 169–178. [Google Scholar] [CrossRef] [Green Version]
- Thomas, C.J.; Kotova, E.; Andrake, M.; Adolf-Bryfogle, J.; Glaser, R.; Regnard, C.; Tulin, A.V. Kinase-mediated changes in nucleosome conformation trigger chromatin decondensation via poly(ADP-ribosyl)ation. Mol. Cell 2014, 53, 831–842. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Chen, J. ADP-ribosylation: Activation, recognition, and removal. Mol. Cells 2014, 37, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Y.; Tulin, A.V. The roles of PARP1 in gene control and cell differentiation. Curr. Opin. Genet. Dev. 2010, 20, 512–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tulin, A.; Stewart, D.; Spradling, A.C. The Drosophila heterochromatic gene encoding poly(ADP-ribose) polymerase (PARP) is required to modulate chromatin structure during development. Genes Dev. 2002, 16, 2108–2119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunderson, C.C.; Moore, K.N. Olaparib: An oral PARP-1 and PARP-2 inhibitor with promising activity in ovarian cancer. Future Oncol. 2015, 11, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Paik, J. Olaparib: A Review as First-Line Maintenance Therapy in Advanced Ovarian Cancer. Target. Oncol. 2021, 16, 847–856. [Google Scholar] [CrossRef]
- Mosca, L.; Rotili, D.; Tempera, I.; Masci, A.; Fontana, M.; Chiaraluce, R.; Mastromarino, P.; d’Erme, M.; Mai, A. Biological effects of MC2050, a quinazoline-based PARP-1 inhibitor, in human neuroblastoma and EBV-positive Burkitt’s lymphoma cells. ChemMedChem 2011, 6, 606–611. [Google Scholar] [CrossRef]
- Menear, K.A.; Adcock, C.; Boulter, R.; Cockcroft, X.L.; Copsey, L.; Cranston, A.; Dillon, K.J.; Drzewiecki, J.; Garman, S.; Gomez, S.; et al. 4-[3-(4-cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phthalazin- 1-one: A novel bioavailable inhibitor of poly(ADP-ribose) polymerase-1. J. Med. Chem. 2008, 51, 6581–6591. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Wu, J.S.; Luo, L. A protocol for dissecting Drosophila melanogaster brains for live imaging or immunostaining. Nat. Protoc. 2006, 1, 2110–2115. [Google Scholar] [CrossRef]
- Ali, Y.O.; Escala, W.; Ruan, K.; Zhai, R.G. Assaying locomotor, learning, and memory deficits in Drosophila models of neurodegeneration. J. Vis. Exp. 2011, 49, 2504. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, R.; Vepuri, V.; Mhatre, S.D.; Paddock, B.E.; Miller, S.; Michelson, S.J.; Delvadia, R.; Desai, A.; Vinokur, M.; Melicharek, D.J.; et al. Characterization of a Drosophila Alzheimer’s disease model: Pharmacological rescue of cognitive defects. PLoS ONE 2011, 6, e20799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crowther, D.C.; Kinghorn, K.J.; Miranda, E.; Page, R.; Curry, J.A.; Duthie, F.A.; Gubb, D.C.; Lomas, D.A. Intraneuronal Abeta, non-amyloid aggregates and neurodegeneration in a Drosophila model of Alzheimer’s disease. Neuroscience 2005, 132, 123–135. [Google Scholar] [CrossRef]
- Finelli, A.; Kelkar, A.; Song, H.J.; Yang, H.; Konsolaki, M. A model for studying Alzheimer’s Abeta42-induced toxicity in Drosophila melanogaster. Mol. Cell. Neurosci. 2004, 26, 365–375. [Google Scholar] [CrossRef]
- Iijima, K.; Liu, H.P.; Chiang, A.S.; Hearn, S.A.; Konsolaki, M.; Zhong, Y. Dissecting the pathological effects of human Abeta40 and Abeta42 in Drosophila: A potential model for Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 6623–6628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenwald, S.H.; Brown, E.E.; Scandura, M.J.; Hennessey, E.; Farmer, R.; Du, J.; Wang, Y.; Pierce, E.A. Mutant Nmnat1 leads to a retina-specific decrease of NAD+ accompanied by increased poly(ADP-ribose) in a mouse model of NMNAT1-associated retinal degeneration. Hum. Mol. Genet. 2021, 30, 644–657. [Google Scholar] [CrossRef] [PubMed]
