
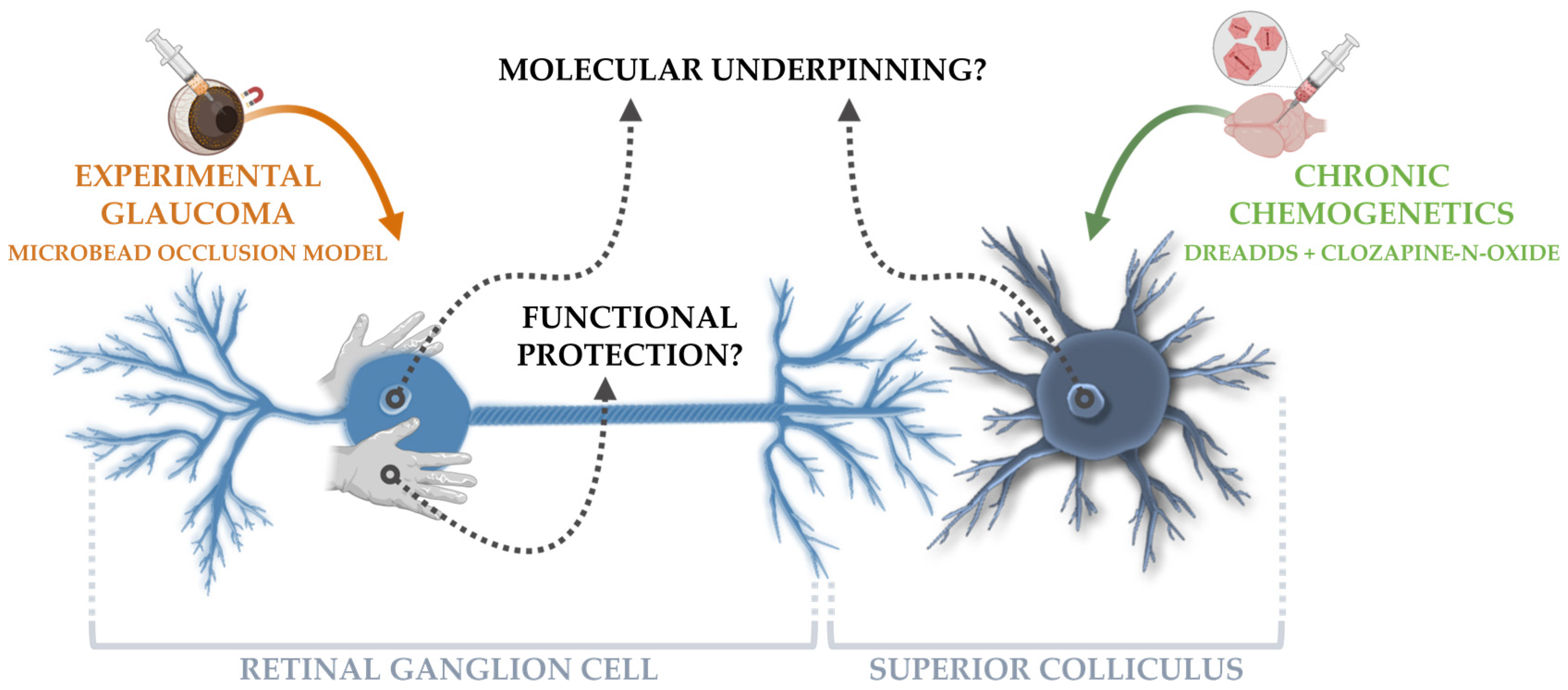
Chronic Chemogenetic Activation of the Superior Colliculus in Glaucomatous Mice: Local and Retrograde Molecular Signature

 ,
, 
 , , and
, , and
Abstract
:1. Introduction
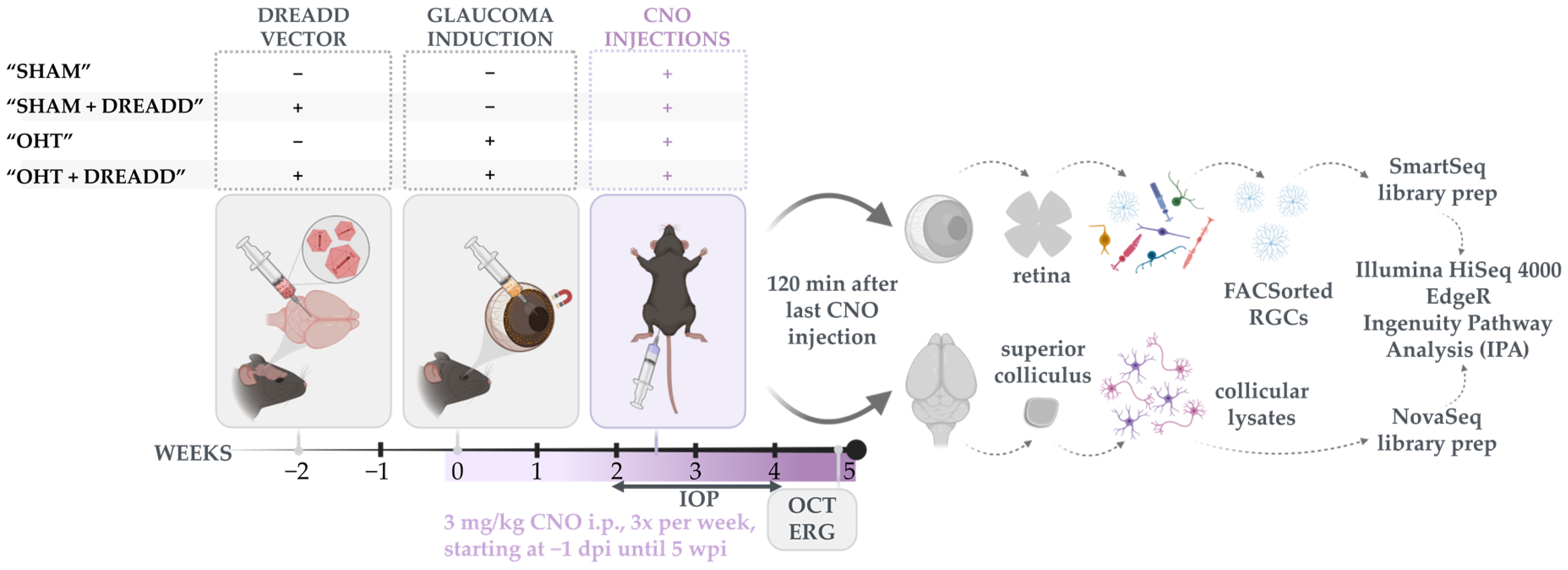
2. Materials and Methods
2.1. Experimental Animals
2.2. Stereotactic Viral Vector Injections
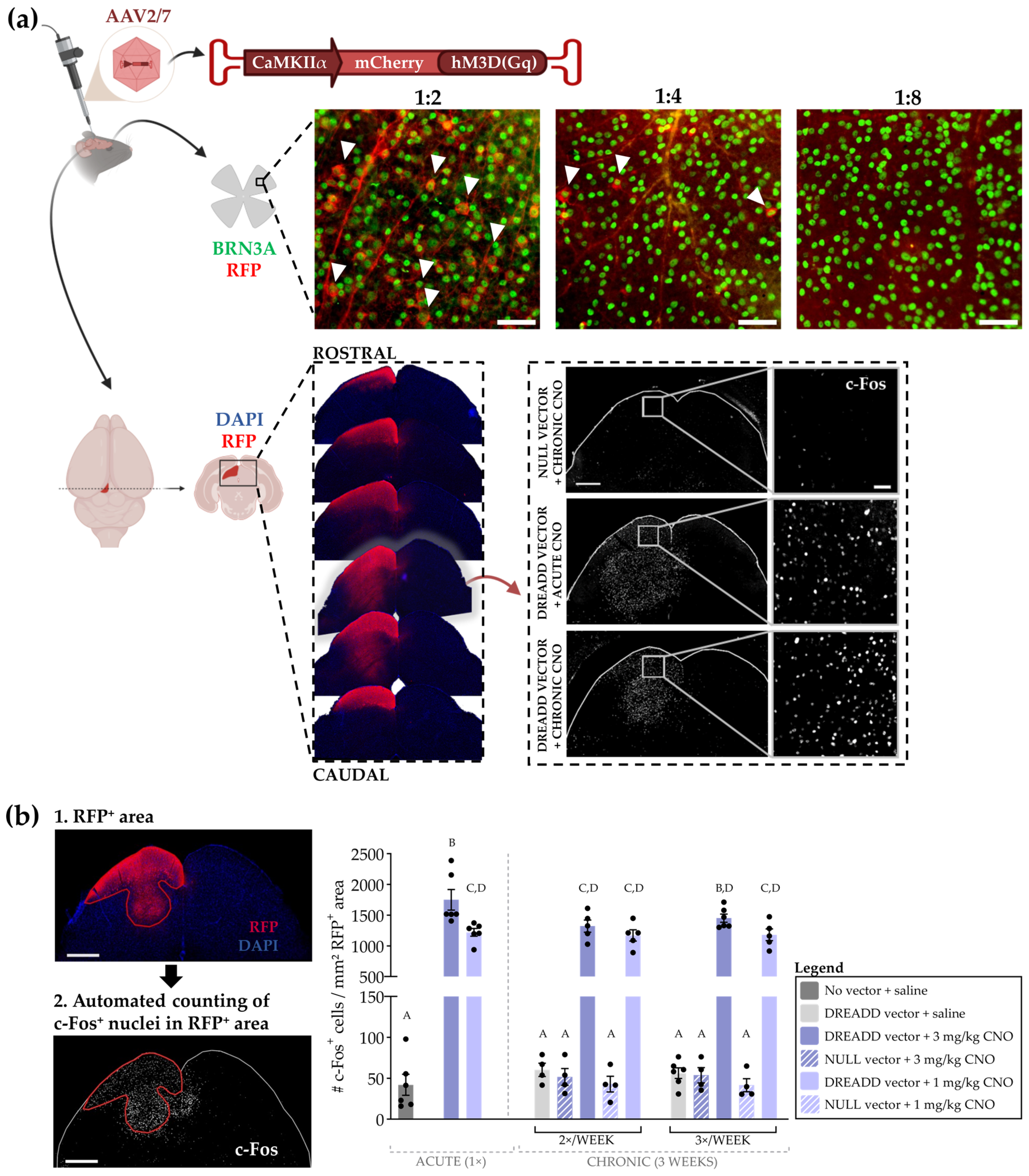
2.3. DREADD Optimization
2.3.1. Exclusion of Viral Vector Transport to the Retina
2.3.2. Neuronal Activation and Receptor Desensitization upon Chronic DREADD Activation
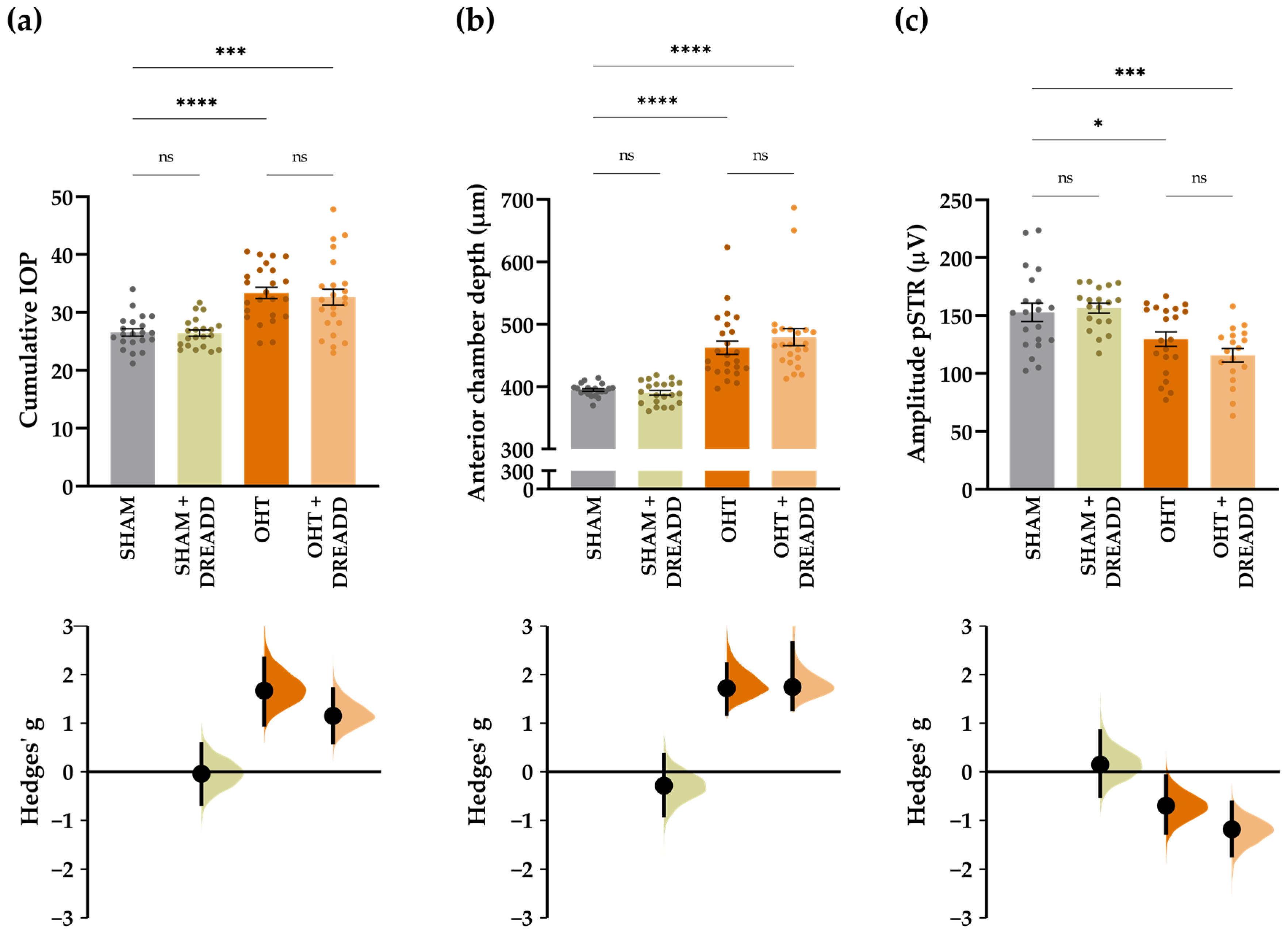
2.4. Experimental Glaucoma Model and In Vivo Read-outs to Probe Glaucoma Induction and Neuroprotection
2.5. Bulk RNA Sequencing on Sorted RGCs and Collicular Lysates
2.6. RNA Sequencing
2.7. Statistics
3. Results
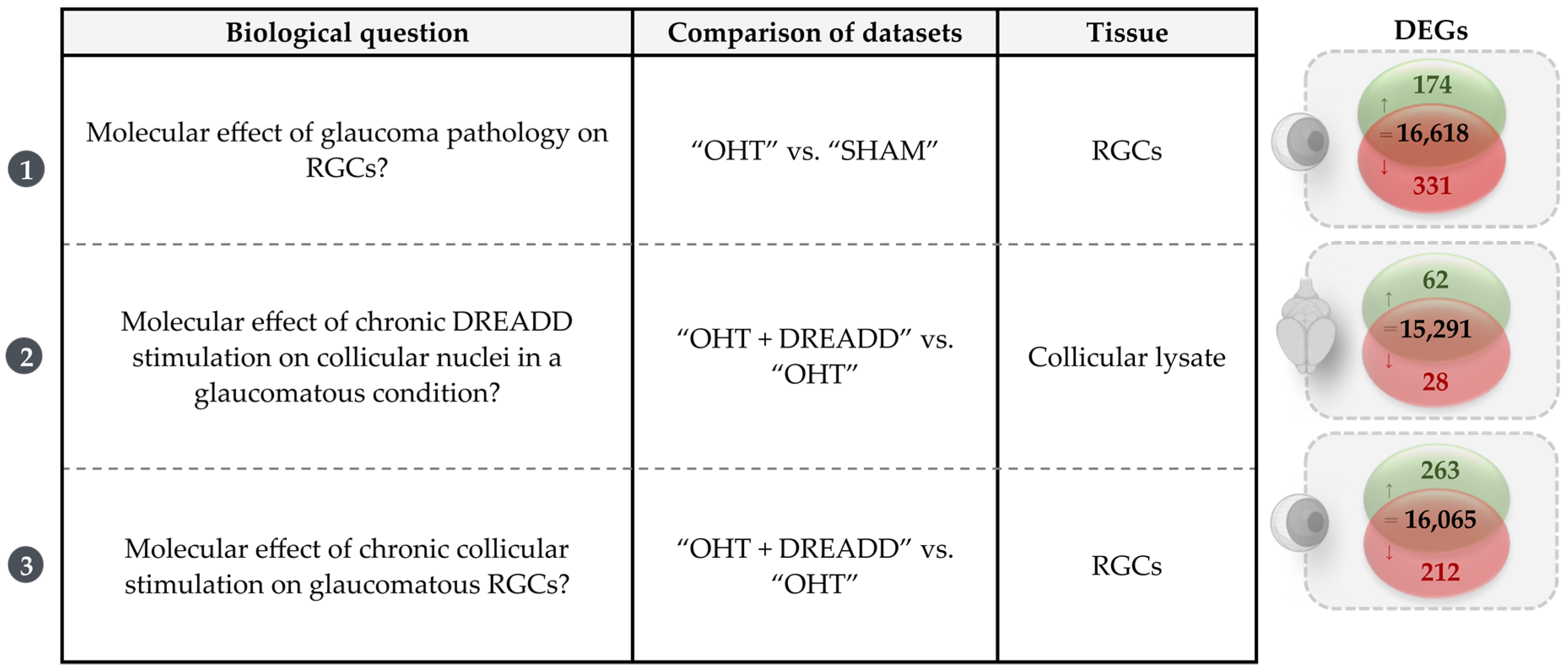
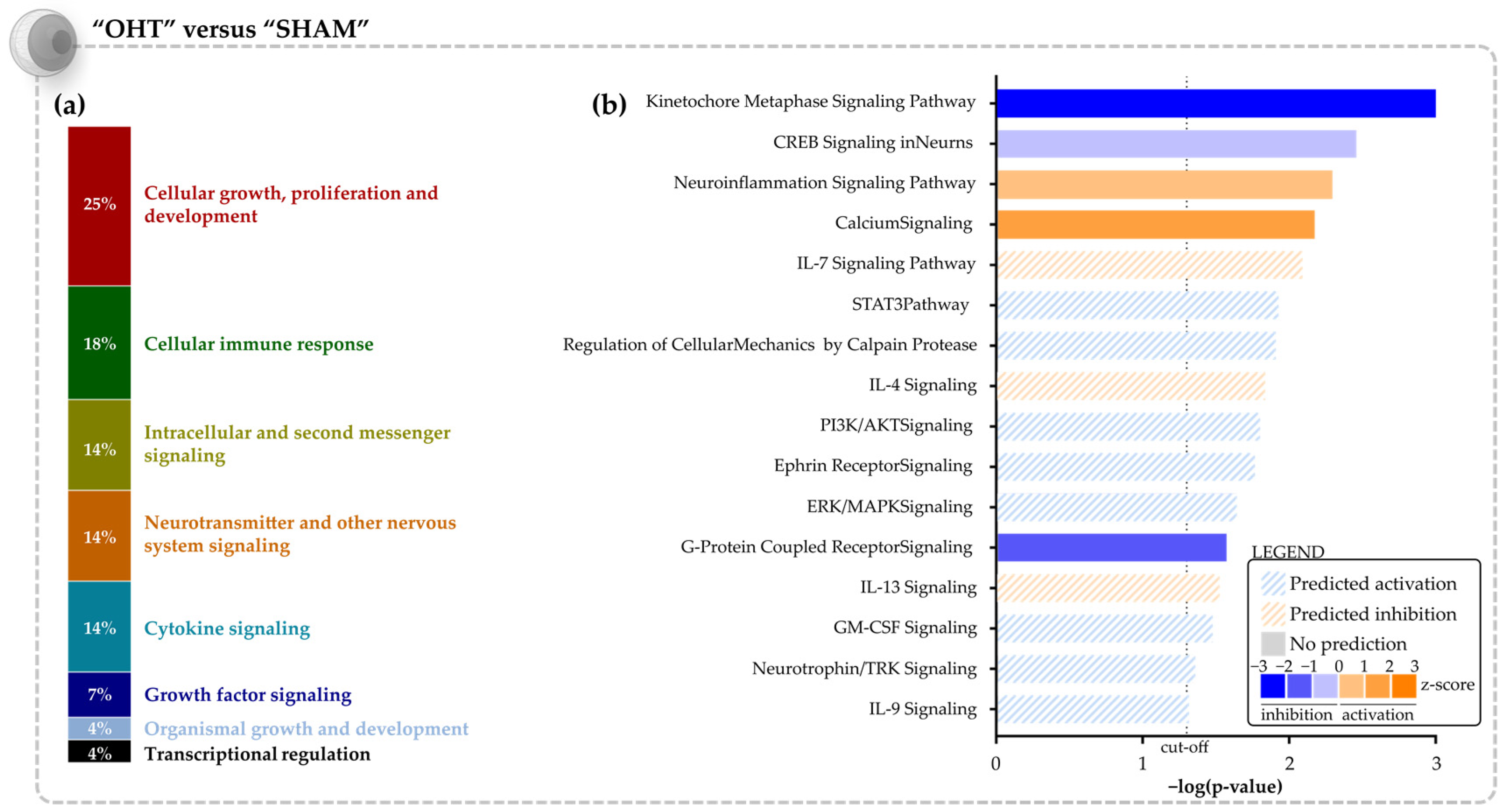
3.1. Molecular Signature of the Microbead Occlusion Model on the Retinocollicular System
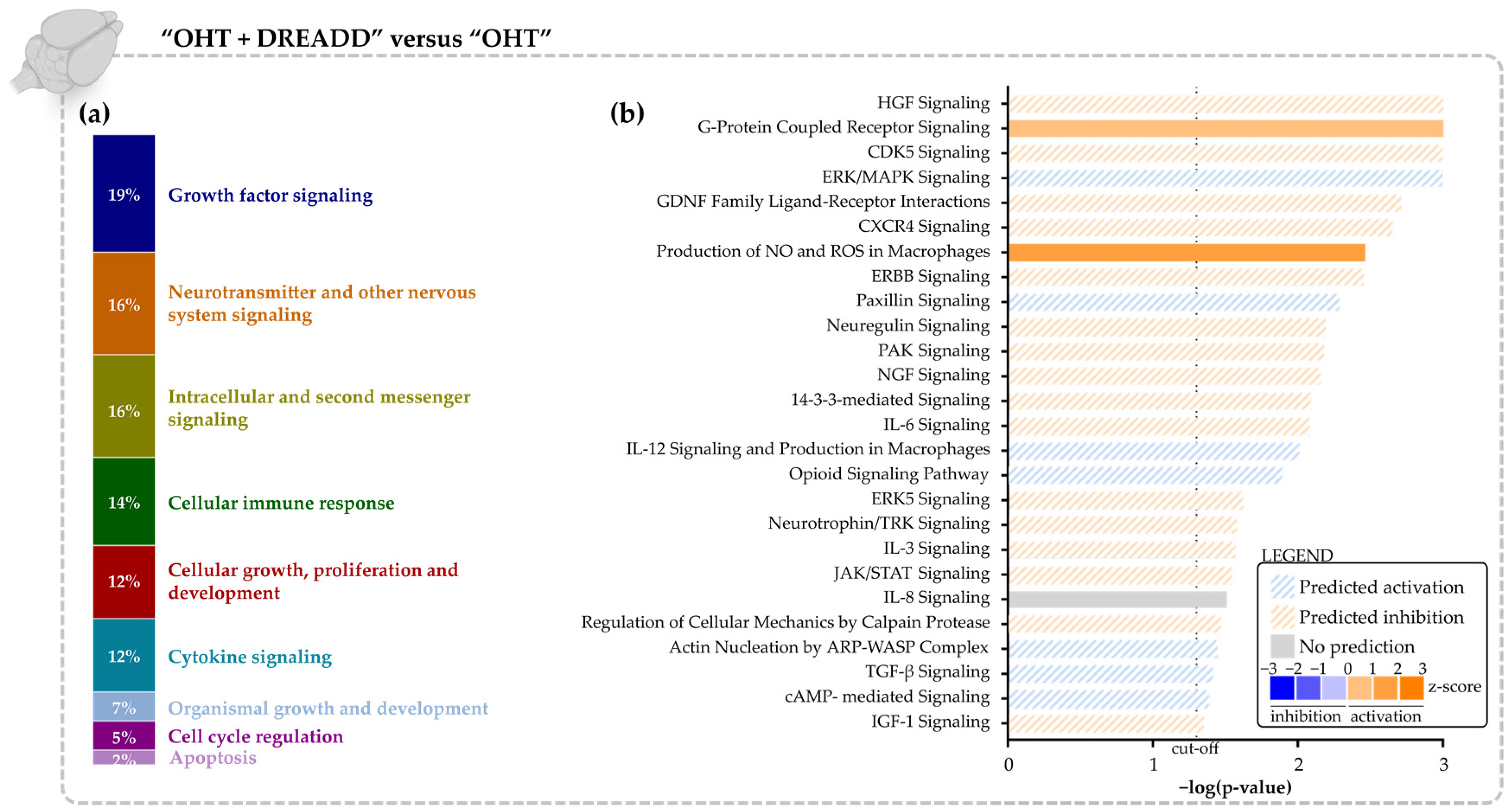
3.2. Chronic DREADD Stimulation in the Superior Colliculus Elicits Neuronal Activation and Is Linked to Growth Factor Signaling on a Molecular Level
3.2.1. Optimization of the Chronic DREADD Toolbox in the Retinocollicular System
3.2.2. Molecular Signature of Chronic DREADD Stimulation in the Superior Colliculus on Collicular Cells
3.3. Collicular Stimulation Does Not Functionally Rescue Glaucomatous Retinal Ganglion Cells, but Induces a Partial Transcriptomic Rescue with Activation of Promising Pro-Survival Pathways
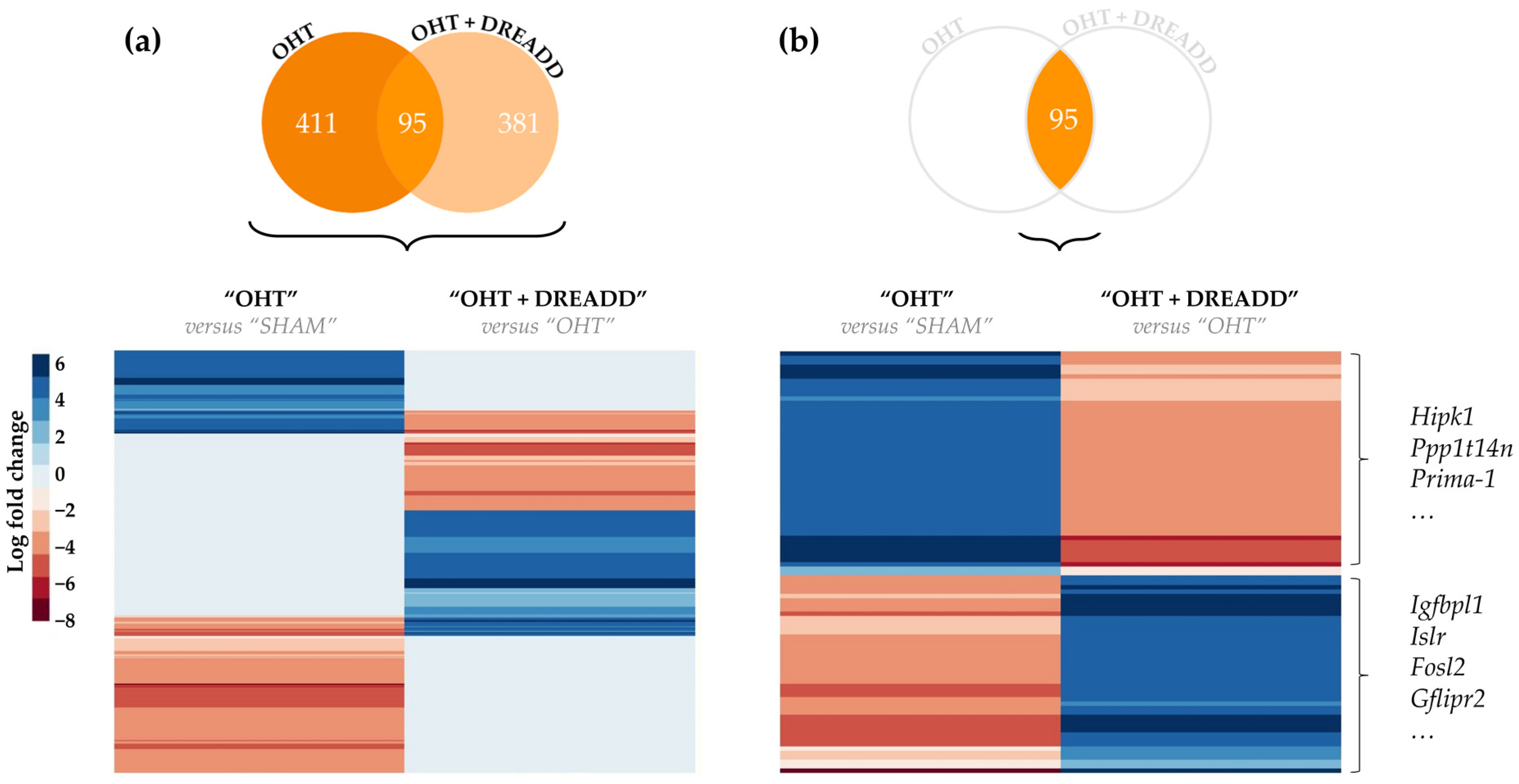
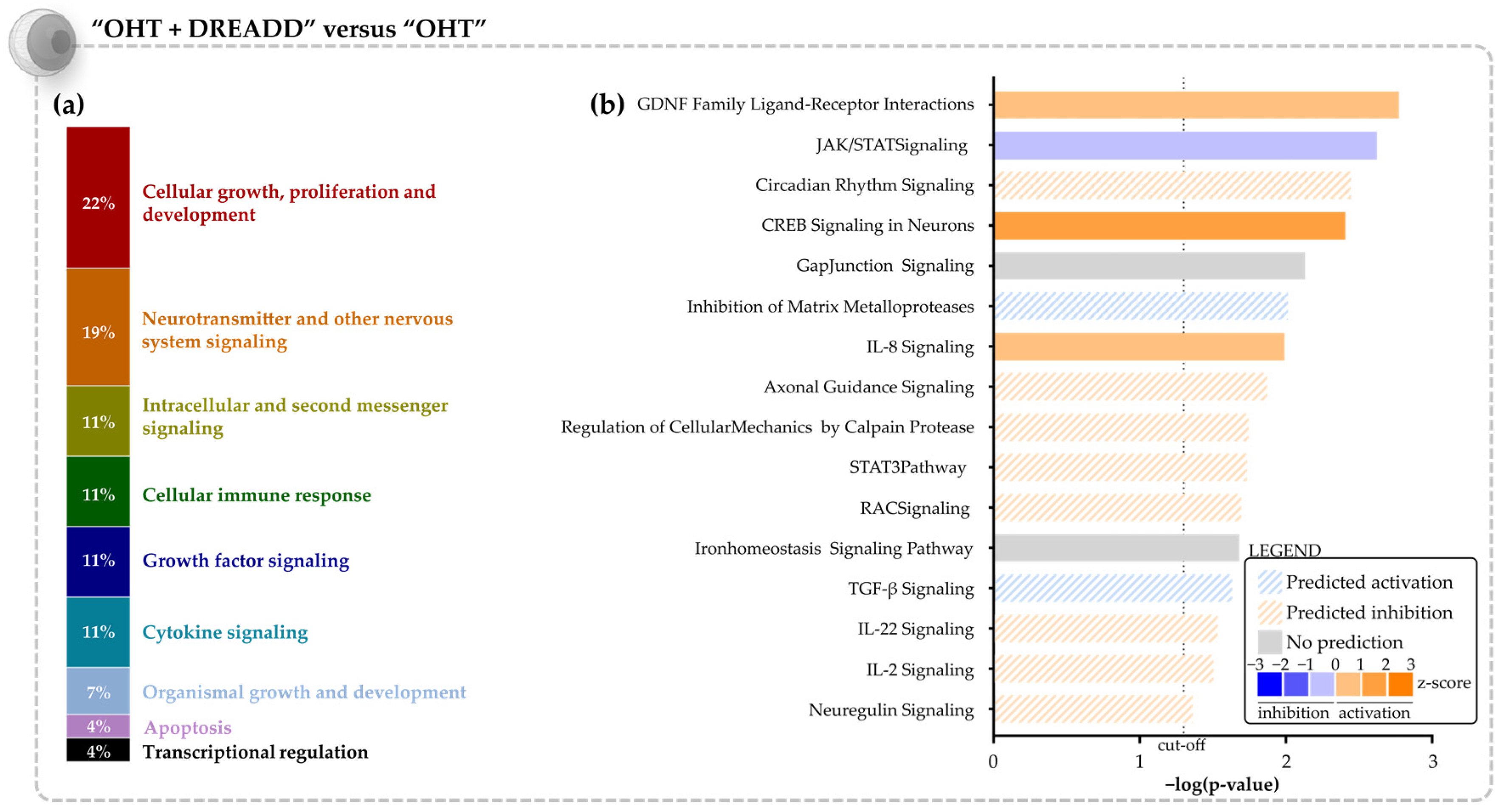
3.3.1. Molecular Effects of Chronic Target Stimulation on Unstimulated, Glaucomatous Retinal Ganglion Cells
3.3.2. Chronic Collicular Stimulation Does Not Alleviate RGC Injury in Glaucomatous Mice
4. Discussion
4.1. Growth Factors Are Key Regulators of the Transcriptomic Changes upon Chronic DREADD Activation
4.2. Transcriptomic Analysis Confirms the Pro-Survival Effects of Chronic Collicular Stimulation on Glaucomatous Retinal Ganglion Cells
