
Target Hopping from Protein Kinases to PXR: Identification of Small-Molecule Protein Kinase Inhibitors as Selective Modulators of Pregnane X Receptor from TüKIC Library

 , ,
, ,  and
and 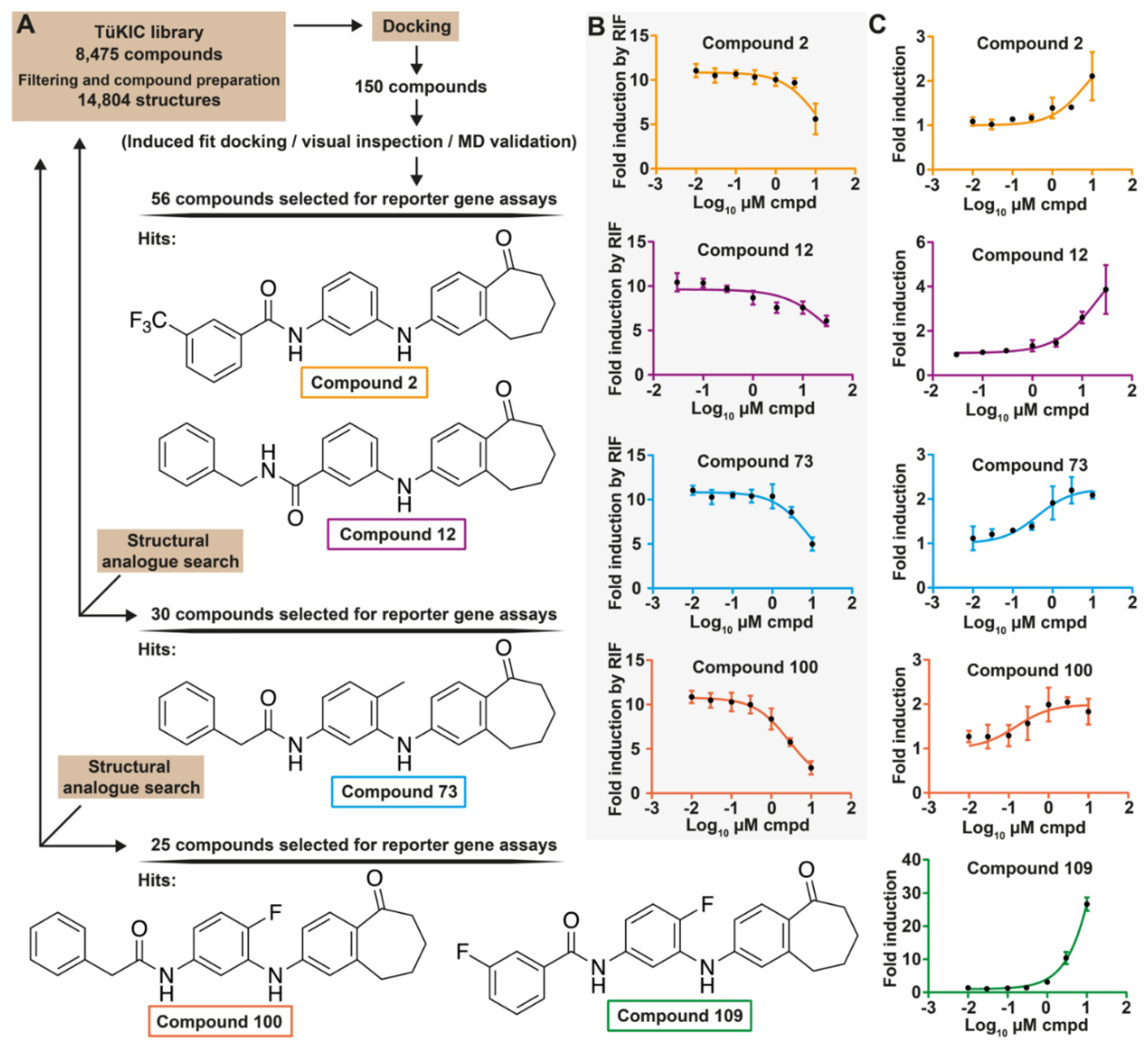{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Origin of Compounds
2.3. Molecular Modeling
2.4. Plasmid Constructs
2.5. Cell Culture
2.6. Cell Viability
2.7. Transient Transfections, Mammalian Two Hybrid, and Reporter Gene Assays
2.8. Limited Proteolytic Digestion
2.9. Competitive Radioligand Binding Assay
2.10. Quantitative Real-Time PCR Analysis
2.11. Kinase Inhibition Profiling
2.12. Statistical Analysis
3. Results
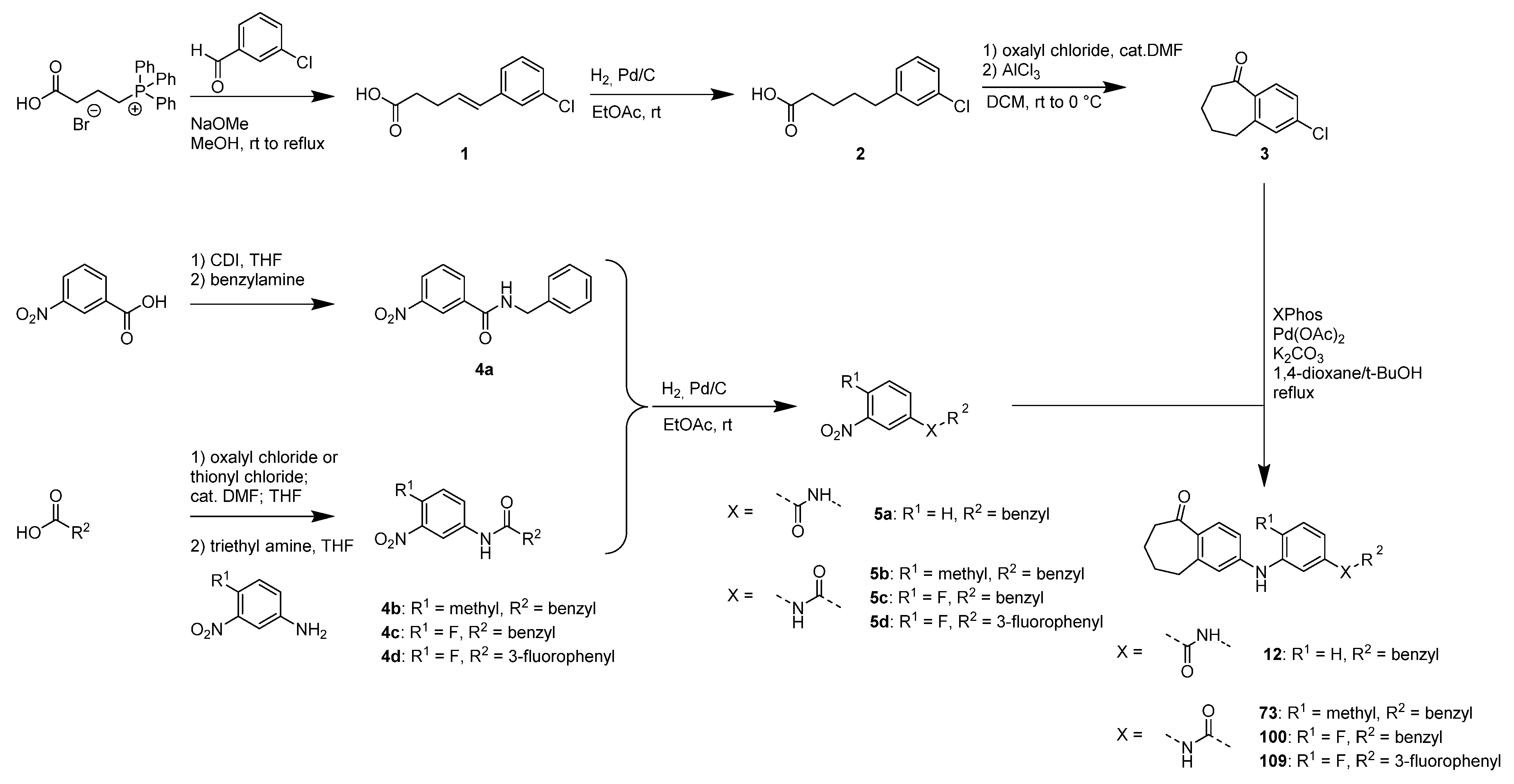
3.1. Identification of Novel PXR Inhibitors with Phenylaminobenzosuberone Scaffold
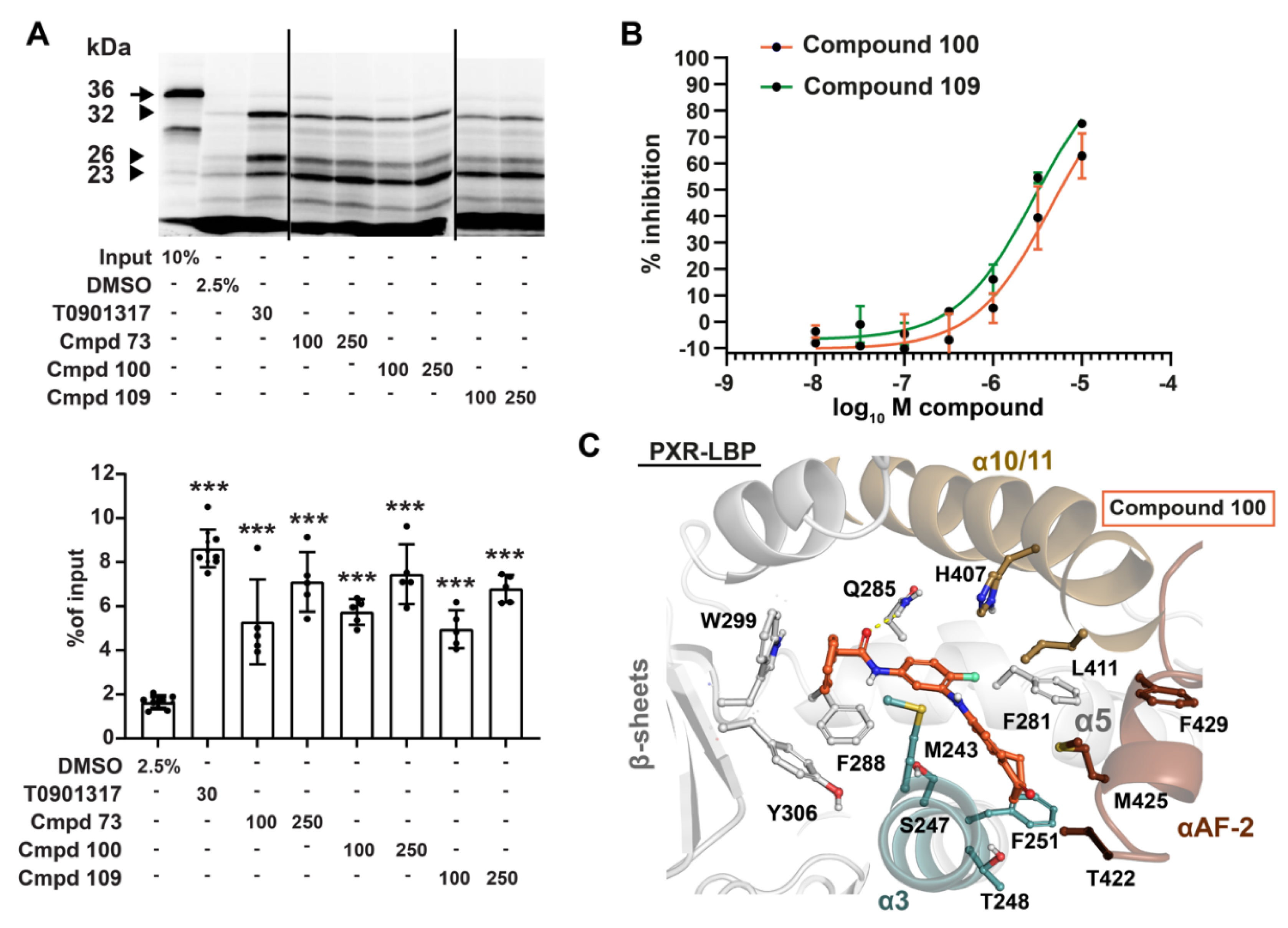
3.2. Compounds 73 and 100 Demonstrate Competitive Antagonism of PXR
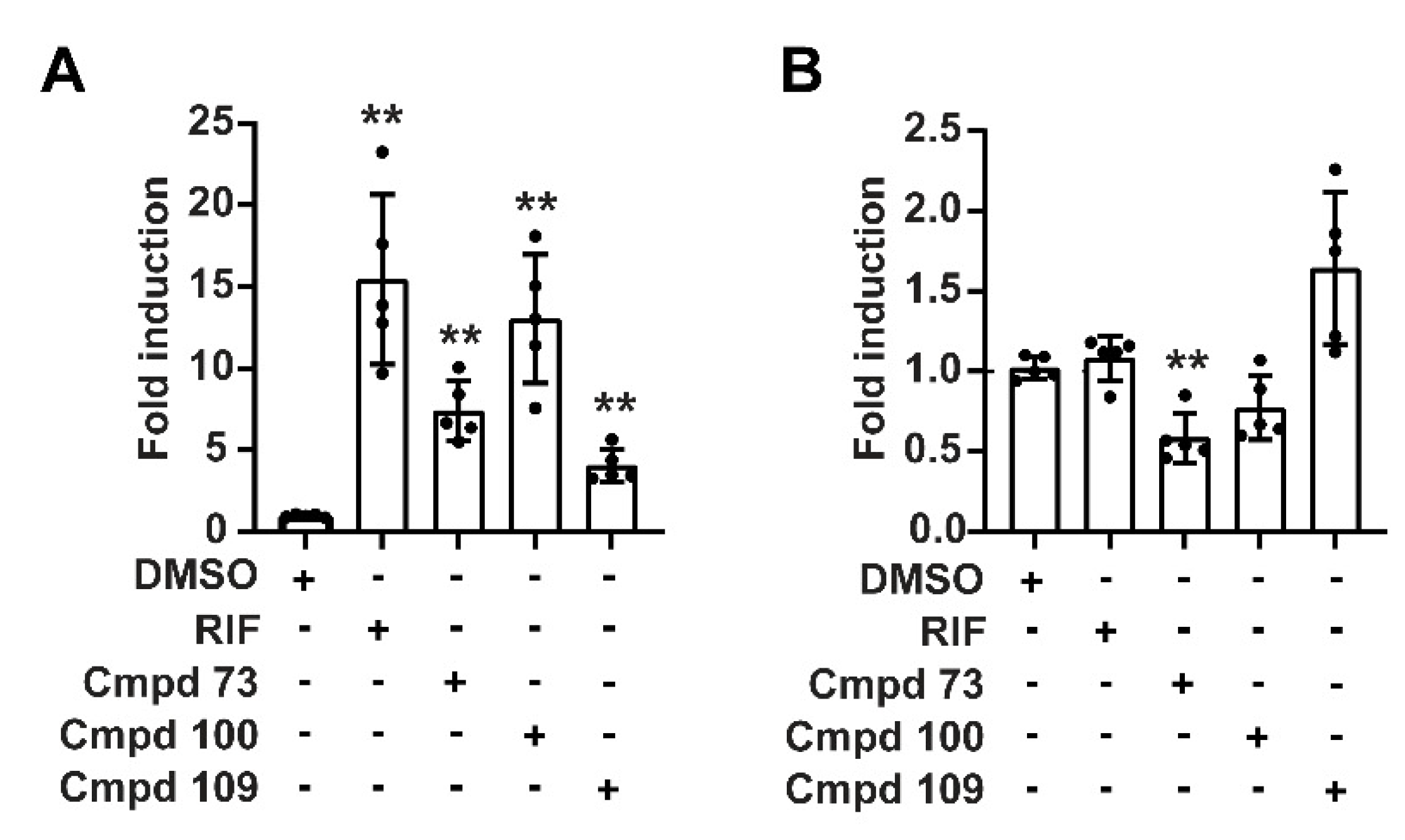
3.3. Compounds 73 and 100 Disrupt PXR’s Coregulatory Protein Interactions
3.4. Phenylaminobenzosuberones Demonstrate Direct Binding to the PXR-LBP
3.5. Kinase Inhibition Profile of 100 and 109
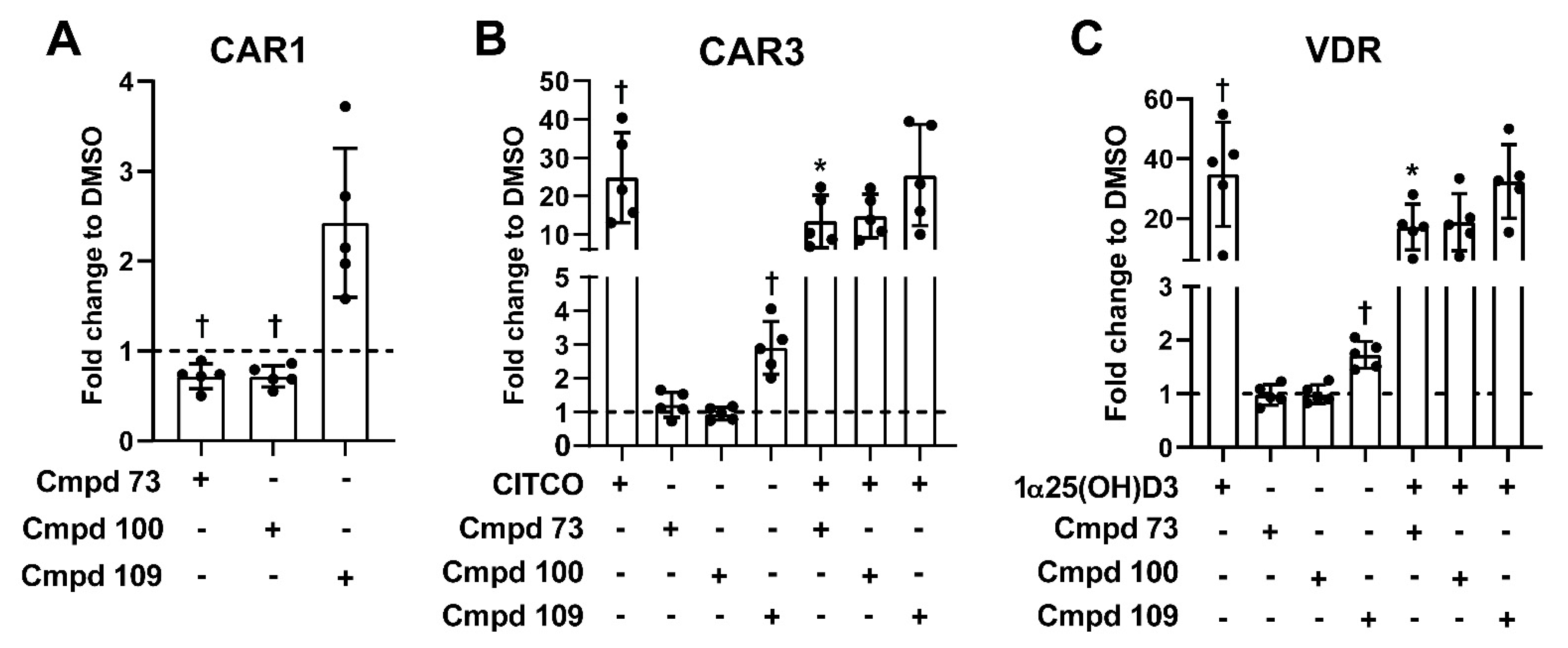
3.6. Nuclear Receptor Selectivity of Novel PXR Ligands
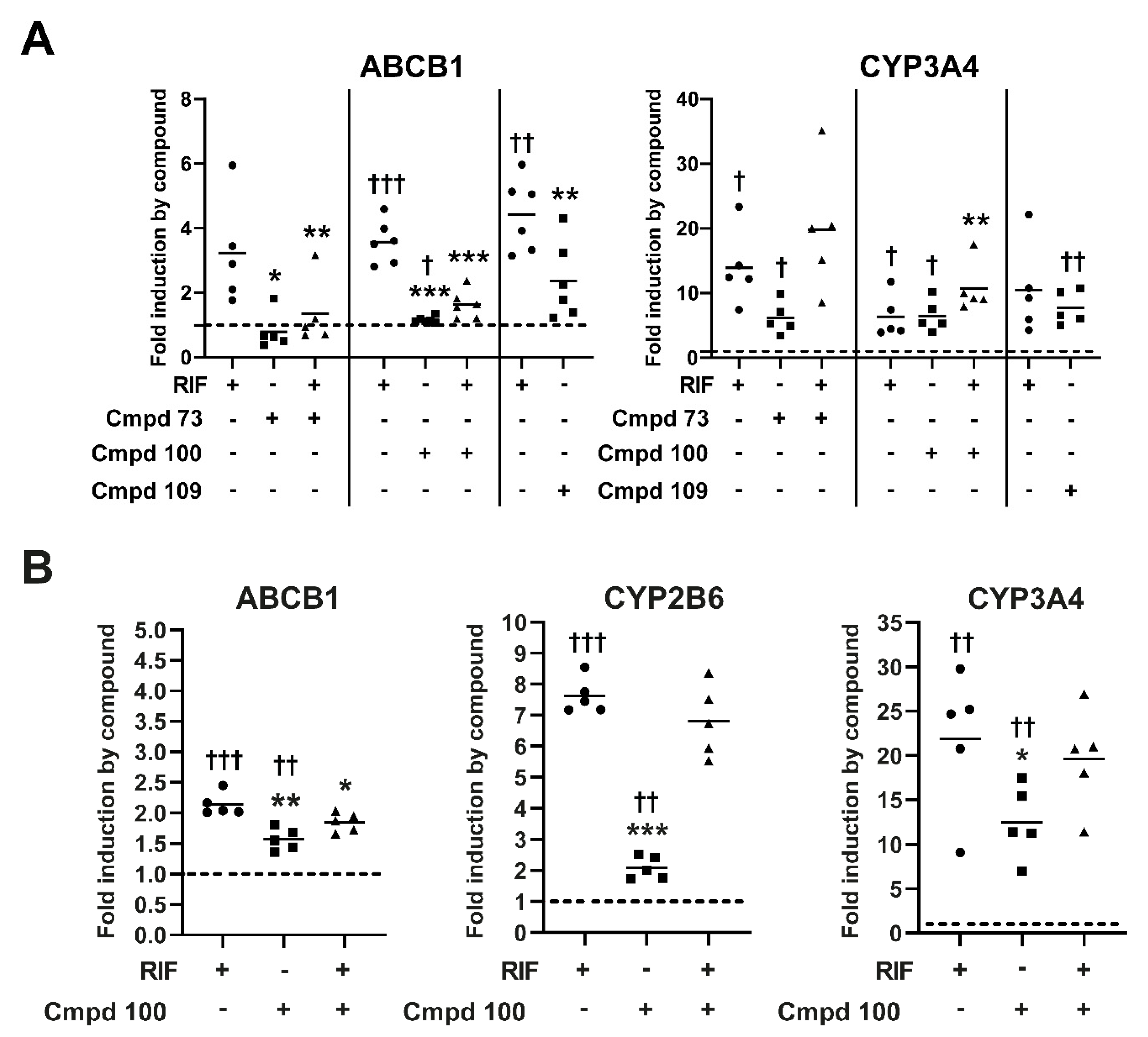
3.7. Expression of Prototypical Endogenous PXR Target Genes Is Differentially Modulated by the Compounds
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kannaiyan, R.; Mahadevan, D. A Comprehensive Review of Protein Kinase Inhibitors for Cancer Therapy. Expert Rev. Anticancer Ther. 2018, 18, 1249–1270. [Google Scholar] [CrossRef] [PubMed]
- Attwood, M.M.; Fabbro, D.; Sokolov, A.V.; Knapp, S.; Schiöth, H.B. Trends in kinase drug discovery: Targets, indications and inhibitor design. Nat. Rev. Drug Discov. 2021, 20, 839–861. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2022 update. Pharmacol. Res. 2022, 175, 106037. [Google Scholar] [CrossRef] [PubMed]
- Munoz, L. Non-kinase targets of protein kinase inhibitors. Nat. Rev. Drug Discov. 2017, 16, 424–440. [Google Scholar] [CrossRef] [PubMed]
- Pollet, M.; Krutmann, J.; Haarmann-Stemmann, T. Commentary: Usage of Mitogen-Activated Protein Kinase Small Molecule Inhibitors: More Than Just Inhibition! Front. Pharmacol. 2018, 9, 935. [Google Scholar] [CrossRef]
- Wang, Y.-M.; Ong, S.S.; Chai, S.C.; Chen, T. Role of CAR and PXR in Xenobiotic Sensing and Metabolism. Expert Opin. Drug Metab. Toxicol. 2012, 8, 803–817. [Google Scholar] [CrossRef] [Green Version]