- Tiraboschi, P.; Sabbagh, M.N.; Hansen, L.A.; Salmon, D.P.; Merdes, A.; Gamst, A.; Masliah, E.; Alford, M.; Thal, L.J.; Corey-Bloom, J. Alzheimer disease without neocortical neurofibrillary tangles: “a second look”. Neurology 2004, 62, 1141–1147. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yang, L.; Zheng, H. Role of APP and Abeta in synaptic physiology. Curr. Alzheimer Res. 2012, 9, 217–226. [Google Scholar] [CrossRef]
- Wolfe, M.S. Processive proteolysis by gamma-secretase and the mechanism of Alzheimer’s disease. Biol. Chem. 2012, 393, 899–905. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Karran, E.; Mercken, M.; De Strooper, B. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef]
- Iijima, K.; Chiang, H.C.; Hearn, S.A.; Hakker, I.; Gatt, A.; Shenton, C.; Granger, L.; Leung, A.; Iijima-Ando, K.; Zhong, Y. Abeta42 mutants with different aggregation profiles induce distinct pathologies in Drosophila. PLoS ONE 2008, 3, e1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonsson, M.E.; Garza, R.; Johansson, P.A.; Jakobsson, J. Transposable Elements: A Common Feature of Neurodevelopmental and Neurodegenerative Disorders. Trends Genet. 2020, 36, 610–623. [Google Scholar] [CrossRef] [PubMed]
- Saleh, A.; Macia, A.; Muotri, A.R. Transposable Elements, Inflammation, and Neurological Disease. Front. Neurol. 2019, 10, 894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tam, C.; Wong, J.H.; Ng, T.B.; Tsui, S.K.W.; Zuo, T. Drugs for Targeted Therapies of Alzheimer’s Disease. Curr. Med. Chem. 2019, 26, 335–359. [Google Scholar] [CrossRef] [PubMed]
- Muotri, A.R.; Marchetto, M.C.; Coufal, N.G.; Oefner, R.; Yeo, G.; Nakashima, K.; Gage, F.H. L1 retrotransposition in neurons is modulated by MeCP2. Nature 2010, 468, 443–446. [Google Scholar] [CrossRef] [PubMed]
- Coufal, N.G.; Garcia-Perez, J.L.; Peng, G.E.; Marchetto, M.C.; Muotri, A.R.; Mu, Y.; Carson, C.T.; Macia, A.; Moran, J.V.; Gage, F.H. Ataxia telangiectasia mutated (ATM) modulates long interspersed element-1 (L1) retrotransposition in human neural stem cells. Proc. Natl. Acad. Sci. USA 2011, 108, 20382–20387. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, H.; Dridi, S.; Tarallo, V.; Gelfand, B.D.; Fowler, B.J.; Cho, W.G.; Kleinman, M.E.; Ponicsan, S.L.; Hauswirth, W.W.; Chiodo, V.A.; et al. DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature 2011, 471, 325–330. [Google Scholar] [CrossRef] [Green Version]
- Lathe, R.; Harris, A. Differential display detects host nucleic acid motifs altered in scrapie-infected brain. J. Mol. Biol. 2009, 392, 813–822. [Google Scholar] [CrossRef]
- Bundo, M.; Toyoshima, M.; Okada, Y.; Akamatsu, W.; Ueda, J.; Nemoto-Miyauchi, T.; Sunaga, F.; Toritsuka, M.; Ikawa, D.; Kakita, A.; et al. Increased l1 retrotransposition in the neuronal genome in schizophrenia. Neuron 2014, 81, 306–313. [Google Scholar] [CrossRef] [Green Version]
- Blaudin de The, F.X.; Rekaik, H.; Peze-Heidsieck, E.; Massiani-Beaudoin, O.; Joshi, R.L.; Fuchs, J.; Prochiantz, A. Engrailed homeoprotein blocks degeneration in adult dopaminergic neurons through LINE-1 repression. EMBO J. 2018, 37, e97174. [Google Scholar] [CrossRef]