Growth Factor Signaling Versus Axonal Regeneration and Dendritic Remodeling
4.3. Why Do These Pro-Survival Cues Not Result in Improved RGC Health?
4.3.1. Hazards of Chronic DREADD Stimulation: Receptor Desensitization and Neuroadaptive Changes
4.3.2. Complexity of Neurotrophic Factor Signaling
4.3.3. Duality in Functioning: Pro-Apoptosis or Pro-Survival?
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Watson, F.L.; Heerssen, H.M.; Bhattacharyya, A.; Klesse, L.; Lin, M.Z.; Segal, R.A. Neurotrophins use the Erk5 pathway to mediate a retrograde survival response. Nat. Neurosci. 2001, 4, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Almasieh, M.; Wilson, A.M.; Morquette, B.; Cueva Vargas, J.L.; Di Polo, A. The molecular basis of retinal ganglion cell death in glaucoma. Prog. Retin. Eye Res. 2012, 31, 152–181. [Google Scholar] [CrossRef] [PubMed]
- Nickells, R.W. From ocular hypertension to ganglion cell death: A theoretical sequence of events leading to glaucoma. Can. J. Ophthalmol. 2007, 42, 278–287. [Google Scholar] [CrossRef]
- Nickells, R.W. The Cell and Molecular Biology of Glaucoma: Mechanisms of Retinal Ganglion Cell Death. Investig. Opthalmol. Vis. Sci. 2012, 53, 2476–2481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vrabec, J.P.; Levin, L.A. The neurobiology of cell death in glaucoma. Eye 2007, 21, S11–S14. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.C.; Guo, Y.; Cepurna, W.O.; Morrison, J.C. Neurotrophin roles in retinal ganglion cell survival: Lessons from rat glaucoma models. Exp. Eye Res. 2009, 88, 808–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lom, B.; Cogen, J.; Sanchez, A.L.; Vu, T.; Cohen-Cory, S. Local and target-derived brain-derived neurotrophic factor exert opposing effects on the dendritic arborization of retinal ganglion cells in vivo. J. Neurosci. 2002, 22, 7639–7649. [Google Scholar] [CrossRef] [Green Version]
- van Oterendorp, C.; Sgouris, S.; Schallner, N.; Biermann, J.; Lagrèze, W.A. Retrograde neurotrophic signaling in rat retinal ganglion cells is transmitted via the ERK5 but not the ERK1/2 pathway. Investig. Ophthalmol. Vis. Sci. 2014, 55, 658–665. [Google Scholar] [CrossRef] [Green Version]