- Pondugula, S.R.; Pavek, P.; Mani, S. Pregnane X Receptor and Cancer: Context-Specificity is Key. Nucl. Recept. Res. 2016, 3, 101198. [Google Scholar] [CrossRef] [Green Version]
- Niu, X.; Wu, T.; Li, G.; Gu, X.; Tian, Y.; Cui, H. Insights into the critical role of the PXR in preventing carcinogenesis and chemotherapeutic drug resistance. Int. J. Biol. Sci. 2022, 18, 742–759. [Google Scholar] [CrossRef]
- Xing, Y.; Yan, J.; Niu, Y. PXR: A center of transcriptional regulation in cancer. Acta Pharm. Sin. B 2020, 10, 197–206. [Google Scholar] [CrossRef]
- Masuyama, H.; Nakamura, K.; Nobumoto, E.; Hiramatsu, Y. Inhibition of pregnane X receptor pathway contributes to the cell growth inhibition and apoptosis of anticancer agents in ovarian cancer cells. Int. J. Oncol. 2016, 49, 1211–1220. [Google Scholar] [CrossRef] [Green Version]
- Staudinger, J.L. Clinical applications of small molecule inhibitors of Pregnane X receptor. Mol. Cell. Endocrinol. 2019, 485, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Chai, S.C.; Wright, W.C.; Chen, T. Strategies for developing pregnane X receptor antagonists: Implications from metabolism to cancer. Med. Res. Rev. 2020, 40, 1061–1083. [Google Scholar] [CrossRef] [PubMed]
- Harmsen, S.; Meijerman, I.; Maas-Bakker, R.F.; Beijnen, J.H.; Schellens, J.H.M. PXR-mediated P-glycoprotein induction by small molecule tyrosine kinase inhibitors. Eur. J. Pharm. Sci. 2013, 48, 644–649. [Google Scholar] [CrossRef] [PubMed]
- Smutny, T.; Bitman, M.; Urban, M.; Dubecka, M.; Vrzal, R.; Dvorak, Z.; Pavek, P. U0126, a mitogen-activated protein kinase kinase 1 and 2 (MEK1 and 2) inhibitor, selectively up-regulates main isoforms of CYP3A subfamily via a pregnane X receptor (PXR) in HepG2 cells. Arch. Toxicol. 2014, 88, 2243–2259. [Google Scholar] [CrossRef]
- Creusot, N.; Gassiot, M.; Alaterre, E.; Chiavarina, B.; Grimaldi, M.; Boulahtouf, A.; Toporova, L.; Gerbal-Chaloin, S.; Daujat-Chavanieu, M.; Matheux, A.; et al. The Anti-Cancer Drug Dabrafenib Is a Potent Activator of the Human Pregnane X Receptor. Cells 2020, 9, 1641. [Google Scholar] [CrossRef]
- Burk, O.; Kuzikov, M.; Kronenberger, T.; Jeske, J.; Keminer, O.; Thasler, W.E.; Schwab, M.; Wrenger, C.; Windshügel, B. Identification of approved drugs as potent inhibitors of pregnane X receptor activation with differential receptor interaction profiles. Arch. Toxicol. 2018, 92, 1435–1451. [Google Scholar] [CrossRef]