- Krug, L.; Chatterjee, N.; Borges-Monroy, R.; Hearn, S.; Liao, W.W.; Morrill, K.; Prazak, L.; Rozhkov, N.; Theodorou, D.; Hammell, M.; et al. Retrotransposon activation contributes to neurodegeneration in a Drosophila TDP-43 model of ALS. PLoS Genet. 2017, 13, e1006635. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Jin, Y.; Prazak, L.; Hammell, M.; Dubnau, J. Transposable elements in TDP-43-mediated neurodegenerative disorders. PLoS ONE 2012, 7, e44099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romano, G.; Klima, R.; Feiguin, F. TDP-43 prevents retrotransposon activation in the Drosophila motor system through regulation of Dicer-2 activity. BMC Biol. 2020, 18, 82. [Google Scholar] [CrossRef]
- Casale, A.M.; Liguori, F.; Ansaloni, F.; Cappucci, U.; Finaurini, S.; Spirito, G.; Persichetti, F.; Sanges, R.; Gustincich, S.; Piacentini, L. Transposable element activation promotes neurodegeneration in a Drosophila model of Huntington’s disease. iScience 2022, 25, 103702. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Jeong, H.H.; Hsieh, Y.C.; Klein, H.U.; Bennett, D.A.; De Jager, P.L.; Liu, Z.; Shulman, J.M. Tau Activates Transposable Elements in Alzheimer’s Disease. Cell Rep. 2018, 23, 2874–2880. [Google Scholar] [CrossRef] [PubMed]
- Misiak, B.; Ricceri, L.; Sasiadek, M.M. Transposable Elements and Their Epigenetic Regulation in Mental Disorders: Current Evidence in the Field. Front. Genet. 2019, 10, 580. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Samimi, H.; Gamez, M.; Zare, H.; Frost, B. Pathogenic tau-induced piRNA depletion promotes neuronal death through transposable element dysregulation in neurodegenerative tauopathies. Nat. Neurosci. 2018, 21, 1038–1048. [Google Scholar] [CrossRef]
- Malik, N.; Smulson, M. A relationship between nuclear poly(adenosine diphosphate ribosylation) and acetylation posttranslational modifications. 1. Nucleosome studies. Biochemistry 1984, 23, 3721–3725. [Google Scholar] [CrossRef]
- Cohen-Armon, M.; Visochek, L.; Rozensal, D.; Kalal, A.; Geistrikh, I.; Klein, R.; Bendetz-Nezer, S.; Yao, Z.; Seger, R. DNA-independent PARP-1 activation by phosphorylated ERK2 increases Elk1 activity: A link to histone acetylation. Mol. Cell 2007, 25, 297–308. [Google Scholar] [CrossRef]
- Chen, H.; Ruiz, P.D.; Novikov, L.; Casill, A.D.; Park, J.W.; Gamble, M.J. MacroH2A1.1 and PARP-1 cooperate to regulate transcription by promoting CBP-mediated H2B acetylation. Nat. Struct. Mol. Biol. 2014, 21, 981–989. [Google Scholar] [CrossRef] [Green Version]
- Martin, K.A.; Cesaroni, M.; Denny, M.F.; Lupey, L.N.; Tempera, I. Global Transcriptome Analysis Reveals That Poly(ADP-Ribose) Polymerase 1 Regulates Gene Expression through EZH2. Mol. Cell. Biol. 2015, 35, 3934–3944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greeve, I.; Kretzschmar, D.; Tschape, J.A.; Beyn, A.; Brellinger, C.; Schweizer, M.; Nitsch, R.M.; Reifegerste, R. Age-dependent neurodegeneration and Alzheimer-amyloid plaque formation in transgenic Drosophila. J. Neurosci. 2004, 24, 3899–3906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martire, S.; Fuso, A.; Mosca, L.; Forte, E.; Correani, V.; Fontana, M.; Scarpa, S.; Maras, B.; d’Erme, M. Bioenergetic Impairment in Animal and Cellular Models of Alzheimer’s Disease: PARP-1 Inhibition Rescues Metabolic Dysfunctions. J. Alzheimer’s. Dis. 2016, 54, 307–324. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.K.L. PARPs. Curr. Biol. 2017, 27, R1256–R1258. [Google Scholar] [CrossRef] [Green Version]