- Claes, M.; De Groef, L.; Moons, L. Target-derived neurotrophic factor deprivation puts retinal ganglion cells on death row: Cold hard evidence and caveats. Int. J. Mol. Sci. 2019, 20, 4314. [Google Scholar] [CrossRef] [Green Version]
- Weber, A.J.; Viswanáthan, S.; Ramanathan, C.; Harman, C.D. Combined application of BDNF to the eye and brain enhances ganglion cell survival and function in the cat after optic nerve injury. Investig. Ophthalmol. Vis. Sci. 2010, 51, 327–334. [Google Scholar] [CrossRef] [Green Version]
- West, A.E.; Pruunsild, P.; Timmusk, T. Neurotrophins: Transcription and translation. Handb. Exp. Pharmacol. 2014, 220, 67–100. [Google Scholar] [CrossRef] [PubMed]
- Corredor, R.G.; Goldberg, J.L. Electrical activity enhances neuronal survival and regeneration. J. Neural Eng. 2009, 6, 055001. [Google Scholar] [CrossRef] [PubMed]
- Kolarow, R.; Kuhlmann, C.R.W.; Munsch, T.; Zehendner, C.; Brigadski, T.; Luhmann, H.J.; Lessmann, V. BDNF-induced nitric oxide signals in cultured rat hippocampal neurons: Time course, mechanism of generation, and effect on neurotrophin secretion. Front. Cell. Neurosci. 2014, 8, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geeraerts, E.; Claes, M.; Dekeyster, E.; Salinas-Navarro, M.; de Groef, L.; van den Haute, C.; Scheyltjens, I.; Baekelandt, V.; Arckens, L.; Moons, L. Optogenetic stimulation of the superior colliculus confers retinal neuroprotection in a mouse glaucoma model. J. Neurosci. 2019, 39, 2313–2325. [Google Scholar] [CrossRef] [Green Version]
- Armbruster, B.N.; Li, X.; Pausch, M.H.; Herlitze, S.; Roth, B.L. Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc. Natl. Acad. Sci. USA 2007, 104, 5163–5168. [Google Scholar] [CrossRef] [Green Version]
- Roth, B.L. DREADDs for Neuroscientists. Neuron 2016, 89, 683–694. [Google Scholar] [CrossRef] [Green Version]
- Claes, M.; De Groef, L.; Moons, L. The DREADDful Hurdles and Opportunities of the Chronic Chemogenetic Toolbox. Cells 2022, 11, 1110. [Google Scholar] [CrossRef]