- Martz, K.E.; Dorn, A.; Baur, B.; Schattel, V.; Goettert, M.I.; Mayer-Wrangowski, S.C.; Rauh, D.; Laufer, S.A. Targeting the Hinge Glycine Flip and the Activation Loop: Novel Approach to Potent P38α Inhibitors. J. Med. Chem. 2012, 55, 7862–7874. [Google Scholar] [CrossRef]
- Roos, K.; Wu, C.; Damm, W.; Reboul, M.; Stevenson, J.M.; Lu, C.; Dahlgren, M.K.; Mondal, S.; Chen, W.; Wang, L.; et al. OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules. J. Chem. Theory Comput. 2019, 15, 1863–1874. [Google Scholar] [CrossRef]
- Greenwood, J.R.; Calkins, D.; Sullivan, A.P.; Shelley, J.C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput. Aided Mol. Des. 2010, 24, 591–604. [Google Scholar] [CrossRef]
- Wallace, B.D.; Betts, L.; Talmage, G.; Pollet, R.M.; Holman, N.S.; Redinbo, M.R. Structural and Functional Analysis of the Human Nuclear Xenobiotic Receptor PXR in Complex with RXRα. J. Mol. Biol. 2013, 425, 2561–2577. [Google Scholar] [CrossRef] [Green Version]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Farid, R.; Day, T.; Friesner, R.A.; Pearlstein, R.A. New insights about HERG blockade obtained from protein modeling, potential energy mapping, and docking studies. Bioorg. Med. Chem. 2006, 14, 3160–3173. [Google Scholar] [CrossRef] [PubMed]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef] [PubMed]
- Sherman, W.; Beard, H.S.; Farid, R. Use of an induced fit receptor structure in virtual screening. Chem. Biol. Drug Des. 2006, 67, 83–84. [Google Scholar] [CrossRef]
- Bochevarov, A.D.; Harder, E.; Hughes, T.F.; Greenwood, J.R.; Braden, D.A.; Philipp, D.M.; Rinaldo, D.; Halls, M.D.; Zhang, J.; Friesner, R.A. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 2013, 113, 2110–2142. [Google Scholar] [CrossRef]
- Burk, O.; Tegude, H.; Koch, I.; Hustert, E.; Wolbold, R.; Glaeser, H.; Klein, K.; Fromm, M.F.; Nuessler, A.K.; Neuhaus, P.; et al. Molecular mechanisms of polymorphic CYP3A7 expression in adult human liver and intestine. J. Biol. Chem. 2002, 277, 24280–24288. [Google Scholar] [CrossRef] [Green Version]
- Geick, A.; Eichelbaum, M.; Burk, O. Nuclear receptor response elements mediate induction of intestinal MDR1 by rifampin. J. Biol. Chem. 2001, 276, 14581–14587. [Google Scholar] [CrossRef] [Green Version]