- Beneke, S.; Burkle, A. Poly(ADP-ribosyl)ation, PARP, and aging. Sci. Aging Knowl. Environ. 2004, 2004, re9. [Google Scholar] [CrossRef] [Green Version]
- Kauppinen, T.M.; Swanson, R.A. The role of poly(ADP-ribose) polymerase-1 in CNS disease. Neuroscience 2007, 145, 1267–1272. [Google Scholar] [CrossRef]
- Alhosaini, K.; Ansari, M.A.; Nadeem, A.; Bakheet, S.A.; Attia, S.M.; Alhazzani, K.; Albekairi, T.H.; Al-Mazroua, H.A.; Mahmood, H.M.; Ahmad, S.F. 5-Aminoisoquinolinone, a PARP-1 Inhibitor, Ameliorates Immune Abnormalities through Upregulation of Anti-Inflammatory and Downregulation of Inflammatory Parameters in T Cells of BTBR Mouse Model of Autism. Brain Sci. 2021, 11, 249. [Google Scholar] [CrossRef]
- Mao, K.; Zhang, G. The role of PARP1 in neurodegenerative diseases and aging. FEBS J. 2021. [Google Scholar] [CrossRef]
- Kam, T.I.; Mao, X.; Park, H.; Chou, S.C.; Karuppagounder, S.S.; Umanah, G.E.; Yun, S.P.; Brahmachari, S.; Panicker, N.; Chen, R.; et al. Poly(ADP-ribose) drives pathologic alpha-synuclein neurodegeneration in Parkinson’s disease. Science 2018, 362, eaat8407. [Google Scholar] [CrossRef] [Green Version]
- Puentes, L.N.; Lengyel-Zhand, Z.; Lee, J.Y.; Hsieh, C.J.; Schneider, M.E., Jr.; Edwards, K.J.; Luk, K.C.; Lee, V.M.; Trojanowski, J.Q.; Mach, R.H. Poly (ADP-ribose) Interacts With Phosphorylated alpha-Synuclein in Post Mortem PD Samples. Front. Aging Neurosci. 2021, 13, 704041. [Google Scholar] [CrossRef]
- Fang, E.F.; Kassahun, H.; Croteau, D.L.; Scheibye-Knudsen, M.; Marosi, K.; Lu, H.; Shamanna, R.A.; Kalyanasundaram, S.; Bollineni, R.C.; Wilson, M.A.; et al. NAD(+) Replenishment Improves Lifespan and Healthspan in Ataxia Telangiectasia Models via Mitophagy and DNA Repair. Cell Metab. 2016, 24, 566–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardinale, A.; Paldino, E.; Giampa, C.; Bernardi, G.; Fusco, F.R. PARP-1 Inhibition Is Neuroprotective in the R6/2 Mouse Model of Huntington’s Disease. PLoS ONE 2015, 10, e0134482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rulten, S.L.; Rotheray, A.; Green, R.L.; Grundy, G.J.; Moore, D.A.; Gomez-Herreros, F.; Hafezparast, M.; Caldecott, K.W. PARP-1 dependent recruitment of the amyotrophic lateral sclerosis-associated protein FUS/TLS to sites of oxidative DNA damage. Nucleic Acids Res. 2014, 42, 307–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Guo, W.; Mitra, J.; Hegde, P.M.; Vandoorne, T.; Eckelmann, B.J.; Mitra, S.; Tomkinson, A.E.; Van Den Bosch, L.; Hegde, M.L. Mutant FUS causes DNA ligation defects to inhibit oxidative damage repair in Amyotrophic Lateral Sclerosis. Nat. Commun. 2018, 9, 3683. [Google Scholar] [CrossRef] [Green Version]
- Farez, M.F.; Quintana, F.J.; Gandhi, R.; Izquierdo, G.; Lucas, M.; Weiner, H.L. Toll-like receptor 2 and poly(ADP-ribose) polymerase 1 promote central nervous system neuroinflammation in progressive EAE. Nat. Immunol. 2009, 10, 958–964. [Google Scholar] [CrossRef]
- Tillement, L.; Lecanu, L.; Papadopoulos, V. Alzheimer’s disease: Effects of beta-amyloid on mitochondria. Mitochondrion 2011, 11, 13–21. [Google Scholar] [CrossRef]
- Chaturvedi, R.K.; Beal, M.F. Mitochondria targeted therapeutic approaches in Parkinson’s and Huntington’s diseases. Mol. Cell. Neurosci. 2013, 55, 101–114. [Google Scholar] [CrossRef]
- Ghavami, S.; Shojaei, S.; Yeganeh, B.; Ande, S.R.; Jangamreddy, J.R.; Mehrpour, M.; Christoffersson, J.; Chaabane, W.; Moghadam, A.R.; Kashani, H.H.; et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog. Neurobiol. 2014, 112, 24–49. [Google Scholar] [CrossRef] [Green Version]