- Tran, N.M.; Shekhar, K.; Whitney, I.E.; Jacobi, A.; Benhar, I.; Hong, G.; Yan, W.; Adiconis, X.; Arnold, M.E.; Lee, J.M.; et al. Single-Cell Profiles of Retinal Ganglion Cells Differing in Resilience to Injury Reveal Neuroprotective Genes. Neuron 2019, 104, 1039–1055.e12. [Google Scholar] [CrossRef]
- Van der Perren, A.; Toelen, J.; Carlon, M.; Van den Haute, C.; Coun, F.; Heeman, B.; Reumers, V.; Vandenberghe, L.H.; Wilson, J.M.; Debyser, Z.; et al. Efficient and stable transduction of dopaminergic neurons in rat substantia nigra by rAAV 2/1, 2/2, 2/5, 2/6.2, 2/7, 2/8 and 2/9. Gene Ther. 2011, 18, 517–527. [Google Scholar] [CrossRef]
- Libbrecht, S.; Van den Haute, C.; Welkenhuysen, M.; Braeken, D.; Haesler, S.; Baekelandt, V. Chronic chemogenetic stimulation of the anterior olfactory nucleus reduces newborn neuron survival in the adult mouse olfactory bulb. J. Neurochem. 2021, 158, 1186–1198. [Google Scholar] [CrossRef]
- Krashes, M.J.; Koda, S.; Ye, C.; Rogan, S.C.; Adams, A.C.; Cusher, D.S.; Maratos-Flier, E.; Roth, B.L.; Lowell, B.B. Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J. Clin. Investig. 2011, 121, 1424–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Gucht, E.; Vandenbussche, E.; Orban, G.A.; Vandesande, F.; Arckens, L. A new cat Fos antibody to localize the immediate early gene c-fos in mammalian visual cortex after sensory stimulation. J. Histochem. Cytochem. 2000, 48, 671–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claes, M.; Santos, J.R.F.; Masin, L.; Cools, L.; Davis, B.M.; Arckens, L.; Farrow, K.; De Groef, L.; Moons, L. A fair assessment of evaluation tools for the murine microbead occlusion model of glaucoma. Int. J. Mol. Sci. 2021, 22, 5633. [Google Scholar] [CrossRef]
- Park, Y.H.; Snook, J.D.; Ostrin, E.J.; Kim, S.; Chen, R.; Frankfort, B.J. Transcriptomic profiles of retinal ganglion cells are defined by the magnitude of intraocular pressure elevation in adult mice. Sci. Rep. 2019, 9, 2594. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/ (accessed on 23 May 2022).