- Mathäs, M.; Burk, O.; Qiu, H.; Nusshag, C.; Gödtel-Armbrust, U.; Baranyai, D.; Deng, S.; Römer, K.; Nem, D.; Windshügel, B.; et al. Evolutionary history and functional characterization of the amphibian xenosensor CAR. Mol. Endocrinol. Baltim. Md 2012, 26, 14–26. [Google Scholar] [CrossRef] [Green Version]
- Burk, O.; Arnold, K.A.; Nussler, A.K.; Schaeffeler, E.; Efimova, E.; Avery, B.A.; Avery, M.A.; Fromm, M.F.; Eichelbaum, M. Antimalarial artemisinin drugs induce cytochrome P450 and MDR1 expression by activation of xenosensors pregnane X receptor and constitutive androstane receptor. Mol. Pharmacol. 2005, 67, 1954–1965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, K.A.; Eichelbaum, M.; Burk, O. Alternative Splicing Affects the Function and Tissue-Specific Expression of the Human Constitutive Androstane Receptor. Nucl. Recept. 2004, 2, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hustert, E.; Zibat, A.; Presecan-Siedel, E.; Eiselt, R.; Mueller, R.; Fuss, C.; Brehm, I.; Brinkmann, U.; Eichelbaum, M.; Wojnowski, L.; et al. Natural protein variants of pregnane X receptor with altered transactivation activity toward CYP3A4. Drug Metab. Dispos. Biol. Fate Chem. 2001, 29, 1454–1459. [Google Scholar] [PubMed]
- Bitter, A.; Rümmele, P.; Klein, K.; Kandel, B.A.; Rieger, J.K.; Nüssler, A.K.; Zanger, U.M.; Trauner, M.; Schwab, M.; Burk, O. Pregnane X receptor activation and silencing promote steatosis of human hepatic cells by distinct lipogenic mechanisms. Arch. Toxicol. 2015, 89, 2089–2103. [Google Scholar] [CrossRef] [PubMed]
- Anthérieu, S.; Chesné, C.; Li, R.; Camus, S.; Lahoz, A.; Picazo, L.; Turpeinen, M.; Tolonen, A.; Uusitalo, J.; Guguen-Guillouzo, C.; et al. Stable expression, activity, and inducibility of cytochromes P450 in differentiated HepaRG cells. Drug Metab. Dispos. Biol. Fate Chem. 2010, 38, 516–525. [Google Scholar] [CrossRef]
- Piedade, R.; Traub, S.; Bitter, A.; Nüssler, A.K.; Gil, J.P.; Schwab, M.; Burk, O. Carboxymefloquine, the major metabolite of the antimalarial drug mefloquine, induces drug-metabolizing enzyme and transporter expression by activation of pregnane X receptor. Antimicrob. Agents Chemother. 2015, 59, 96–104. [Google Scholar] [CrossRef] [Green Version]
- Jeske, J.; Windshügel, B.; Thasler, W.E.; Schwab, M.; Burk, O. Human pregnane X receptor is activated by dibenzazepine carbamate-based inhibitors of constitutive androstane receptor. Arch. Toxicol. 2017, 91, 2375–2390. [Google Scholar] [CrossRef]