- Kesidou, E.; Lagoudaki, R.; Touloumi, O.; Poulatsidou, K.N.; Simeonidou, C. Autophagy and neurodegenerative disorders. Neural. Regen. Res. 2013, 8, 2275–2283. [Google Scholar] [CrossRef]
- Chiarugi, A.; Moskowitz, M.A. Poly(ADP-ribose) polymerase-1 activity promotes NF-kappaB-driven transcription and microglial activation: Implication for neurodegenerative disorders. J. Neurochem. 2003, 85, 306–317. [Google Scholar] [CrossRef]
- Berger, N.A.; Besson, V.C.; Boulares, A.H.; Burkle, A.; Chiarugi, A.; Clark, R.S.; Curtin, N.J.; Cuzzocrea, S.; Dawson, T.M.; Dawson, V.L.; et al. Opportunities for the repurposing of PARP inhibitors for the therapy of non-oncological diseases. Br. J. Pharmacol. 2018, 175, 192–222. [Google Scholar] [CrossRef] [PubMed]
- Bitler, B.G.; Watson, Z.L.; Wheeler, L.J.; Behbakht, K. PARP inhibitors: Clinical utility and possibilities of overcoming resistance. Gynecol. Oncol. 2017, 147, 695–704. [Google Scholar] [CrossRef] [Green Version]
- Zeng, K.W.; Wang, X.M.; Ko, H.; Kwon, H.C.; Cha, J.W.; Yang, H.O. Hyperoside protects primary rat cortical neurons from neurotoxicity induced by amyloid beta-protein via the PI3K/Akt/Bad/Bcl(XL)-regulated mitochondrial apoptotic pathway. Eur. J. Pharm. 2011, 672, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Czapski, G.A.; Cieslik, M.; Wencel, P.L.; Wojtowicz, S.; Strosznajder, R.P.; Strosznajder, J.B. Inhibition of poly(ADP-ribose) polymerase-1 alters expression of mitochondria-related genes in PC12 cells: Relevance to mitochondrial homeostasis in neurodegenerative disorders. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 281–288. [Google Scholar] [CrossRef]
- Gao, C.Z.; Dong, W.; Cui, Z.W.; Yuan, Q.; Hu, X.M.; Wu, Q.M.; Han, X.; Xu, Y.; Min, Z.L. Synthesis, preliminarily biological evaluation and molecular docking study of new Olaparib analogues as multifunctional PARP-1 and cholinesterase inhibitors. J. Enzym. Inhib. Med. Chem. 2019, 34, 150–162. [Google Scholar] [CrossRef]
- Altmeyer, M.; Neelsen, K.J.; Teloni, F.; Pozdnyakova, I.; Pellegrino, S.; Grofte, M.; Rask, M.D.; Streicher, W.; Jungmichel, S.; Nielsen, M.L.; et al. Liquid demixing of intrinsically disordered proteins is seeded by poly(ADP-ribose). Nat. Commun. 2015, 6, 8088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, E.O.; Ramirez, P.; Hyman, B.T.; Ray, W.J.; Frost, B. Testing the neuroinflammatory role of tau-induced transposable elements in tauopathy. Alzheimer’s Dement. 2021, 17 (Suppl. S2), e058664. [Google Scholar] [CrossRef]
- Frost, B.; Hemberg, M.; Lewis, J.; Feany, M.B. Tau promotes neurodegeneration through global chromatin relaxation. Nat. Neurosci. 2014, 17, 357–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quenet, D.; El Ramy, R.; Schreiber, V.; Dantzer, F. The role of poly(ADP-ribosyl)ation in epigenetic events. Int. J. Biochem. Cell Biol. 2009, 41, 60–65. [Google Scholar] [CrossRef]
- Bordet, G.; Lodhi, N.; Guo, D.; Kossenkov, A.; Tulin, A.V. Poly(ADP-ribose) polymerase 1 in genome-wide expression control in Drosophila. Sci. Rep. 2020, 10, 21151. [Google Scholar] [CrossRef]
- He, J.; Fu, X.; Zhang, M.; He, F.; Li, W.; Abdul, M.M.; Zhou, J.; Sun, L.; Chang, C.; Li, Y.; et al. Transposable elements are regulated by context-specific patterns of chromatin marks in mouse embryonic stem cells. Nat. Commun. 2019, 10, 34. [Google Scholar] [CrossRef] [PubMed]