- Aronesty, E. ea-utils: Command-line tools for processing biological sequencing data. Expr. Anal. Durham 2011. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq--A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Putri, G.; Anders, S.; Pyl, P.; Pimanda, J.; Zanini, F. Analysing high-throughput sequencing data with HTSeq 2.0. arXiv 2021, arXiv:2112.00939. [Google Scholar] [CrossRef]
- Chen, Y.; Lun, A.T.L.; Smyth, G.K. From reads to genes to pathways: Differential expression analysis of RNA-Seq experiments using Rsubread and the edgeR quasi-likelihood pipeline. F1000Research 2016, 5, 1438. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krämer, A.; Green, J.; Pollard, J.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.; Tumkaya, T.; Aryal, S.; Choi, H.; Claridge-Chang, A. Moving beyond P values: Data analysis with estimation graphics. Nat. Methods 2019, 16, 565–566. [Google Scholar] [CrossRef]
- da Penha Berzaghi, M.; Cooper, J.; Castren, E.; Zafra, F.; Sofroniew, M.; Thoenen, H.; Lindholm, D. Cholinergic regulation of brain-derived neurotrophic factor (BDNF) and nerve growth factor (NGF) but not neurotrophin-3 (NT-3) mRNA levels in the developing rat hippocampus. J. Neurosci. 1993, 13, 3818–3826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palomer, E.; Carretero, J.; Benvegnù, S.; Dotti, C.G.; Martin, M.G. Neuronal activity controls Bdnf expression via Polycomb de-repression and CREB/CBP/JMJD3 activation in mature neurons. Nat. Commun. 2016, 7, 11081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, A.; Carnahan, J.; Greenberg, M. Requirement for BDNF in activity-dependent survival of cortical neurons. Science 1994, 263, 1618–1623. [Google Scholar] [CrossRef]
- Tao, X.; Finkbeiner, S.; Arnold, D.B.; Shaywitz, A.J.; Greenberg, M.E. Ca2+ Influx Regulates BDNF Transcription by a CREB Family Transcription Factor-Dependent Mechanism. Neuron 1998, 20, 709–726. [Google Scholar] [CrossRef] [Green Version]
- Nagappan, G.; Zaitsev, E.; Senatorov, V.V.; Yang, J.; Hempstead, B.L.; Lu, B. Control of extracellular cleavage of ProBDNF by high frequency neuronal activity. Proc. Natl. Acad. Sci. USA 2009, 106, 1267–1272. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, N.; Lu, H.; Fukata, Y.; Noritake, J.; Gao, H.; Mukherjee, S.; Nemoto, T.; Fukata, M.; Poo, M.M. Differential activity-dependent secretion of brain-derived neurotrophic factor from axon and dendrite. J. Neurosci. 2009, 29, 14185–14198. [Google Scholar] [CrossRef] [Green Version]
- Kuczewski, N.; Porcher, C.; Gaiarsa, J.L. Activity-dependent dendritic secretion of brain-derived neurotrophic factor modulates synaptic plasticity. Eur. J. Neurosci. 2010, 32, 1239–1244. [Google Scholar] [CrossRef]
- Kim, C.K.; Sanchez, M.I.; Hoerbelt, P.; Fenno, L.E.; Malenka, R.C.; Deisseroth, K.; Ting, A.Y. A Molecular Calcium Integrator Reveals a Striatal Cell Type Driving Aversion. Cell 2020, 183, 2003–2019.e16. [Google Scholar] [CrossRef] [PubMed]
- Magby, J.P.; Bi, C.; Chen, Z.-Y.; Lee, F.S.; Plummer, M.R. Single-Cell Characterization of Retrograde Signaling by Brain-Derived Neurotrophic Factor. J. Neurosci. 2006, 26, 13531–13536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer-Franke, A.; Wilkinson, G.A.; Kruttgen, A.; Hu, M.; Munro, E.; Hanson, M.G.; Reichardt, L.F.; Barres, B.A. Depolarization and cAMP elevation rapidly recruit TrkB to the plasma membrane of CNS neurons. Neuron 1998, 21, 681–693. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Feng, L.; Yang, F.; Lu, B. Activity- and Ca2+-dependent modulation of surface expression of brain-derived neurotrophic factor receptors in hippocampal neurons. J. Cell Biol. 2000, 150, 1423–1434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagappan, G.; Lu, B. Activity-dependent modulation of the BDNF receptor TrkB: Mechanisms and implications. Trends Neurosci. 2005, 28, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Hallock, H.L.; Quillian, H.M.; Maynard, K.R.; Mai, Y.; Chen, H.-Y.; Hamersky, G.R.; Shin, J.H.; Maher, B.J.; Jaffe, A.E.; Martinowich, K. Molecularly Defined Hippocampal Inputs Regulate Population Dynamics in the Prelimbic Cortex to Suppress Context Fear Memory Retrieval. Biol. Psychiatry 2020, 88, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Milibari, L.; Pan, J.-X.; Ren, X.; Yao, L.-L.; Zhao, Y.; Shen, C.; Chen, W.-B.; Tang, F.-L.; Lee, D.; et al. Critical Roles of Embryonic Born Dorsal Dentate Granule Neurons for Activity-Dependent Increases in BDNF, Adult Hippocampal Neurogenesis, and Antianxiety-like Behaviors. Biol. Psychiatry 2021, 89, 600–614. [Google Scholar] [CrossRef]
- Xia, S.-H.; Hu, S.-W.; Ge, D.-G.; Liu, D.; Wang, D.; Zhang, S.; Zhang, Q.; Yuan, L.; Li, Y.-Q.; Yang, J.-X.; et al. Priority Communication Chronic Pain Impairs Memory Formation via Disruption of Neurogenesis Mediated by Mesohippocampal Brain-Derived Neurotrophic Factor Signaling. Biol. Psychiatry 2020, 88, 597–610. [Google Scholar] [CrossRef]
- Blázquez, C.; Chiarlone, A.; Bellocchio, L.; Resel, E.; Pruunsild, P.; García-Rincón, D.; Sendtner, M.; Timmusk, T.; Lutz, B.; Galve-Roperh, I.; et al. The CB1 cannabinoid receptor signals striatal neuroprotection via a PI3K/Akt/mTORC1/BDNF pathway. Cell Death Differ. 2015, 22, 1618–1629. [Google Scholar] [CrossRef]
- Xiu, J.; Han, R.; Liu, Z.; Li, J.; Liu, S.; Shen, Y.; Ding, Y.-Q.; Xu, Q. Hijacking Dorsal Raphe to Improve Metabolism and Depression-Like Behaviors via BDNF Gene Transfer in Mice. Diabetes 2021, 70, 1780–1793. [Google Scholar] [CrossRef]
- Zweifel, L.S.; Kuruvilla, R.; Ginty, D.D. Functions and mechanisms of retrograde neurotrophin signalling. Nat. Rev. Neurosci. 2005, 6, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Harrington, A.W.; Ginty, D.D. Long-distance retrograde neurotrophic factor signalling in neurons. Nat. Rev. Neurosci. 2013, 14, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Vermehren-Schmaedick, A.; Olah, M.J.; Ramunno-Johnson, D.; Lidke, K.A.; Cohen, M.S.; Vu, T.Q. Molecular-Scale Dynamics of Long Range Retrograde Brain-Derived Neurotrophic Factor Transport Shaped by Cellular Spatial Context. Front. Neurosci. 2022, 16, 835815. [Google Scholar] [CrossRef]
- Moya-Alvarado, G.; Guerra, M.V.; Wu, C.; Mobley, W.C.; Perlson, E.; Bronfman, F.C. BDNF/TrkB signaling endosomes in axons coordinate CREB/mTOR activation and protein synthesis in the cell body to induce dendritic growth in cortical neurons. bioRxiv 2022. [Google Scholar] [CrossRef]
- Takihara, Y.; Inatani, M.; Hayashi, H.; Adachi, N.; Iwao, K.; Inoue, T.; Iwao, M.; Tanihara, H. Dynamic imaging of axonal transport in living retinal ganglion cells in vitro. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3039–3045. [Google Scholar] [CrossRef]
- Pease, M.E.; McKinnon, S.J.; Quigley, H.A.; Kerrigan-Baumrind, L.A.; Zack, D.J. Obstructed axonal transport of BDNF and its receptor TrkB in experimental glaucoma. Investig. Ophthalmol. Vis. Sci. 2000, 41, 764–774. [Google Scholar]