- Zhu, Z.; Kim, S.; Chen, T.; Lin, J.-H.; Bell, A.; Bryson, J.; Dubaquie, Y.; Yan, N.; Yanchunas, J.; Xie, D.; et al. Correlation of high-throughput pregnane X receptor (PXR) transactivation and binding assays. J. Biomol. Screen. 2004, 9, 533–540. [Google Scholar] [CrossRef] [Green Version]
- Hoffart, E.; Ghebreghiorghis, L.; Nussler, A.K.; Thasler, W.E.; Weiss, T.S.; Schwab, M.; Burk, O. Effects of atorvastatin metabolites on induction of drug-metabolizing enzymes and membrane transporters through human pregnane X receptor. Br. J. Pharmacol. 2012, 165, 1595–1608. [Google Scholar] [CrossRef] [Green Version]
- Schoch, G.A.; D’Arcy, B.; Stihle, M.; Burger, D.; Bär, D.; Benz, J.; Thoma, R.; Ruf, A. Molecular Switch in the Glucocorticoid Receptor: Active and Passive Antagonist Conformations. J. Mol. Biol. 2010, 395, 568–577. [Google Scholar] [CrossRef]
- Allan, G.F.; Leng, X.; Tsai, S.Y.; Weigel, N.L.; Edwards, D.P.; Tsai, M.J.; O’Malley, B.W. Hormone and antihormone induce distinct conformational changes which are central to steroid receptor activation. J. Biol. Chem. 1992, 267, 19513–19520. [Google Scholar] [CrossRef]
- Rashidian, A.; Mustonen, E.-K.; Kronenberger, T.; Schwab, M.; Burk, O.; Laufer, S.A.; Pantsar, T. Discrepancy in interactions and conformational dynamics of pregnane X receptor (PXR) bound to an agonist and a novel competitive antagonist. Comput. Struct. Biotechnol. J. 2022, 2022. submitted. [Google Scholar]
- Pissios, P.; Tzameli, I.; Kushner, P.; Moore, D.D. Dynamic stabilization of nuclear receptor ligand binding domains by hormone or corepressor binding. Mol. Cell 2000, 6, 245–253. [Google Scholar] [CrossRef]
- Lin, W.; Wang, Y.-M.; Chai, S.C.; Lv, L.; Zheng, J.; Wu, J.; Zhang, Q.; Wang, Y.-D.; Griffin, P.R.; Chen, T. SPA70 is a potent antagonist of human pregnane X receptor. Nat. Commun. 2017, 8, 741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Tang, Y.; Robbins, G.T.; Nie, D. Camptothecin attenuates cytochrome P450 3A4 induction by blocking the activation of human pregnane X receptor. J. Pharmacol. Exp. Ther. 2010, 334, 999–1008. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Wang, H.; Sinz, M.; Zoeckler, M.; Staudinger, J.; Redinbo, M.R.; Teotico, D.G.; Locker, J.; Kalpana, G.V.; Mani, S. Inhibition of drug metabolism by blocking the activation of nuclear receptors by ketoconazole. Oncogene 2007, 26, 258–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Li, H.; Moore, L.B.; Johnson, M.D.L.; Maglich, J.M.; Goodwin, B.; Ittoop, O.R.R.; Wisely, B.; Creech, K.; Parks, D.J.; et al. The Phytoestrogen Coumestrol Is a Naturally Occurring Antagonist of the Human Pregnane X Receptor. Mol. Endocrinol. 2008, 22, 838–857. [Google Scholar] [CrossRef]