- Pinnola, A.; Naumova, N.; Shah, M.; Tulin, A.V. Nucleosomal core histones mediate dynamic regulation of poly(ADP-ribose) polymerase 1 protein binding to chromatin and induction of its enzymatic activity. J. Biol. Chem. 2007, 282, 32511–32519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotova, E.; Lodhi, N.; Jarnik, M.; Pinnola, A.D.; Ji, Y.; Tulin, A.V. Drosophila histone H2A variant (H2Av) controls poly(ADP-ribose) polymerase 1 (PARP1) activation in chromatin. Proc. Natl. Acad. Sci. USA 2011, 108, 6205–6210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, G.P.; Gurung, S.K.; Inam, A.; Nigam, L.; Bist, A.; Mohapatra, D.; Senapati, S.; Subbarao, N.; Azam, A.; Mondal, N. CID-6033590 inhibits p38MAPK pathway and induces S-phase cell cycle arrest and apoptosis in DU145 and PC-3 cells. Toxicol. Vitr. 2019, 60, 420–436. [Google Scholar] [CrossRef] [PubMed]
- Caruso, L.B.; Martin, K.A.; Lauretti, E.; Hulse, M.; Siciliano, M.; Lupey-Green, L.N.; Abraham, A.; Skorski, T.; Tempera, I. Poly(ADP-ribose) Polymerase 1, PARP1, modifies EZH2 and inhibits EZH2 histone methyltransferase activity after DNA damage. Oncotarget 2018, 9, 10585–10605. [Google Scholar] [CrossRef] [Green Version]
- Rudolph, J.; Roberts, G.; Luger, K. Histone Parylation factor 1 contributes to the inhibition of PARP1 by cancer drugs. Nat. Commun. 2021, 12, 736. [Google Scholar] [CrossRef]
- Bartlett, E.; Bonfiglio, J.J.; Prokhorova, E.; Colby, T.; Zobel, F.; Ahel, I.; Matic, I. Interplay of Histone Marks with Serine ADP-Ribosylation. Cell Rep. 2018, 24, 3488–3502.e5. [Google Scholar] [CrossRef] [Green Version]
- Aguilar-Quesada, R.; Munoz-Gamez, J.A.; Martin-Oliva, D.; Peralta, A.; Valenzuela, M.T.; Matinez-Romero, R.; Quiles-Perez, R.; Menissier-de Murcia, J.; de Murcia, G.; Ruiz de Almodovar, M.; et al. Interaction between ATM and PARP-1 in response to DNA damage and sensitization of ATM deficient cells through PARP inhibition. BMC Mol. Biol. 2007, 8, 29. [Google Scholar] [CrossRef] [Green Version]
- Mekhaeil, M.; Dev, K.K.; Conroy, M.J. Existing Evidence for the Repurposing of PARP-1 Inhibitors in Rare Demyelinating Diseases. Cancers 2022, 14, 687. [Google Scholar] [CrossRef]
- Puentes, L.N.; Lengyel-Zhand, Z.; Reilly, S.W.; Mach, R.H. Evaluation of a Low-Toxicity PARP Inhibitor as a Neuroprotective Agent for Parkinson’s Disease. Mol. Neurobiol. 2021, 58, 3641–3652. [Google Scholar] [CrossRef]
- Sahaboglu, A.; Miranda, M.; Canjuga, D.; Avci-Adali, M.; Savytska, N.; Secer, E.; Feria-Pliego, J.A.; Kayik, G.; Durdagi, S. Drug repurposing studies of PARP inhibitors as a new therapy for inherited retinal degeneration. Cell. Mol. Life. Sci. 2020, 77, 2199–2216. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maggiore, A.; Casale, A.M.; Toscanelli, W.; Cappucci, U.; Rotili, D.; Grieco, M.; Gagné, J.-P.; Poirier, G.G.; d’Erme, M.; Piacentini, L. Neuroprotective Effects of PARP Inhibitors in Drosophila Models of Alzheimer’s Disease. Cells 2022, 11, 1284. https://doi.org/10.3390/cells11081284
Maggiore A, Casale AM, Toscanelli W, Cappucci U, Rotili D, Grieco M, Gagné J-P, Poirier GG, d’Erme M, Piacentini L. Neuroprotective Effects of PARP Inhibitors in Drosophila Models of Alzheimer’s Disease. Cells. 2022; 11(8):1284. https://doi.org/10.3390/cells11081284
Chicago/Turabian StyleMaggiore, Anna, Assunta Maria Casale, Walter Toscanelli, Ugo Cappucci, Dante Rotili, Maddalena Grieco, Jean-Philippe Gagné, Guy G. Poirier, Maria d’Erme, and Lucia Piacentini. 2022. "Neuroprotective Effects of PARP Inhibitors in Drosophila Models of Alzheimer’s Disease" Cells 11, no. 8: 1284. https://doi.org/10.3390/cells11081284