- Quigley, H.A.; McKinnon, S.J.; Zack, D.J.; Pease, M.E.; Kerrigan-Baumrind, L.A.; Kerrigan, D.F.; Mitchell, R.S. Retrograde axonal transport of BDNF in retinal ganglion cells is blocked by acute IOP elevation in rats. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3460–3466. [Google Scholar]
- Yan, Q.; Wang, J.; Matheson, C.R.; Urich, J.L. Glial cell line-derived neurotrophic factor (GDNF) promotes the survival of axotomized retinal ganglion cells in adult rats: Comparison to and combination with brain-derived neurotrophic factor (BDNF). J. Neurobiol. 1999, 38, 382–390. [Google Scholar] [CrossRef]
- Du, J.L.; Poo, M.M. Rapid BDNF-induced retrograde synaptic modification in a developing retinotectal system. Nature 2004, 429, 878–883. [Google Scholar] [CrossRef]
- Chou, T.-H.; Park, K.K.; Luo, X.; Porciatti, V. Retrograde Signaling in the Optic Nerve Is Necessary for Electrical Responsiveness of Retinal Ganglion Cells. Investig. Opthalmol. Vis. Sci. 2013, 54, 1236. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Cai, Q.; Xie, Y.; Sheng, Z.-H. Snapin Recruits Dynein to BDNF-TrkB Signaling Endosomes for Retrograde Axonal Transport and Is Essential for Dendrite Growth of Cortical Neurons. Cell Rep. 2012, 2, 42–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, F.L.; Heerssen, H.M.; Moheban, D.B.; Lin, M.Z.; Sauvageot, C.M.; Bhattacharyya, A.; Pomeroy, S.L.; Segal, R.A. Rapid Nuclear Responses to Target-Derived Neurotrophins Require Retrograde Transport of Ligand–Receptor Complex. J. Neurosci. 1999, 19, 7889–7900. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Poo, M. Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 2013, 14, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Boia, R.; Ruzafa, N.; Aires, I.D.; Pereiro, X.; Ambrósio, A.F.; Vecino, E.; Santiago, A.R. Neuroprotective Strategies for Retinal Ganglion Cell Degeneration: Current Status and Challenges Ahead. Int. J. Mol. Sci. 2020, 21, 2262. [Google Scholar] [CrossRef] [Green Version]
- Shibuki, H.; Katai, N.; Kuroiwa, S.; Kurokawa, T.; Arai, J.; Matsumoto, K.; Nakamura, T.; Yoshimura, N. Expression and neuroprotective effect of hepatocyte growth factor in retinal ischemia-reperfusion injury. Investig. Ophthalmol. Vis. Sci. 2002, 43, 528–536. [Google Scholar]
- Tönges, L.; Ostendorf, T.; Lamballe, F.; Genestine, M.; Dono, R.; Koch, J.C.; Bähr, M.; Maina, F.; Lingor, P. Hepatocyte growth factor protects retinal ganglion cells by increasing neuronal survival and axonal regeneration in vitro and in vivo. J. Neurochem. 2011, 117, 892–903. [Google Scholar] [CrossRef]
- Wong, W.K.; Cheung, A.W.S.; Yu, S.W.; Sha, O.; Cho, E.Y.P. Hepatocyte Growth Factor Promotes Long-Term Survival and Axonal Regeneration of Retinal Ganglion Cells after Optic Nerve Injury: Comparison with CNTF and BDNF. CNS Neurosci. Ther. 2014, 20, 916–929. [Google Scholar] [CrossRef]
- Jaiswal, P.; Mistretta, O.; Ward, P.; English, A. Chemogenetic Enhancement of Axon Regeneration Following Peripheral Nerve Injury in the SLICK-A Mouse. Brain Sci. 2018, 8, 93. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.H.A.; Stafford, B.K.; Nguyen, P.L.; Lien, B.V.; Wang, C.; Zukor, K.; He, Z.; Huberman, A.D. Neural activity promotes long-distance, target-specific regeneration of adult retinal axons. Nat. Neurosci. 2016, 19, 1073–1084. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Jin, Y.; Shapiro, T.M.; Hinduja, A.; Baas, P.W.; Tom, V.J. Chronic neuronal activation increases dynamic microtubules to enhance functional axon regeneration after dorsal root crush injury. Nat. Commun. 2020, 11, 6131. [Google Scholar] [CrossRef]
- Varadarajan, S.G.; Dhande, O.S.; Le, P.; Huberman, A.D. Probing the role of retinorecipient target cells in visual circuit regeneration. In Proceedings of the 2021 Neuroscience Meeting Planner, Chicago, IL, USA, 8–11 November 2021. [Google Scholar]
- Fischer, D. Hyper-IL-6: A potent and efficacious stimulator of RGC regeneration. Eye 2017, 31, 173–178. [Google Scholar]
- Chidlow, G.; Wood, J.P.M.; Ebneter, A.; Casson, R.J. Interleukin-6 is an efficacious marker of axonal transport disruption during experimental glaucoma and stimulates neuritogenesis in cultured retinal ganglion cells. Neurobiol. Dis. 2012, 48, 568–581. [Google Scholar] [CrossRef] [PubMed]
- Leibinger, M.; Müller, A.; Gobrecht, P.; Diekmann, H.; Andreadaki, A.; Fischer, D. Interleukin-6 contributes to CNS axon regeneration upon inflammatory stimulation. Cell Death Dis. 2013, 4, e609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wareham, L.K.; Echevarria, F.D.; Sousa, J.L.; Konlian, D.O.; Dallas, G.; Formichella, C.R.; Sankaran, P.; Goralski, P.J.; Gustafson, J.R.; Sappington, R.M. Interleukin-6 promotes microtubule stability in axons via Stat3 protein–protein interactions. iScience 2021, 24, 103141. [Google Scholar] [CrossRef] [PubMed]
- Leibinger, M.; Andreadaki, A.; Diekmann, H.; Fischer, D. Neuronal STAT3 activation is essential for CNTF- and inflammatory stimulation-induced CNS axon regeneration. Cell Death Dis. 2013, 4, e805. [Google Scholar] [CrossRef] [PubMed]
- Pernet, V.; Joly, S.; Jordi, N.; Dalkara, D.; Guzik-Kornacka, A.; Flannery, J.G.; Schwab, M.E. Misguidance and modulation of axonal regeneration by Stat3 and Rho/ROCK signaling in the transparent optic nerve. Cell Death Dis. 2013, 4, e734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenzetto, E.; Ettorre, M.; Pontelli, V.; Bolomini-Vittori, M.; Bolognin, S.; Zorzan, S.; Laudanna, C.; Buffelli, M. Rac1 Selective Activation Improves Retina Ganglion Cell Survival and Regeneration. PLoS ONE 2013, 8, e64350. [Google Scholar] [CrossRef]
- Morquette, B.; Morquette, P.; Agostinone, J.; Feinstein, E.; McKinney, R.A.; Kolta, A.; Di Polo, A. REDD2-mediated inhibition of mTOR promotes dendrite retraction induced by axonal injury. Cell Death Differ. 2015, 22, 612–625. [Google Scholar] [CrossRef]
- Feng, L.; Zhao, Y.; Yoshida, M.; Chen, H.; Yang, J.F.; Kim, T.S.; Cang, J.; Troy, J.B.; Liu, X. Sustained Ocular Hypertension Induces Dendritic Degeneration of Mouse Retinal Ganglion Cells That Depends on Cell Type and Location. Investig. Opthalmol. Vis. Sci. 2013, 54, 1106. [Google Scholar] [CrossRef]
- Agostinone, J.; Alarcon-Martinez, L.; Gamlin, C.; Yu, W.-Q.; Wong, R.O.L.; Di Polo, A. Insulin signalling promotes dendrite and synapse regeneration and restores circuit function after axonal injury. Brain 2018, 141, 1963–1980. [Google Scholar] [CrossRef]
- Kalesnykas, G.; Oglesby, E.N.; Zack, D.J.; Cone, F.E.; Steinhart, M.R.; Tian, J.; Pease, M.E.; Quigley, H.A. Retinal Ganglion Cell Morphology after Optic Nerve Crush and Experimental Glaucoma. Investig. Opthalmol. Vis. Sci. 2012, 53, 3847. [Google Scholar] [CrossRef]
- Leung, C.K.; Weinreb, R.N.; Li, Z.W.; Liu, S.; Lindsey, J.D.; Choi, N.; Liu, L.; Cheung, C.Y.; Ye, C.; Qiu, K.; et al. Long-Term In Vivo Imaging and Measurement of Dendritic Shrinkage of Retinal Ganglion Cells. Investig. Opthalmol. Vis. Sci. 2011, 52, 1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mak, H.K.; Ng, S.H.; Ren, T.; Ye, C.; Leung, C.K. Impact of PTEN/SOCS3 deletion on amelioration of dendritic shrinkage of retinal ganglion cells after optic nerve injury. Exp. Eye Res. 2020, 192, 107938. [Google Scholar] [CrossRef] [PubMed]