- Ding, X.; Staudinger, J.L. Repression of PXR-mediated induction of hepatic CYP3A gene expression by protein kinase C. Biochem. Pharmacol. 2005, 69, 867–873. [Google Scholar] [CrossRef]
- Lichti-Kaiser, K.; Xu, C.; Staudinger, J.L. Cyclic AMP-dependent protein kinase signaling modulates pregnane x receptor activity in a species-specific manner. J. Biol. Chem. 2009, 284, 6639–6649. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.; Wu, J.; Dong, H.; Bouck, D.; Zeng, F.-Y.; Chen, T. Cyclin-dependent kinase 2 negatively regulates human pregnane X receptor-mediated CYP3A4 gene expression in HepG2 liver carcinoma cells. J. Biol. Chem. 2008, 283, 30650–30657. [Google Scholar] [CrossRef] [Green Version]
- Pondugula, S.R.; Brimer-Cline, C.; Wu, J.; Schuetz, E.G.; Tyagi, R.K.; Chen, T. A phosphomimetic mutation at threonine-57 abolishes transactivation activity and alters nuclear localization pattern of human pregnane x receptor. Drug Metab. Dispos. Biol. Fate Chem. 2009, 37, 719–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taneja, G.; Chu, C.; Maturu, P.; Moorthy, B.; Ghose, R. Role of c-Jun-N-Terminal Kinase in Pregnane X Receptor-Mediated Induction of Human Cytochrome P4503A4 In Vitro. Drug Metab. Dispos. Biol. Fate Chem. 2018, 46, 397–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, B.; Li, S.; Dong, D. 3D structures and ligand specificities of nuclear xenobiotic receptors CAR, PXR and VDR. Drug Discov. Today 2013, 18, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Aninat, C.; Piton, A.; Glaise, D.; Le Charpentier, T.; Langouët, S.; Morel, F.; Guguen-Guillouzo, C.; Guillouzo, A. Expression of Cytochromes P450, Conjugating Enzymes and Nuclear Receptors in Human Hepatoma HepaRG Cells. Drug Metab. Dispos. 2006, 34, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, A.; Chanteux, H.; Ménochet, K.; Ledecq, M.; Schulze, M.-S.E.D. Designing Out PXR Activity on Drug Discovery Projects: A Review of Structure-Based Methods, Empirical and Computational Approaches. J. Med. Chem. 2021, 64, 6413–6522. [Google Scholar] [CrossRef] [PubMed]
- Pantsar, T.; Poso, A. Binding Affinity via Docking: Fact and Fiction. Molecules 2018, 23, 1899. [Google Scholar] [CrossRef] [Green Version]