- Jakobs, T.C.; Libby, R.T.; Ben, Y.; John, S.W.M.; Masland, R.H. Retinal ganglion cell degeneration is topological but not cell type specific in DBA/2J mice. J. Cell Biol. 2005, 171, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.T.; Faulkner-Jones, B.E.; Sanes, J.R.; Wong, R.O.L. Rapid Dendritic Remodeling in the Developing Retina: Dependence on Neurotransmission and Reciprocal Regulation by Rac and Rho. J. Neurosci. 2000, 20, 5024–5036. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, R.; Luo, D.; Park, S.-J.; Wang, Q.; Kim, Y.; Min, W. Tumor Necrosis Factor α-induced Desumoylation and Cytoplasmic Translocation of Homeodomain-interacting Protein Kinase 1 Are Critical for Apoptosis Signal-regulating Kinase 1-JNK/p38 Activation. J. Biol. Chem. 2005, 280, 15061–15070. [Google Scholar] [CrossRef] [Green Version]
- Izetti, P.; Hautefeuille, A.; Abujamra, A.L.; de Farias, C.B.; Giacomazzi, J.; Alemar, B.; Lenz, G.; Roesler, R.; Schwartsmann, G.; Osvaldt, A.B.; et al. PRIMA-1, a mutant p53 reactivator, induces apoptosis and enhances chemotherapeutic cytotoxicity in pancreatic cancer cell lines. Investig. New Drugs 2014, 32, 783–794. [Google Scholar] [CrossRef]
- Guo, C.; Cho, K.-S.; Li, Y.; Tchedre, K.; Antolik, C.; Ma, J.; Chew, J.; Utheim, T.P.; Huang, X.A.; Yu, H.; et al. IGFBPL1 Regulates Axon Growth through IGF-1-mediated Signaling Cascades. Sci. Rep. 2018, 8, 2054. [Google Scholar] [CrossRef] [Green Version]
- Panza, P.; Sitko, A.A.; Maischein, H.-M.; Koch, I.; Flötenmeyer, M.; Wright, G.J.; Mandai, K.; Mason, C.A.; Söllner, C. The LRR receptor Islr2 is required for retinal axon routing at the vertebrate optic chiasm. Neural Dev. 2015, 10, 23. [Google Scholar] [CrossRef] [Green Version]
- Mandai, K.; Guo, T.; St. Hillaire, C.; Meabon, J.S.; Kanning, K.C.; Bothwell, M.; Ginty, D.D. LIG Family Receptor Tyrosine Kinase-Associated Proteins Modulate Growth Factor Signals during Neural Development. Neuron 2009, 63, 614–627. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, K.A.; Harder, J.M.; Kim, J.; Libby, R.T. JUN regulates early transcriptional responses to axonal injury in retinal ganglion cells. Exp. Eye Res. 2013, 112, 106–117. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Liu, F.; Niu, Q.; Li, Y.; Liu, C.; Zhang, L.; Ni, D.; Pu, X. GLIPR-2 Overexpression in HK-2 Cells Promotes Cell EMT and Migration through ERK1/2 Activation. PLoS ONE 2013, 8, e58574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Pernet, V.; Hauswirth, W.W.; Di Polo, A. Activation of the Extracellular Signal-Regulated Kinase 1/2 Pathway by AAV Gene Transfer Protects Retinal Ganglion Cells in Glaucoma. Mol. Ther. 2005, 12, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Boyden, E.S.; Zhang, F.; Bamberg, E.; Nagel, G.; Deisseroth, K. Millisecond-timescale, genetically targeted optical control of neural activity. Nat. Neurosci. 2005, 8, 1263–1268. [Google Scholar] [CrossRef] [PubMed]
- Bockaert, J.; Pin, J.P. Molecular tinkering of G protein-coupled receptors: An evolutionary success. EMBO J. 1999, 18, 1723–1729. [Google Scholar] [CrossRef] [Green Version]
- Atasoy, D.; Sternson, S.M. Chemogenetic tools for causal cellular and neuronal biology. Physiol. Rev. 2018, 98, 391–418. [Google Scholar] [CrossRef]
- Rajagopal, S.; Shenoy, S.K. GPCR desensitization: Acute and prolonged phases. Cell. Signal. 2018, 41, 9–16. [Google Scholar] [CrossRef]
- Gainetdinov, R.R.; Premont, R.T.; Bohn, L.M.; Lefkowitz, R.J.; Caron, M.G. DESENSITIZATION OF G PROTEIN–COUPLED RECEPTORS AND NEURONAL FUNCTIONS. Annu. Rev. Neurosci. 2004, 27, 107–144. [Google Scholar] [CrossRef] [Green Version]
- Schöneberg, T. Tolerance and Desensitization. In Encyclopedia of Molecular Pharmacology; Springer: Berlin/Heidelberg, Germany, 2008; pp. 1203–1207. [Google Scholar]
- Black, J.B.; Premont, R.T.; Daaka, Y. Feedback regulation of G protein-coupled receptor signaling by GRKs and arrestins. Semin. Cell Dev. Biol. 2016, 50, 95–104. [Google Scholar] [CrossRef] [Green Version]
- Dekeyster, E.; Geeraerts, E.; Buyens, T.; Van den Haute, C.; Baekelandt, V.; De Groef, L.; Salinas-Navarro, M.; Moons, L. Tackling Glaucoma from within the Brain: An Unfortunate Interplay of BDNF and TrkB. PLoS ONE 2015, 10, e0142067. [Google Scholar] [CrossRef]
- Di Polo, A.; Aigner, L.J.; Dunn, R.J.; Bray, G.M.; Aguayo, A.J. Prolonged delivery of brain-derived neurotrophic factor by adenovirus-infected Muller cells temporarily rescues injured retinal ganglion cells. Proc. Natl. Acad. Sci. USA 1998, 95, 3978–3983. [Google Scholar] [CrossRef] [Green Version]
- Osborne, A.; Khatib, T.Z.; Songra, L.; Barber, A.C.; Hall, K.; Kong, G.Y.X.; Widdowson, P.S.; Martin, K.R. Neuroprotection of retinal ganglion cells by a novel gene therapy construct that achieves sustained enhancement of brain-derived neurotrophic factor/tropomyosin-related kinase receptor-B signaling. Cell Death Dis. 2018, 9, 1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, L.; Sapieha, P.; Kittlerová, P.; Hauswirth, W.W.; Di Polo, A. TrkB Gene Transfer Protects Retinal Ganglion Cells from Axotomy-Induced Death In Vivo. J. Neurosci. 2002, 22, 3977–3986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuda, M.; Tanaka, Y.; Ryu, M.; Tsuda, S.; Nakazawa, T. RNA Sequence Reveals Mouse Retinal Transcriptome Changes Early after Axonal Injury. PLoS ONE 2014, 9, e93258. [Google Scholar] [CrossRef]
- Ueki, Y.; Reh, T.A. Activation of BMP-Smad1/5/8 Signaling Promotes Survival of Retinal Ganglion Cells after Damage In Vivo. PLoS ONE 2012, 7, e38690. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G. TNF-α signaling in glaucomatous neurodegeneration. Prog. Brain Res. 2008, 173, 409–421. [Google Scholar]
- Cheng, L.; Yu, H.; Yan, N.; Lai, K.; Xiang, M. Hypoxia-Inducible Factor-1α Target Genes Contribute to Retinal Neuroprotection. Front. Cell. Neurosci. 2017, 11, 20. [Google Scholar] [CrossRef]
- Lim, J.C.; Lu, W.; Beckel, J.M.; Mitchell, C.H. Neuronal Release of Cytokine IL-3 Triggered by Mechanosensitive Autostimulation of the P2X7 Receptor Is Neuroprotective. Front. Cell. Neurosci. 2016, 10, 270. [Google Scholar] [CrossRef]
- Perígolo-Vicente, R.; Ritt, K.; Gonçalves-De-Albuquerque, C.F.; Castro-Faria-Neto, H.C.; Paes-De-Carvalho, R.; Giestal-De-Araujo, E. IL-6, A1 and A2aR: A crosstalk that modulates BDNF and induces neuroprotection. Biochem. Biophys. Res. Commun. 2014, 449, 477–482. [Google Scholar] [CrossRef] [Green Version]
- Schiapparelli, L.M.; Shah, S.H.; Ma, Y.; McClatchy, D.B.; Sharma, P.; Li, J.; Yates, J.R.; Goldberg, J.L.; Cline, H.T. The Retinal Ganglion Cell Transportome Identifies Proteins Transported to Axons and Presynaptic Compartments in the Visual System In Vivo. Cell Rep. 2019, 28, 1935–1947.e5. [Google Scholar] [CrossRef] [Green Version]
- Schiapparelli, L.M.; Sharma, P.; He, H.-Y.; Li, J.; Shah, S.H.; McClatchy, D.B.; Ma, Y.; Liu, H.-H.; Goldberg, J.L.; Yates, J.R.; et al. Proteomic screen reveals diverse protein transport between connected neurons in the visual system. Cell Rep. 2022, 38, 110287. [Google Scholar] [CrossRef]
- Edgar, R. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Upstream Regulator | Expr Log Ratio | Molecule Type | Activation z-Score | p-Value of Overlap | Target Molecules in Dataset |
|---|---|---|---|---|---|
| BMP2 | growth factor | −2.58 | 3.71 × 10−3 | Bmp4, Col10a1, Fgfr2, Grem1, Ifi16, Nfatc1, Pth1r, Tagln | |
| STAT3 | 0.19 | transcription regulator | −2.50 | 2.12 × 10−4 | Alas2, Bcl3, Cd274, Cd46, Ciita, Ect2, Fcgr1a, Ffar2, Hla-A, Ifi16 |
| SMARCB1 | 0.02 | transcription regulator | −2.24 | 7.12 × 10−3 | Bmp4, Fcgr1a, Fcgr2a, Fgfr2, Ifi16, Il12b, Sgcg |
| IFNAR1 | −0.23 | transmembrane receptor | −2.21 | 8.66 × 10−3 | Cd274, Ciita, Hla-A, Ifi16, Il12b, Socs1 |
| IL12B | −4.31 | cytokine | −2.20 | 5.00 × 10−4 | Il12b, Lta, Pik3ap1, S100a9, Socs1 |
| IL4 | −0.11 | cytokine | −2.12 | 2.29 × 10−4 | Bcl3, Cd274, Cd8a, Ciita, Col6a3, Csf2rb, Cybb, Ephb4, Fcgr1a, Fcgr2a |
| IL6 | cytokine | −2.02 | 2.77 × 10−5 | Afp, Avp, Bcl3, Cd274, Cd46, Ces1, Ciita, Csf2rb, Cybb, Fcgr1a | |
| BRD4 | kinase | −1.98 | 4.40 × 10−5 | Bcl3, Cd274, Fosl2, Insm1, Itga1, Loxl2, Map4k1, Mmachc, Pole2 |
| Upstream Regulator | Expr Log Ratio | Molecule Type | Activation z-Score | p-Value of Overlap | Target Molecules in Dataset |
|---|---|---|---|---|---|
| CREB1 | −0.23 | transcription regulator | 3.91 | 9.08 × 10−11 | Apold1, Csrnp1, Dio2, Dusp1, Egr1, Egr4, Errfi1, Fos, Fosb, Gpr3, Junb, Nr4a1, Nr4a3, Per1, Sik1, Tiparp |
| EGF | growth factor | 3.63 | 4.62 × 10−9 | Cxcl13, Dio2, Dusp1, Dusp4, Egr1, Errfi1, Fos, Fosb, Fosl2, Junb, Nr4a1, Nr4a3, Per1, Serpina3, Sh2b3 | |
| IL1B | 0.40 | cytokine | 3.51 | 7.74 × 10−5 | Csrnp1, Cxcl13, Ddit4, Dusp1, Egr1, Errfi1, Fos, Fosb, Junb, Nr4a1, Nr4a3, Serpina3, Slc2a1 |
| Insulin | group | 3.12 | 9.86 × 10−8 | Cox6a2, Ddit4, Dusp1, Dusp4, Egr1, Errfi1, Fos, Fosb, Junb, Lpin1, Nr4a1, Nr4a3, Per1, Slc2a1 | |
| CREB | group | 3.02 | 1.71 × 10−9 | Cox6a2, Dusp1, Dusp4, Egr1, Egr4, Fos, Fosb, Junb, Nr4a1, Nr4a3, Per1, Slc2a1 | |
| GPER1 | 0.28 | G-protein coupled receptor | 2.91 | 6.92 × 10−13 | Ddit4, Dusp1, Dusp4, Egr1, Errfi1, Fos, Fosb, Rasd1, Slc2a1 |
| IGF1 | 0.16 | growth factor | 2.90 | 2.10 × 10−4 | Ddit4, Dusp1, Egr1, Errfi1, Fos, Fosb, Junb, Nr4a1, Slc2a1 |
| TGFB1 | 0.31 | growth factor | 2.88 | 2.81 × 10−5 | Cxcl13, Cyp3a7, Ddit4, Dusp1, Dusp4, Egr1, Fos, Fosb, Fosl2, Junb, Klf2, Mboat2, Nr4a1, Nr4a3, Rasl11b, Rbpms, Serpina3, Slc2a1, Srsf2 |
| MAPK3 | −0.08 | kinase | 2.77 | 2.97 × 10−9 | Dusp4, Egr1, Egr4, Fos, Fosb, Fosl2, Junb, Nr4a1 |
| F2 | peptidase | 2.72 | 4.79 × 10−6 | Cxcl13, Dusp1, Egr1, Fos, Fosb, Fosl2, Junb, Nr4a3 | |
| TNF | 0.36 | cytokine | 2.66 | 9.25 × 10−5 | Cxcl13, Depp1, Dpys, Dusp1, Dusp4, Egr1, Fos, Fosb, Fosl2, Junb, Klf2, Nr4a1, Nr4a3, Rasd1, Rbpms, Serpina3, Slc2a1, Tst |
| FOXO3 | −0.13 | transcription regulator | 2.63 | 2.55 × 10−5 | Ddit4, Depp1, Egr1, Egr4, Fos, Fosb, Junb, Sh2b3 |
| RELA | 0.27 | transcription regulator | 2.61 | 2.34 × 10−4 | Dio2, Dusp1, Egr1, Fos, Fosb, Junb, Nr4a1, Nr4a3 |
| GNAS | enzyme | 2.60 | 9.85× 10−9 | Arl4d, Fos, Fosl2, Gdf9, Nr4a1, Nr4a3, Rasd1 | |
| IL6 | cytokine | 2.60 | 6.17× 10−3 | Cxcl13, Dusp1, Egr1, Errfi1, Fos, Junb, Lyz, Serpina3 | |
| HGF | 0.01 | growth factor | 2.58 | 1.63× 10−3 | Dusp4, Egr1, Errfi1, Fos, Fosb, Nr4a1, Nr4a3 |
| Pkc(s) | group | 2.57 | 1.07 × 10−5 | Dusp1, Egr1, Fos, Junb, Nr4a1, Nr4a3, Per1 | |
| BDNF | 0.03 | growth factor | 2.57 | 2.35 × 10−6 | Dusp1, Dusp4, Egr1, Fos, Fosb, Nr4a1, Nr4a3, Sik1, Tiparp |
| CSF1 | 0.24 | cytokine | 2.54 | 3.36 × 10−5 | Dusp1, Dusp4, Egr1, Fos, Junb, Klf2, Slc2a1 |
| NGF | −0.27 | growth factor | 2.48 | 3.04 × 10−7 | Dusp1, Dusp4, Egr1, Egr4, Errfi1, Fos, Fosl2, Junb, Nr4a1 |
| Upstream Regulator | Expr Log Ratio | Molecule Type | Activation z-Score | p-Value of Overlap | Target Molecules in Dataset |
|---|---|---|---|---|---|
| HIF1A | 0.47 | transcription regulator | 2.88 | 1.12 × 10−3 | Bmp2, Cavin2, Ccn3, Cdkn1a, Egfr, Eng, Fos, Gck, H4c15, Il17ra |
| BDNF | 0.26 | growth factor | 2.60 | 8.93 × 10−3 | Bmp2, Cdkn1a, Cebpb, Egr1, Egr2, Fos, Limk1, Mag, Nr4a1, Ryr1 |
| AGT | 1.70 | growth factor | 2.56 | 3.54 × 10−4 | Adam12, Avp, Bmp2, C5ar1, Ccn3, Cdc20, Cdkn1a, Cox7a1, Crlf1, Egfr |
| TGFBR2 | kinase | 2.55 | 7.71 × 10−3 | Bcat1, Bcl3, C15orf48, C5ar1, Cdkn1a, Cdkn2b, Runx2, Sdc1, Sh3bp2, Stc2 | |
| TNF | cytokine | 2.53 | 2.76 × 10−6 | Arrdc3, Avp, Bcl3, Bmp2, C15orf48, C5ar1, Card6, Cbr3, Ccn3, Cd55 | |
| FOXO3 | −0.31 | transcription regulator | 2.52 | 1.05 × 10−4 | Bmp2, Cdc20, Cdkn1a, Cdkn2b, Dok5, Egr1, Egr2, Fos, Lcp2, Mbnl1 |
| IL6 | cytokine | 2.45 | 1.44 × 10−4 | Avp, Bcl3, Bmp2, C5ar1, Cd74, Cdc20, Cdkn1a, Cdkn2b,Cebpb, Crlf1 | |
| NGF | growth factor | 2.40 | 1.59 × 10−3 | Cdkn1a, Dusp8, Egfr, Egr1, Egr2, Fos, Fosl2, Hrk, Nr4a1, Tnfr, sf12a | |
| MAPK3 | 0.01 | kinase | 2.39 | 3.20 × 10−4 | Cdkn1a, Cdkn2b, Egr1, Egr2, Fos, Fosl2, Nr4a1 |
| TGFB3 | 0.04 | growth factor | 2.36 | 1.65 × 10−5 | Adam12, Ccn3, Cdkn1a, Cdkn2b, Eng, Fos, Fosl2, Htra1, Sox9 |
| CSF2 | cytokine | 2.34 | 3.46 × 10−5 | Anln, Bcl3, C5ar1, Card6, Cd74, Cdc20, Cdkn1a, Cdkn2b,Dscc1, Egr1 | |
| CHUK | −0.16 | kinase | 2.21 | 6.41 × 10−5 | Bmp2, Ccn3, Cdkn1a, Cebpb, Crlf1, Fos, Hla-A, Il22, Mt1, Mt2 |
| Ngf | group | 2.21 | 1.44 × 10−3 | Cdkn1a, Egr1, Fos, Hrk, Nr4a1 | |
| FGF21 | growth factor | 2.21 | 1.09 × 10−3 | Avp, Cox7a1, Cpt1a, Egr1, Fos | |
| EP300 | 1.00 | transcription regulator | 2.20 | 1.26 × 10−4 | Adam12, Card6, Cdkn1a, Cdkn2b, Cpt1a, Egr1, Egr2, Fndc3b, Fos, Fosl2 |
| IKBKB | −0.44 | kinase | 2.16 | 9.56 × 10−4 | Bmp2, Cdkn1a, Cebpb, Egr1, Fos, Hla-A, Mt1, Mt2, Ogn, S100a4 |
| HGF | 1.12 | growth factor | 2.06 | 3.51 × 10−3 | Bmp2, Calcrl, Cdc20, Cdkn1a, Cdkn2b, Crlf1, Cry1, Dok5,Egr1, Emp2 |
| CXCL12 | 0.36 | cytokine | 2.05 | 1.41 × 10−6 | Avp, Bcl3, Bmp2, Egfr, Egr1, Fos, Itgax, Lcp2, Mmp11, Nr4a1 |
| IL3 | cytokine | 2.01 | 1.17 × 10−6 | Bmp2, Ccn3, Cdkn1a, Cpt1a, Egr1, Egr2, Fcer2, Fcgr2a, Fos, Fosl2 | |
| IKBKG | −0.42 | kinase | 1.97 | 3.25 × 10−4 | Bmp2, Cebpb, Fcer2, Fos, Hla-A, Mt1, Mt2, Ogn |
| NTRK1 | 0.38 | kinase | 1.96 | 1.88 × 10−4 | Cdkn1a, Egr1, Fos, Nr4a1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Claes, M.; Geeraerts, E.; Plaisance, S.; Mentens, S.; Van den Haute, C.; De Groef, L.; Arckens, L.; Moons, L. Chronic Chemogenetic Activation of the Superior Colliculus in Glaucomatous Mice: Local and Retrograde Molecular Signature. Cells 2022, 11, 1784. https://doi.org/10.3390/cells11111784
Claes M, Geeraerts E, Plaisance S, Mentens S, Van den Haute C, De Groef L, Arckens L, Moons L. Chronic Chemogenetic Activation of the Superior Colliculus in Glaucomatous Mice: Local and Retrograde Molecular Signature. Cells. 2022; 11(11):1784. https://doi.org/10.3390/cells11111784
Chicago/Turabian StyleClaes, Marie, Emiel Geeraerts, Stéphane Plaisance, Stephanie Mentens, Chris Van den Haute, Lies De Groef, Lut Arckens, and Lieve Moons. 2022. "Chronic Chemogenetic Activation of the Superior Colliculus in Glaucomatous Mice: Local and Retrograde Molecular Signature" Cells 11, no. 11: 1784. https://doi.org/10.3390/cells11111784