- Burk, O.; Piedade, R.; Ghebreghiorghis, L.; Fait, J.T.; Nussler, A.K.; Gil, J.P.; Windshügel, B.; Schwab, M. Differential effects of clinically used derivatives and metabolites of artemisinin in the activation of constitutive androstane receptor isoforms. Br. J. Pharmacol. 2012, 167, 666–681. [Google Scholar] [CrossRef]
- Li, Y.; Lin, W.; Wright, W.C.; Chai, S.C.; Wu, J.; Chen, T. Building a Chemical Toolbox for Human Pregnane X Receptor Research: Discovery of Agonists, Inverse Agonists, and Antagonists Among Analogs Based on the Unique Chemical Scaffold of SPA70. J. Med. Chem. 2021, 64, 1733–1761. [Google Scholar] [CrossRef]
- Burk, O.; Kronenberger, T.; Keminer, O.; Lee, S.M.L.; Schiergens, T.S.; Schwab, M.; Windshügel, B. Nelfinavir and Its Active Metabolite M8 Are Partial Agonists and Competitive Antagonists of the Human Pregnane X Receptor. Mol. Pharmacol. 2021, 99, 184–196. [Google Scholar] [CrossRef]
- Johnson, A.B.; O’Malley, B.W. Steroid receptor coactivators 1, 2, and 3: Critical regulators of nuclear receptor activity and steroid receptor modulator (SRM)-based cancer therapy. Mol. Cell. Endocrinol. 2012, 348, 430–439. [Google Scholar] [CrossRef] [Green Version]
- Hendriks, D.F.G.; Vorrink, S.U.; Smutny, T.; Sim, S.C.; Nordling, Å.; Ullah, S.; Kumondai, M.; Jones, B.C.; Johansson, I.; Andersson, T.B.; et al. Clinically Relevant Cytochrome P450 3A4 Induction Mechanisms and Drug Screening in Three-Dimensional Spheroid Cultures of Primary Human Hepatocytes. Clin. Pharmacol. Ther. 2020, 108, 844–855. [Google Scholar] [CrossRef] [PubMed]
- Dohse, M.; Scharenberg, C.; Shukla, S.; Robey, R.W.; Volkmann, T.; Deeken, J.F.; Brendel, C.; Ambudkar, S.V.; Neubauer, A.; Bates, S.E. Comparison of ATP-binding cassette transporter interactions with the tyrosine kinase inhibitors imatinib, nilotinib, and dasatinib. Drug Metab. Dispos. Biol. Fate Chem. 2010, 38, 1371–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Multi-Disciplinary Review and Evaluation NDA 210496 BRAFTOVI™ (encorafenib); U.S. Food & Drug Administration: Silver Spring, MD, UDA. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/210496Orig1s000MultidisciplineR.pdf (accessed on 6 April 2022).
- Multi-Disciplinary Review and Evaluation NDA 210868 LORBRENATM (lorlatinib); U.S. Food & Drug Administration: Silver Spring, MD, UDA. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/210868Orig1s000MultidisciplineR.pdf (accessed on 6 April 2022).
- Multi-Disciplinary Review and Evaluation NDA 208772 ALUNBRIGTM (brigatinib); U.S. Food & Drug Administration; U.S. Food & Drug Administration: Silver Spring, MD, UDA. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/208772Orig1s000MultidisciplineR.pdf (accessed on 6 April 2022).
- Wang, X.; Zhang, X.; Huang, X.; Li, Y.; Wu, M.; Liu, J. The drug-drug interaction of sorafenib mediated by P-glycoprotein and CYP3A4. Xenobiotica Fate Foreign Compd. Biol. Syst. 2016, 46, 651–658. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, A.K.; McLaughlin, L.A.; Henderson, C.J.; Wolf, C.R. Activation status of the Pregnane X Receptor (PXR) influences Vemurafenib availability in humanized mouse models. Cancer Res. 2015, 75, 4573–4581. [Google Scholar] [CrossRef] [Green Version]
- Schneider, M.; Delfosse, V.; Gelin, M.; Grimaldi, M.; Granell, M.; Heriaud, L.; Pons, J.-L.; Cohen Gonsaud, M.; Balaguer, P.; Bourguet, W.; et al. Structure-Based and Knowledge-Informed Design of B-Raf Inhibitors Devoid of Deleterious PXR Binding. J. Med. Chem. 2022, 65, 1552–1566. [Google Scholar] [CrossRef]
- Feng, F.; Jiang, Q.; Cao, S.; Cao, Y.; Li, R.; Shen, L.; Zhu, H.; Wang, T.; Sun, L.; Liang, E.; et al. Pregnane X receptor mediates sorafenib resistance in advanced hepatocellular carcinoma. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 1017–1030. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mustonen, E.-K.; Pantsar, T.; Rashidian, A.; Reiner, J.; Schwab, M.; Laufer, S.; Burk, O. Target Hopping from Protein Kinases to PXR: Identification of Small-Molecule Protein Kinase Inhibitors as Selective Modulators of Pregnane X Receptor from TüKIC Library. Cells 2022, 11, 1299. https://doi.org/10.3390/cells11081299
Mustonen E-K, Pantsar T, Rashidian A, Reiner J, Schwab M, Laufer S, Burk O. Target Hopping from Protein Kinases to PXR: Identification of Small-Molecule Protein Kinase Inhibitors as Selective Modulators of Pregnane X Receptor from TüKIC Library. Cells. 2022; 11(8):1299. https://doi.org/10.3390/cells11081299
Chicago/Turabian StyleMustonen, Enni-Kaisa, Tatu Pantsar, Azam Rashidian, Juliander Reiner, Matthias Schwab, Stefan Laufer, and Oliver Burk. 2022. "Target Hopping from Protein Kinases to PXR: Identification of Small-Molecule Protein Kinase Inhibitors as Selective Modulators of Pregnane X Receptor from TüKIC Library" Cells 11, no. 8: 1299. https://doi.org/10.3390/cells11081299